Abstract
The evaporation of N-Methyl-2-Pyrrolidone (NMP) solvent during the large-scale production of LiNiMnCoO (NMC) cathodes usually occurs in convection ovens. This paper aims to close the gap between the industrial convection drying method and the conventional vacuum oven typically used at the laboratory scale. Multiple studies focus on modeling convection dryers to reduce energy consumption, but few have studied their impact on the cathode quality experimentally and compared them to vacuum-dried cathodes. A convection oven designed for LIB electrode drying was developed to investigate the influence of drying kinetics on the formation of small electrode surface cracks (<1400 μm) and binder migration. The drying kinetics were revealed through thermogravimetric analysis (TGA) at drying temperatures of 50 and 100 °C and hot air velocities of 0.5 and 1 m/s. Even at these relatively low drying rates, structural differences were detected when comparing the two drying methods, illustrating the importance of implementing drying conditions that represent the industry process in laboratories. Surface cracking increased with drying rates, and cathodes with multiple cracks after calendering obtained a higher discharge capacity at discharge currents >C/2. An alternative surface analysis with less sample preparation was sufficient for determining the relative change in binder migration.
1. Introduction
Being the most used battery in mobile devices and battery electric vehicles (BEV’s), the global market for lithium-ion batteries (LIB) is expanding rapidly [1]. Several life cycle assessments (LCAs) have been conducted on BEVs to mitigate their greenhouse gas emissions (GHGs) [2]. The findings suggest that the production of LIBs contribute to a significant part of the BEV’s overall carbon footprint [3,4] and costs [1,5,6]. The main contributor to the GHGs associated with the LIBs is the electrode manufacturing process, where the drying of cathodes is amongst the most energy-demanding [7].
For decades, new battery materials and alternative battery technologies have been the main research focus for providing high-energy-density LIBs [8,9,10]. Since the LIBs produced for BEVs are starting to reach the theoretical capacity limit of the electrode materials [8], more attention is being directed towards the production route, such as alternatives to the wet-slurry electrode production processes [7,11,12,13]. Extrusion mixing is considered a cost-efficient mixing method as it allows for solvent reduction, whereas alternative drying methods such as laser-drying and infrared (IR) methods have recently been implemented on production lines for time efficiency. Furthermore, electrode spray drying (ESD) [14,15,16,17] and freeze-drying methods [18] have been explored as rapid production methods that enable a high-porosity electrode. More recent research [19,20,21,22] has investigated the possibility of the complete elimination of solvent to enable a dry electrode manufacturing route. This method eliminates the drying and recovery step of NMP and consequently reduces the energy consumption during production drastically. An overview of the challenges and advantages related to these state-of-the-art methods can be found in a recent review conducted by the authors [7].
The most common electrode drying methods in laboratories and factories are vacuum and convective drying, respectively [7,12]. Extensive research has been conducted on the convection drying processes in order to achieve high energy efficiency; however, little experimental work has been conducted on the drying kinetics for electrodes dried under convection at the laboratory scale [7]. Since the industry largely utilizes convective dryers, implementing such methods at the laboratory scale should be considered to avoid mismatches between production scales [7,12].
Studies have revealed that a small variation in drying conditions, such as temperature and air velocity, controls the solvent evaporation and dictates the final structure of the electrode. The major structural changes includes the crack formation [23,24] and binder migration [25,26,27]. These electrode quality parameters determine the electrode’s mechanical strength, and eventually, the specific discharge capacity and capacity loss [25].
There is a significant challenge when comparing the scientific results due to large variations in production parameters, such as the drying conditions, coating thickness, mass loading, and solvent-to-powder ratio. For example, the temperature typically varies between 80–130 °C during vacuum drying, whereas research on the convective drying method usually ranges between 20–180 °C [7,23,25]. The solvent-to-powder content usually varies from 30 to 60% depending on the mixing method [7]. A higher drying temperature or solvent content usually promotes cracking and binder migration, which results in a decrease in the cathode quality. Moreover, most studies have investigated cathodes with mass loading between 11–25 mg/cm [28]. For those with the highest loading, significant solvent gradients are usually detected during drying, which causes intensive binder migration and cracking even at drying temperatures of 20 °C [23]. Extensive research has been conducted to understand the underlying mechanism during the drying of anodes [24,29,30,31,32]; however, there is a lack of studies conducted on the underlying mechanisms during the drying of cathodes. Since the anode and cathode usually have a unique production route due to the different solvent chemistries and materials utilized, the chemical and physical interactions between the components during the production are expected to vary [6,30].
The drying mechanism during the solvent removal of a porous electrode coating shown in Figure 1 may be revealed through drying kinetics and is often represented in a drying rate curve [7]. The drying process includes the alteration of heat and mass transfer as the solvent evaporates, and consists of three stages. The first stage comprises the heating of the solvent, surface evaporation, and film shrinkage. The next stage is termed the constant drying rate and commences where the solvent evaporation is capillary-driven and no longer limited to the surface. At the end of the constant drying rate period, the critical moisture content (X) can be estimated. This is followed by the third stage, called the falling rate or pore-emptying, which is where the solvent evaporation is kinetically limited by the pores.
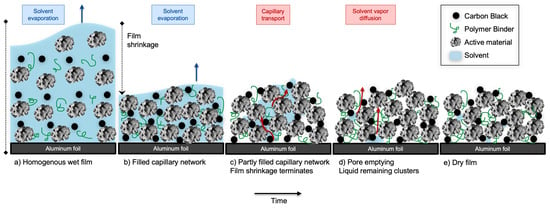
Figure 1.
The cross-section of electrode coating, and the drying mechanisms taking place during convection drying, inspired by Jaiser et al. [30] and copied from Bryntesen et al. [7]. First, the heating of the solvent takes place. Thereafter, the solvent evaporation from the surface and the inner mass flow is equal. The drying rate is constant, and film shrinkage occurs. After the solvent is removed from the surface, the dry layer formed on the surface interrupts the internal mass transfer. Pore-emptying takes place, which results in a falling drying rate.
Multiple studies have modeled the drying mechanism and altered the drying kinetics to optimize the energy efficiency related to the drying process. Oppegård et al. [33] modeled the first drying stage of the solvent evaporation; however, it is problematic to create an accurate estimation of the mechanism within these complex porous electrode structures. Furthermore, binder migration and cracking occur in two different stages, and these quality aspects complicate the optimization process. The binder migration mainly occurs in the first stage. As the solvent evaporates rapidly, a mass flow occurs in the upwards direction and causes the binder to migrate. The cracking mainly occurs at the end of the falling drying stage by the capillary pressure created in the channels during pore emptying. Westphal et al. [25,29] reported that the binder migration occurs in two subsequent mechanisms: the mass flow in the upwards direction due to solvent evaporation and the gravitational force in the downwards direction. These competing mechanisms depend on the binder’s molecular weight, the length of its polymer chains, and the drying rate.
Some studies have analyzed the electrode’s surface crack size as a quality measurement to understand its influence on the mechanical and electrochemical performance [34]. For example, Rollag et al. [23] studied the crack formation of aqueous processed cathodes with extensive cracks (80,000 μm) and neglected the small voids <10 μm. However, few studies have quantitatively investigated the effect of small crack sizes (<1400 μm) and classified them according to the length and width for NMP-processed NMC cathodes with low mass loading (<6 mg/cm). It is important to determine the minimal crack size at which cracking starts having a positive influence on the porosity and Li-diffusion, as this can potentially allow for a higher capacity when increasing the charge/discharge currents.
The analysis of binder migration normally takes place across the electrode’s cross-section, and typically includes advanced sample preparation methods [27,32]. When comparing cathodes of similar chemistries dried at different temperatures, a surface scan would simplify this quality control step and be a time- and cost-effective alternative in battery factories. Since thin electrodes with low mass loading tend to show a lower concentration of binder migration, the proof of principle for detecting the PVDF binder migration through the surface analysis method is tested on thin electrodes (<5.7 mg/cm). Additionally, few have studied the effect of small cracks on the cathodes’ rate capability and long-term performance. Larger cracks and more mechanically unstable coatings are often formed when fabricating electrodes with high mass loading, thus this study was conducted on relatively thin samples.
In the present work, we aim to understand the effect of drying kinetics on the surface cracking and binder migration of convection-dried cathodes and compare them to those dried under vacuum. This will help to develop a laboratory approach to studying the NMC cathode fabrication process typically used at industrial scale and help to define the limiting factors that influence the electrode’s quality. To provide a qualitative and quantitative analysis of the impact of cracks, they were classified according to size (μm), surface coverage (%), and quantity. The electrochemical performance was determined by the long-term capacity loss over 100 cycles and rate capability (at discharge currents up to 5 C). Additionally, PVDF binder migration was analyzed through the cross-section and over the surface of the electrodes using SEM/EDX. This was to propose a simplified, time-efficient, and cost-efficient method for detecting binder migration when comparing multiple cathodes. The experimental results were compared with a reference cathode dried in a vacuum at 90 °C, which is considered the most common laboratory method and is optimal in terms of electrode performance.
2. Materials and Methods
The materials and experimental procedures used for electrode manufacturing, drying kinetic analysis, and coin cell assembly are described below. The characterization methods used for the microstructural and electrochemical analysis of cathodes are presented.
2.1. Cathode Preparation
A pre-made solution of NMP/PVDF (20:1 wt%) was mixed (THINKY Corp. ARE-250, Lindberg & Lund AS, Vestby, Norway) with carbon black (CB), and NMC111 powder in an NMC111:CB:PVDF 85:10:5 wt% ratio for 25 min at 1500 rpm and 5 min at 2000 rpm to form a slurry. The slurry was coated at room temperature (22 °C) onto an aluminum foil (Al-foil, 15 μm thick) current collector with a doctor blade gap of 200 μm using a tape caster (MSK-AFA-HC100, MTI Corp., Richmond, CA, USA) Detailed information about the chemicals used is presented in Table 1.

Table 1.
Detailed list of materials and their suppliers used for the cathode production. Avg.Mw = average molecular weight, GPC = gel permeation chromatography.
2.2. Drying Setup
The electrodes were dried in the custom-designed convection drying chamber presented in Figure 2. Dried compressed air (5 bar, 0% moisture) was heated by an electrical heater (2 kW). The drying temperature (T-type thermocouple) and air velocity (Mass flow meter Alicat Scientific, Marana, AZ, USA) were controlled and logged (LabVIEW 20.0 Development System), while the weight of the electrode during solvent evaporation was measured (Mettler PM1200, METTLER TOLEDO) and logged (LabVIEW 20.0 Development System) to study the drying kinetics.
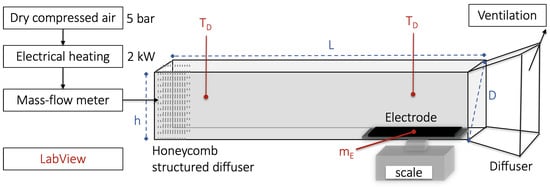
Figure 2.
Setup of the custom-made lab-scale convection oven. Dry compressed air (5 bar) was heated to a pre-defined temperature (T) using an electrical heater (2 kW) and blown with a pre-defined air velocity (V controlled by a mass flow meter. The honeycomb structured flow normalizer and the diffuser at the end ensure a uniform air-flow distribution at the inlet. The diameter (D) and length (L) of the oven were 17.15 cm and 100 cm. The height (h) was adjustable. The electrode was placed on an analytical scale and the mass loss (m) was recorded during solvent evaporation.
A temperature of 50 and 100 °C was tested, and the air velocity was kept low (1 m/s and 0.5 m/s) relative to large-scale driers (25 m/s). These drying conditions were chosen as they were amongst the upper and lower values within the temperature range commonly used industrially and in laboratories that provided the least fluctuations during the TGA measurement. This resulted in the five samples presented in Table 2. The mass reduction in NMP measured during drying was calculated relative to the dry basis (CB, PVDF, and NMC111) and the drying rate was calculated using Equation (1).
where m is the mass of dry solid (kg), W is the drying rate (kg/ms), X is the NMP content (kg of solvent per kg of dry electrode material, kg/kg), A is the top planar surface area of the electrode (m), and dt is the drying time (seconds) [35,36].
Table 2.
The drying conditions tested, including the respective sample names.
The Reynolds number was calculated using Equation (2) [37], where is the density of the fluid, V is the velocity of the fluid flow, is the viscosity of the fluid, and D is the diameter of the pipe that the fluid flows through. In the present study, the fluid was air, and electrodes were dried under turbulent (1 m/s) and transient (0.5 m/s) modes. The reference cathode (V0T90) production was conducted in a vacuum (3–10 mbar) using a dryer (Heraeus VT5042EK) and a vacuum pump (Pfeiffer Asslar PKD41029C). The drying temperature was set to 90 °C for 5 h.
2.3. Electrode Analysis
After drying, the electrode surface was analyzed for cracks using a field emission scanning electron microscopy (FESEM) apparatus (Zeiss Ultra 55VP) with an Everheart-Thornley Secondary Electron Detector, and a Bruker EDX/NORDIF EBSD system. The accelerating voltage was 10 kV, the working distance was 17–44 mm, and the magnitude was 300×. ImageJ (version 1.53) was used for surface analysis of the SEM images as illustrated in Figure 3. The threshold varied (between 50–100) and the particle analysis function was used to measure the crack area, which was categorized according to its size. The share of cracks for each size was calculated over the total crack area (%). For each temperature and air velocity, three parallel electrode coatings were tested. An area of ∼700 × 500 μm was analyzed for cracks in SEM at three different locations for each coating. The standard deviation between each drying condition was then calculated.

Figure 3.
SEM analysis of an electrode surface. The original scan is shown to the left, and the crack area (blue areas) as analyzed using ImageJ is shown to the right.
The scans were considered as representative for the entire electrode surface as two magnitudes larger and two magnitudes smaller than 300× were tested at three different locations on the V1T100 and V05T50 electrodes, and the chosen magnitude (300×) was the lowest magnitude at which there was no significant difference between the scanned locations. At a lower magnitude, similar results were obtained, but the smaller cracks were not detected. At higher magnitudes, the large cracks were not detected and there were larger deviations across scans in the same coating. These two electrodes were also analyzed three or five times at three different locations on the surface, and no significant difference was detected between the results when scanned three or five times. Three scan points were therefore considered sufficient.
Energy-dispersive X-ray spectroscopy (EDX) was conducted on the same apparatus with a Bruker XFlash EDX detector, and Bruker software version 1.9 using an accelerating voltage of 15 kV and a working distance of 8.5 mm. Elemental mapping (8–10 min scanning time) of fluorine (F), carbon (C), oxygen (O), and manganese (Mn) was conducted over the electrode top surface (2000× magnitude) and cross-section (2500× magnitude) at three different places. For the cross-section analysis, the cathode was cut in two on three different locations using a ceramic knife and the three pieces were mounted onto a sample holder using carbon tape and analyzed. The average of those three locations were calculated and presented. In order to ensure a representative scan and area, higher and lower magnitudes and scanning times were tested. The chosen magnitude and scanning time were the minimum at which the measurements resulted in similar values.
The electrodes were then calendered (Electric Hot Press, MSK-HRP-01, MTI Corp., Richmond, CA, USA) four times at room temperature down to ∼70% of its original thickness. The calendering process smoothed the electrode surfaces, and cracks disappeared from the surface, but not necessarily from the inner areas. Therefore, the surface crack analysis was performed before and after calendering. Further work should include the analysis of the inner cracks and cracking depth as these also likely relate to the rate capability. In this report, the inner voids/depth of cracks are only accounted for in the porosity calculation.
The dried cathodes were weighed, and the thickness was measured before and after calendering using a micrometer (VWR ± 0.001 μm). The calculated mass loading, thickness, and porosity before and after calendering are presented in Table 3. The porosity was defined as the ratio between the volume of the pores in the electrode and the total volume of the electrode itself, and was calculated according to:
where V is the total volume of the electrode, C is the ratio of each material in the electrode, W is the weight per unit area (loading), and is the density of each material. The porosity of the dried electrodes was calculated using the coating thickness and the theoretical material density of NMC111 (2.3 g/cm), CB (1.9 g/cm), and PVDF (1.7 g/cm). The cathodes were punched into 15 mm diameter cathode discs (MSK-T-12, MTI Corp., Richmond, CA, USA), and the amount of active material in each cathode was calculated from the electrode’s mass. The cathodes were post-dried at 120 °C for 4 h and transferred into a glove box for cell assembly.
Table 3.
The average mass loading (mg/cm), thickness (μm), and porosity (%) of the dried cathode coatings before and after calendering. There is no statistical difference between samples with identical labels (a,b).
2.4. Coin Cell Assembly
The coin cell assembly took place inside a glove box (MBraun, MB-Labmaster Pro SP 1250/780, Germany) filled with argon (Ar) atmosphere that maintained a level of moisture and oxygen < 0.1 ppm. Electrodes were tested in a coin cell, type CR2016 (MTI Corp., Richmond, CA, USA). Pre-cut lithium (Li) metal chips (99.9%, Tmax Battery Equipment, Xiamen, China) with a diameter and thickness of 15.6 mm and 0.25 mm, respectively, were cleaned with a plastic brush and used as the counter electrode (referred to as the anode). A porous polypropylene sheet (Celgard 2320, Charlotte, NC, USA) was used as a separator and placed between the anode and the cathode. The electrolyte was 1 M LiPF salt in a mixture of ethylene carbonate, ethyl-methyl carbonate, and diethyl carbonate (EC:DMC:DEC, 1:1:1). To achieve good contact and obtain uniform current distribution, a stainless steel spacer and spring were placed on top of the Li anode before crimping the cell using an automatic gas-driven coin cell crimper (MSK-PN110-S, MTI Corp., Richmond, CA, USA).
2.5. Electrochemical Analysis
All coin cells were cycled galvanostatically at different currents (C-rates) using an Arbin Battery Cycler (LBT Cell HC, model LBT211084) at 20 °C between 3.0–4.3 V vs. Li/Li according to the program presented in Table 4, where 1 C = 160 mA/g. Generally, cycling at discharging rates up to C/2 was combined with equal charging rates, whereas cycling at discharging rates above C/2 was combined with C/2 charging rates. All of the charging processes included a constant voltage step (CV) at the upper cut-off potential until the current decayed to C/10.
Table 4.
Galvanostatic cycling program with cut-off voltages of 3–4.3 V vs. Li/Li. Some cells had two additional cycles at C/10 before the 100 last cycles; however, this was considered to have a negligible effect on the results.
2.6. Statistical Analysis
The analysis of variance (ANOVA: single test and two-factor test with replication) was applied to analyze the effects of temperature and air velocity on the drying kinetics, structural changes, and electrochemical performance of the cathodes. The difference was considered significant at p < 0.05. The standard deviation was also calculated.
3. Results and Discussion
This section presents the drying kinetics, surface cracking analysis, binder migration, and electrochemical performance of four different cathodes dried using a convective drying method. These will be compared with the reference cathode dried under standard drying conditions for laboratory cathodes.
3.1. Drying Kinetics
A drying curve was obtained by recording the weight loss of NMP over time for the four different convection conditions. Figure 4 presents the typical drying curve recorded during the electrode drying used for drying kinetic analysis.
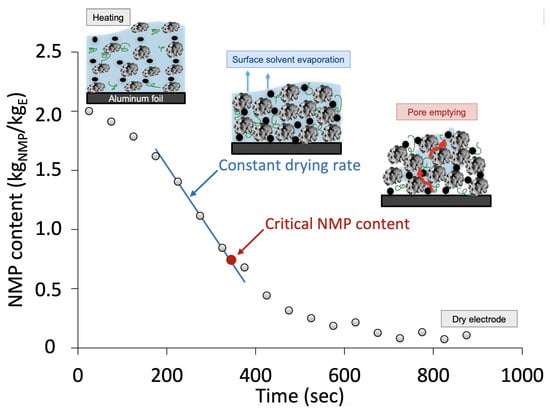
Figure 4.
A typical drying curve used for drying kinetic analysis. The critical NMP content (red) and linear regression revealing the constant drying rate (blue) are presented. This sample was dried at an air velocity of 1 m/s and temperature of 100 °C (V1T100).
The experiments showed a rapid temperature rise, meaning that the electrode drying was controlled by mass transfer rather than heat transfer. This was mainly because of the very thin electrode layer [38]. Three periods of drying were detected. The first period was characterized by an acceleration in the drying rate. When the drying rate reached its maximum value, the constant drying rate period began. The falling period was found at the end of drying and resembled the mass-transfer-controlled pore-emptying step. This is the time-consuming process shown in Figure 4, which occupied over 60% of the drying time. The same trend was observed for all of the experiments. Susarla et al. [38] analyzed the drying of cathodes with similar powder:solvent ratios (1:2) and wet thickness (158 μm) and found that approximately half of the drying time was dedicated to removing the last 10% of the solvent.
The drying kinetics parameters are presented in Table 5. The constant drying rate represented the fastest drying rate observed during electrode drying. From an industrial production point of view, it is beneficial to maintain a high constant drying rate during the whole drying process. Increasing the drying temperature from 50 to 100 °C and the air velocity from 0.5 to 1 m/s increased the drying rate (p < 0.05). Furthermore, an air velocity of 1 m/s resulted in a high standard deviation of the average values of the constant drying rate due to the weight-scale fluctuations.
Table 5.
The experimental drying kinetics for each convection drying condition and the percentage of the crack area on the electrode surface area (%) for each drying condition before and after calendering. There is no statistical difference between samples with identical labels (a).
The highest constant drying rate (2.93 × 10 kg/ms) was observed for the V1T100 cathode, which is lower than those normally reported from industry and other scientific studies [23,31,34]. The constant drying rate value for an industrial line varies between 5.0–11.9 × 10 kg/ms [31]. The critical NMP content is the remaining NMP concentration when the constant drying rate period is completed, and is where the internal and external mass transfer processes are equal. After reaching the critical NMP content, a dry layer is formed on the surface of the electrode. This reduces the internal mass transfer of NMP, and the drying rate gradually decreases below this NMP concentration. A low value of critical NMP content indicates a uniform NMP distribution throughout the electrode during drying. According to Table 5, the critical NMP content increases with the drying temperature and air velocity (p < 0.05). In other words, intensive drying will shift the critical NMP content to higher values. This was only true for cathodes dried at 100 °C; however, the air velocity did not impact the critical NMP content at 50 °C.
According to Font et al. [27], the Peclet number (Pe) can be calculated for each of the convective drying conditions from the experimental drying rates in Table 5, the measured electrode thickness in Table 3), the diffusion coefficient (D) for PVDF (1.14 × 10 cm/s), and the density of NMP (1.03 g/cm at 75 °C). Font et al. [27] estimated that binder migration occurs in the electrodes between Pe = 10 and 0.1. At the lowest drying rate (Pe = 0.1), they estimated a 10% concentration difference in PVDF between the upper and lower part of the electrode cross-section. In this study, lower drying rates were investigated, as the Pe was calculated to be between 0.11 and 0.01. For Pe = 0.11, the EDX analysis revealed a concentration difference of 40% between the upper and lower electrode area, whereas no concentration difference was detected for the electrodes dried at a Pe < 0.11. The experimental values for binder migration in these low-thickness cathodes were, therefore, four magnitudes higher compared to the values reported earlier [27].
3.2. Structure Analysis
The cathodes’ mass loading, thickness, and porosity are presented in Table 3. When comparing the V05T50 cathode with the V05T100 and V1T50 cathodes, there was a statistically significant difference in the mass loading and porosity (p < 0.05); however, the thickness was constant (p > 0.05). The difference in mass loading arose during tape-casting due to the low-precision doctor-blade, which caused an in-homogeneous coating. Since the thickness was constant, the cathodes with a high porosity (i.e., V05T100 and V1T100) should theoretically perform better at high cycling rates compared to those with a lower porosity (V1T50 and V05T50). The pores will be filled with electrolytes and improve the diffusion path of Li, which is often the kinetically limiting step during high charge/discharge rates. The high porosity calculated for the electrodes dried at 100 °C may be a sign of a large amount of inner and deep cracks, which are typically formed at high drying temperatures.
The SEM images used for surface analysis in ImageJ are presented in Appendix A Figure A1. Due to the difficulties in visualizing the details of those images, an enlarged example of the crack analysis before and after calendering is presented Figure 5, and an example of the crack sizes analyzed in SEM are shown in Figure 3. The percentage of the crack area covering the electrode surfaces (%) before and after calendering calculated from these images are presented in Table 5. The highest area of cracks was detected on the V1T100 cathode. This was in agreement with the results reported by Rollag et al. [23], where an increasing drying temperature from 20–70 °C resulted in a subsequent increasing crack area on the electrode surface. When we decreased the drying temperature to 50 °C (V05T50 and V1T50), the air velocity no longer had a statistically significant impact on the total crack area (p > 0.05). As mentioned, the same trend was reported for the critical NMP content, indicating that these two phenomena might be interconnected. The reference cathode (V0T90) had a higher crack coverage on the cathode surface when compared with all convective dried cathodes, except for the V1T100 cathode.

Figure 5.
An enlarged example of the SEM analysis of the surface of uncalendered and calendered reference V0T90 cathode dried in vacuum at 90 °C, and the converted image used for crack analysis in ImageJ.
After calendering, the area of cracks decreased for all cathodes, while the crack area became more uniform as shown in Table 5. The largest reduction in surface cracks after calendering was observed for the cathodes dried at 100 °C. Indeed, the percentage of cracks covering the cathode surface area for the V1T100 and V05T100 cathodes was reduced by ∼64% and ∼65%, respectively. The crack area for V05T50, V1T50, and V0T90 cathodes was reduced by ∼10%, ∼31%, and ∼39%, respectively. Inner voids/cracks are likely still present within the volume of the cathodes; however, these voids will only be accounted for in the porosity calculation. The SEM analysis also revealed that the NMC111 particles remained structurally intact during calendering, as no morphological difference was observed between the uncalendered and calendered cathodes.
The number of cracks per total electrode area (cm) was estimated and classified according to their size, ranging from 10 μm to 300 μm, and presented in Figure 6a. When increasing the drying temperature from 50 to 100 °C, the number of cracks increased significantly (p < 0.05). However, the drying air velocity did not have a significant impact on the crack formation when the drying temperature was high (100 °C) (p > 0.05). On the other hand, when decreasing the temperature to 50 °C, the air velocity did not have a significant impact on the number of cracks >300 μm and 100–200 μm (p < 0.05). Neither the temperature or air velocity had a significant impact on the number of small cracks (10–20 μm) (p > 0.05).
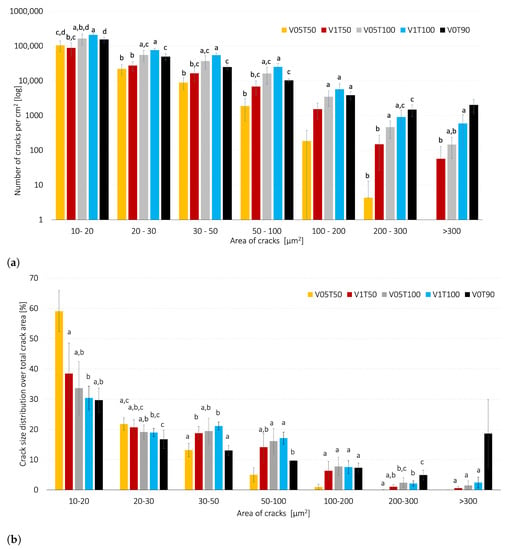
Figure 6.
The crack size distribution for electrode surfaces dried at air velocities (V) of 0, 0.5 m/s, or 1 m/s, and temperatures of T = 50 °C, T = 90, or T = 100 °C. (a) The number of cracks is classified according to their crack size. (b) The crack area over the total area of cracks (%) is classified according to the crack size. There is no statistical difference between samples with identical labels (a–d).
Figure 6a reveal that the reference cathode (V0T90) obtained the highest amount of large cracks > 300 μm when compared to the convection-dried cathodes (p < 0.05), although its mass loading was lower. The same trend was observed for the crack size distribution over the total crack area in Figure 6b. Therefore, the vacuum drying procedure was considered a more extreme drying method when compared with the investigated convective drying conditions.
Figure 6b shows that the small cracks (10–20 μm) dominated in all of the investigated cases. Furthermore, the fraction of small cracks was significantly higher (59.05 ± 6.79%) when the cathodes were dried gently (V05T50), compared to the other cathodes (<40%). The V05T50 cathodes obtained the lowest share of cracks >50 μm (p < 0.05), whereas there was no statistically significant difference for the cracks > 20 μm (p > 0.05) between the V1T50, V05T100, and V1T100 cathodes. These observations agree with previous studies where the high drying rate promotes crack formation [23,28,38]; however, the number of cracks and the size of cracks were not specified in those studies.
The V1T50 cathodes formed a significantly larger number of cracks, with an area >300 μm, whereas the V05T50 did not show any cracks of such size. The V1T50 cathodes also had the highest mass loading and the lowest porosity. Studies have shown that a high mass loading increases crack formation, irrespective of the air velocity and drying temperature [23]. One can therefore assume that factors other than the drying rate and critical NMP content, such as porosity and mass loading, could have influenced the formation of large cracks on the V1T50 cathodes. Furthermore, the falling drying rate period observed during the drying of the V1T50 cathodes was shorter than that observed for V05T50 cathodes (p < 0.05). Thus, the formation of these large cracks on the V1T50 cathodes possibly occurred after the cathodes had reached the critical NMP content (i.e., during the falling rate period).
To summarize, the V05T50 cathodes displayed the lowest drying rate (0.39 × 10 kg/ms) with the longest constant drying rate period, and the lowest critical NMP content (X = 0.27 kg/kg). These conditions resulted in smaller-sized surface cracks, as the V05T50 cathode had the smallest area covered by cracks (2.5 ± 0.71%). On the contrary, the V1T100 with the most extreme convective drying conditions led to the highest drying rate (2.93 × 10 kg/ms) and critical NMP content (X = 0.51 kg/kg), and the largest area covered by cracks (9.82 ± 1.62). The V05T100 cathode experienced a drying rate of 1.55 × 10 kg/ms and an X of 0.37 kg/kg.
3.3. Elemental Mapping
An elemental mapping of fluorine (F) inspired by Westphal et al. [25] was conducted using EDX mapping to confirm the PVDF binder migration for the cathodes dried under the most extreme conditions (V05T50, V1T100, and V0T90). The EDX mapping was performed on both the top surface and over the cross-section as illustrated in Figure 7. The scan conducted on the cathode’s cross-section was divided into two parts: the upper area near the electrode surface and the lower area near the Al current collector.

Figure 7.
An example of the elemental EDX mapping of fluorine (F, yellow), manganese (Mn, purple), and carbon (C, orange) on the cathode coating’s surface (left) and cross-section (right) dried at T = 50 °C (V05T50) or V = 1 m/s and T = 100 °C (V1T100).
Table 6 summarizes the normalized wt% ratio between F, Mn, and C, from the surface and cross-section mapping. These elements represented the concentration of PVDF, NMC111 particles, and carbon black, respectively. The V05T50 and V0T90 did not show a statistically significant difference in F concentration between the upper and lower cross-section areas (p > 0.05). However, the migration of the PVDF binder occurred under the most extreme convective drying condition (V1T100), where the difference between the lower and upper levels was 40%. This agrees with other reports that observed a significant difference in the F distribution across the cross-section of the cathode after drying at a temperature of 90 °C [25,39].
Table 6.
EDX data from the electrode’s top surface. The values are the normalized concentration (wt%) of each component and the error between the analyzed images (%). F, Mn, and C represent the PVDF, NMC111, and carbon black, respectively. There is no statistical difference between samples with identical labels (a–d).
When analyzing the electrode surface, a statistically significant higher F concentration was detected on the surface of the V1T100 cathode when compared to the V05T50 and V0T90 (p < 0.05). The surface scan results are consistent with the F distribution detected in the cross-section scan. Such a clear trend was not observed for the distribution of carbon (C) and NMC111 (Mn) over the cross-section and on the surface.
Complicated cross-section analyses, where the cathode is cut using a focused ion beam (FIB) such as in Figure A2, are often implemented in laboratories and at the industrial scale to analyze binder migration [40]. The results confirm that both the surface and cross-section mapping may be used as a method for detecting PVDF binder migration. This study proposes surface analysis as a straightforward and low-cost method that is sufficient for comparing binder migration in cathodes using a PVDF binder. Assumptions are often made that electrodes with low mass loading (3.5–5.4 mg/cm) have no significant binder migration [25]. Since binder migration was detected on the surface scan for cathodes of low mass loading, the results imply that the sensitivity of such a surface analysis method is suitable for cathodes of higher mass loading, where binder migration is presumably more extreme, and may be implemented at the industrial scale.
Figure A3 shows the cross-section of the two cathodes dried under the most extreme conditions in this study: V05T50 and V1T100. Figure A3b shows that the V1T100 cathode coating with binder migration delaminated from the Al current collector (Al-foil) in some places, whereas the other cathodes did not show such detachment. This dependence between the binder migration and the delamination of the coating from the Al-foil was reported previously by Jaiser et al. [32]. The disruption in the electrical contact between the active particles, conductive additive, and the Al-foil may result in a loss of electronic/ionic conductivity and eventually in a capacity decline during long-term cycling [41].
3.4. Cycling Performance
The cathodes dried under the five different drying conditions are cycled according to the cycling program shown in Table 4. The specific discharge capacities obtained at different discharge rates (up to 5 C) are presented in Figure 8. Rollag et al. [23] found that the drying temperature (45 to 70 °C) did not have a significant impact on the initial specific capacity (C/10) for electrodes with a mass loading of 11 mg/cm. However, at C/2, the specific discharge capacity decreased with a higher drying temperature due to extensive crack formation. The largest crack area was approximately 80,000 μm. In this study, there was no clear correlation between the influence of cracks and discharge capacity at C/10 or C/2 for the convection-dried cathodes. This indicated that the crack size of 10–1400 μm observed here was too small to influence the cycling performance at these low discharge currents [23]. Moreover, the reference cathode (V0T90) obtained an initial specific capacity of 151.4 mAh/g at C/10, which was lower than that observed for the convection-dried cathodes (156.8–164.8 at C/10). This may be caused by the significantly higher fraction of large cracks > 300 μm in the V0T90 coating, which interrupts the electronic network and lowers the initial discharge capacity.
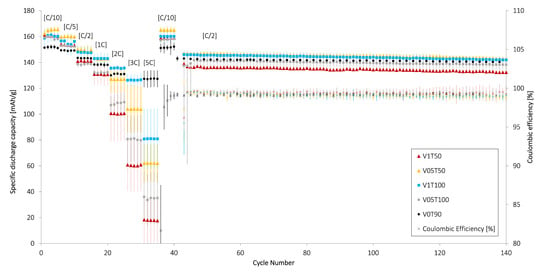
Figure 8.
The average specific discharge capacity (mAh/g) and Coulombic efficiency (%) of cathodes dried under different drying conditions. The cathodes were tested as coin cells vs. a Li metal anode over 40 cycles at various discharge C-rates [in brackets], followed by 100 cycles at charge and discharge C/2. The capacity loss over 100 cycles at C/2 was 2.62% (V05T50), 2.85% (V1T50), 2.24% (V05T100), 2.82% (V1T100), and 0.99% (V0T90).
The lowest specific discharge capacity amongst the cathodes was achieved at 5 C, varying between 7.8 and 39.5 mAh/g. A higher current density decreased the capacity since parts of the coatings were not accessible due to Li-diffusion limitations. Amongst the convective dried cathodes, the V05T50 cathodes obtained the highest specific discharge capacity at low C-rates < C/5, whereas V1T100 cathodes showed the highest specific discharge capacity at high C-rates > 2 C. As mentioned, the V1T100 cathodes had the highest number of cracks and the highest total area of cracks after calendering (Figure 6). These cracks are filled by the electrolyte and acted as pores, which likely created an effective Li-diffusion pathway, allowing Li to diffuse faster through these disrupted electrode parts. This was also true for the reference cathode (V0T90), which had the highest amount of large cracks of all samples (p < 0.05) and outperformed all of the convective-dried cathodes in terms of the discharge capacity at C-rates > 2 C. The high rate capability observed for the V0T90 electrode was also likely partly caused by the low mass loading (3.5 mg/cm).
The V1T100 cathodes exhibited a high standard deviation when cycling at 5 C, which can be caused by the local differences within the coating created by the partial delamination from the Al-foil seen in Figure A3b. According to earlier research [23,28], large cracks may also initiate further crack propagation and result in a fragile electrode.
The V1T50 cathodes showed the lowest specific discharge capacity at high C-rates > 2 C. The V1T50 cathode also had a lower area of cracks on the surface before calendering (∼3.25%) when compared with the V1T100 (∼9.82%), V05T100 (∼6.73%), and V0T90 (∼7.11%) cathodes. However, the area of cracks covering the surface after the calendering of V1T50 (2.24%) was similar to V05T50 (2.26%) and V05T100 (2.43%). Therefore, it can be concluded that the poor rate capability at high C-rates for V1T50 was caused by a combination of the high mass loading (∼5.7 mg/cm) and low porosity (∼48%) relative to the other convection-dried cathodes (∼4.7–5.2 mg/cm and ∼54–60%). Other studies on NMC111-based cathodes revealed that the rate capability declined with the porosity, although the mass loading was constant [42]. All of the electrodes restored their capacity after the high C-rate test, i.e., the electrodes did not face any damage during the cycling. Post-mortem analyses were therefore excluded in this study.
The capacity loss over 100 cycles at C/2 was independent of the convective drying conditions as all cathodes showed a capacity loss in the range of 2.24–2.85%. This indicated that the cracks may have been too small to initiate an extensive crack propagation, which decreased the long-term cyclability for cathodes in other studies [23]. In fact, the V0T90 cathode with a high number of large surface cracks obtained a minimal loss of 0.99%. The cathodes dried at 100 °C showed a higher standard deviation than those dried at 50 °C. Indeed, the V05T50 cathodes, which also had the lowest surface crack area before (∼2.50%) and after (∼2.26%) calendering, showed the lowest standard deviation upon cycling.
Ahmad et al. [43] reported a capacity reduction after 100 cycles at C/10 between 3–5%. The electrode load was higher than in this study (18 mg/cm), and the drying temperature was 115 °C. Xu et al. [44] tested 45 μm thick electrodes dried at 80 °C in a vacuum. The capacity loss was approximately 19% after 100 cycles at C/5. Al-Shroofy et al. [21] studied NMC111 electrodes with a mass loading of 15 mg/cm, with a thickness reduced from 80 μm to 55 μm after calendering. These electrodes were cycled over 160 cycles at C/2 from 3–4.3 V vs. Li/Li, reporting an initial specific capacity of 148 mAh/g and a capacity loss after 100 cycles of ∼6.8%. The relatively large capacity fading was explained by extensive surface cracking during cycling. At 2 C, the discharge capacity was 126 mAh/g, whereas, in the current study, the capacity varied between 136.5–107.5 mAh/g, depending on the drying conditions. Since the above-mentioned reports studied electrodes with mass loading two to three times as high as the current study (4.8–5.7 mg/cm), lower discharge capacities were expected.
3.5. Future Work
Some drawbacks were associated with the convective oven used in this study, such as the relatively low temperatures and air velocities compared to the industrial values. Future work should include the testing of even more intensive drying conditions and higher drying rates by increasing the air velocity and temperature up to industrial values (around 25 m/s and 180 °C). Additional quality parameters for binder migration and crack propagation, such as the binder’s thermal stability, should be analyzed as they can define the binder’s upper limiting temperature [45].
Alternative solvents and binders to the PVDF and NMP should also be tested under these intensive drying conditions to analyze their influence on the quality parameters (i.e., binder migration and crack size). For example, a water-based slurry is expected to intensify the crack formation due to the high capillary forces [45]; thus, the optimization of drying kinetics to minimize the crack formation is of high importance. Since extensive research is dedicated to finding sustainable, fluorine-free binders such as CMC and lignin [7,45,46], new methods for detecting carbon migration should be explored. Furthermore, the EDX surface mapping protocol should be tested for electrodes with reduced binder contents as the industry is moving towards a PVDF binder content of around 2% [7].
Electrochemical impedance spectroscopy (EIS) should be included in future work to estimate the influence of cracks on the electrode’s electronic and ionic conductivity. This analysis was not included here as coin-cells were utilized for all measurements, and a third reference electrode is needed to deconvolute the impedance responses.
4. Conclusions
The study confirmed that an increase in the drying rate intensified the crack formation for convective dried cathodes. This trend was observed even at low drying rates (relative to industrial values) and for cathodes with low mass loadings (<5.7 mg/cm). The electrode surface mapping in SEM/EDX is presented as a simplified alternative to the cross-section method when comparing binder migration in multiple electrodes. A drying temperature of 100 °C and air velocity of 1 m/s (V1T100) resulted in an uneven binder distribution over the electrode’s cross section, where the difference between the upper and lower area reached 40%. The delamination of the cathode coating from the Al current collector was related to the binder migration.
The investigated drying conditions were successful for the production of high-quality cathodes with a low capacity loss (<3%) over 100 cycles at C/2. Interestingly, if the surface cracks are relatively small (<1400 μm), so the fraction of the crack area over the total electrode surface range between 2.50–9.82 %, the cracks does not impair the electrochemical performance at low discharge rates (< C/2). In fact, the cathodes with the highest amount of large surface cracks (V0T90 and V1T100) and the most extensive coverage of cracks on the surface area obtained the highest specific discharge capacity at discharge rates > C/2. The large crack coverage and voids helped to maintain a high porosity at the surface after calendering. This allowed for electrolyte penetration and fast Li-transport through the cathodes during cycling.
The highest discharge capacity loss at high C-rates and over 100 cycles was found for the V1T50 cathode with the highest mass loading and low porosity. Furthermore, the reference cathode dried in a vacuum (V0T90) with a relatively low porosity (39%) and mass loading of 3.5 mg/cm obtained the highest capacity. Therefore, the optimization of the drying process of cathodes should include not only the drying rate and temperature but also the mass loading and porosity. Despite the low mass loading for the V0T90 cathode, it obtained the highest amount of large cracks (>300 μm), indicating that vacuum drying was more intense for crack propagation compared to the convective drying method.
The crack analysis depended on the applied analysis method (number of cracks, crack size distribution, and total coverage of cracks on the surface). For example, the statistical analysis revealed no difference in the crack size distribution between the V1T100, V1T50, and V05T100 cathodes, whereas the number of cracks per cm on the surface was different. The number of cracks and crack distribution did not show a clear correlation between the cracks and cycling performance for convective-dried cathodes of low mass loading when the crack area was below 1400 μm. However, the analysis of the total crack area after calendering revealed that the best rate capability was detected for the cathodes with the highest total area of surface cracks. The surface cracks improved the electrolyte diffusion, and thus the Li transport through the cathodes. The results indicated that analyzing the total surface crack area on cathodes after calendering can be sufficient for predicting the rate performance if the maximum crack size is below 1400 m.
Author Contributions
Conceptualization, S.N.B., O.S.B. and J.J.L.; methodology, I.T. and S.N.B.; formal analysis, S.N.B., I.T. and A.K.; investigation, S.N.B.; data curation, S.N.B. and I.T.; writing—original draft preparation, S.N.B. and I.T.; writing—review and editing, O.S.B. and J.J.L.; visualization, S.N.B. and I.T.; supervision and resources, O.S.B., J.J.L. and I.T.; project administration, O.S.B.; funding acquisition, O.S.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Freyr Battery (Project number: 90492502), ENERSENSE, and EIT InnoEnergy.
Data Availability Statement
Not applicable.
Acknowledgments
The authors gratefully acknowledge the funding support from the Norwegian battery company Freyr Battery, and the ENERSENSE 68024013 research initiative at NTNU.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| X | Critical NMP content (kg/kg) |
| Air viscosity (Pa·s) | |
| V | Air flow velocity (m/s) |
| E | Cell potential (V vs. Li/Li) |
| A | Electrode surface area (μm) |
| Molar density of the surrounding gas flow (moles/m) | |
| p | Operating pressure (Pa) |
| Pe | Peclet number (-) |
| Re | Reynolds number (-) |
| T | Drying temperature (°C) |
| m | Mass of dry solid (kg) |
| W | Drying rate (kg/ms) |
| D | Drying time (s) |
| t | time (s) |
| Density of air (kg/m) | |
| Thick | Electrode coating thickness (μm) |
| P | Electrode coating porosity (%) |
| wt% | Weight relative to NMC111 (%) |
| n | Cycle number (-) |
| CE | Coulombic efficiency (%) |
| IDC | Initial specific discharge capacity (mAh/g) |
| ML | Mass loading of electrode coating (mg/cm) |
| Water | |
| H | Hydrogen |
| O | Oxygen |
| Li | Lithium |
| Li | Lithium ion |
| LIB | Lithium-ion battery |
| CB | Carbon black |
| PVDF | Polyvinyliden Fluorid |
| NMP | N-Methyl-2-Pyrrolidone |
| NMC | LiNiMnCoO |
| NMC111 | LiNiMnCoO |
| NMC811 | LiNiMnCoO |
| LFP | LiFePO |
| CC | Current collector |
| Al-foil | Aluminum foil current collector |
| C-Al | Carbon-coated aluminum foil current collector |
| RT | Room temperature (°C) |
| dis | Discharge |
| char | Charge |
| o | Outlet |
| i | Inlet |
| t | Time |
| g | Gas |
| a | Air |
| E | Electrode |
| d | Drying |
| c | Critical |
| bat.cap. | Battery capacity |
Appendix A
Appendix A.1. SEM

Figure A1.
The SEM analysis of the surface of uncalendered and calendered cathodes, and the converted image used for crack analysis in ImageJ.

Figure A2.
An electrode cross-section cut with a focused ion beam (FIB) to show the distribution of species across the coating. EDX was used to perform an elemental scan for nickel (Ni), cobalt (Co), manganese (Mn), oxygen (NMC particles), carbon (C) (carbon black), aluminum (Al) (current collector), and fluorine (F) (PVDF). The cathode was dried at 90 °C in a vacuum (0 air flow) consisting of 85% NMC111, 10% carbon black, and 5% PVDF.

Figure A3.
The cross-section of a cathode coating dried with the lowest (V05T50) (a) and highest (V1T100) (b) air velocities (V) and temperatures (T). (a) is the V05T50 cathode (V = 0.5 m/s and T = 50 °C), which is continuously attached to the current collector. (b) shows the V1T100 cathode (V = 1 m/s and T = 100 °C) with a detached area between the cathode and current collector. Note that the scale bars vary between these images.
References
- Tsiropoulos, I.; Tarvydas, D.; Lebedeva, N. Li-Ion Batteries for Mobility and Stationary Storage Applications; Technical report; EESC: European Economic and Social Committee: Brussels, Belgium, 2018. [Google Scholar] [CrossRef]
- Larcher, D.; Tarascon, J.M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2015, 7, 19–29. [Google Scholar] [CrossRef]
- Kurland, S.D. Energy use for GWh-scale lithium-ion battery production. Environ. Res. Commun. 2020, 2, 012001. [Google Scholar] [CrossRef]
- Ellingsen, L.A.W.; Singh, B.; Strømman, A.H. The size and range effect: Life-cycle greenhouse gas emissions of electric vehicles. Environ. Res. Lett. 2016, 11, 054010. [Google Scholar] [CrossRef]
- Ahmed, S.; Nelson, P.A.; Gallagher, K.G.; Dees, D.W. Energy impact of cathode drying and solvent recovery during lithium-ion battery manufacturing. J. Power Sources 2016, 322, 169–178. [Google Scholar] [CrossRef]
- Wood, D.L.; Quass, J.D.; Li, J.; Ahmed, S.; Ventola, D.; Daniel, C. Technical and economic analysis of solvent-based lithium-ion electrode drying with water and NMP. Dry. Technol. 2018, 36, 234–244. [Google Scholar] [CrossRef]
- Bryntesen, S.N.; Strømman, A.H.; Tolstorebrov, I.; Shearing, P.R.; Lamb, J.J.; Stokke Burheim, O. Opportunities for the State-of-the-Art Production of LIB Electrodes—A Review. Energies 2021, 14, 1406. [Google Scholar] [CrossRef]
- Ding, Y.; Cano, Z.P.; Yu, A.; Lu, J.; Chen, Z. Automotive Li-Ion Batteries: Current Status and Future Perspectives. Electrochem. Energy Rev. 2019, 2, 1–28. [Google Scholar] [CrossRef]
- Scrosati, B.; Garche, J. Lithium batteries: Status, prospects and future. J. Power Sources 2010, 195, 2419–2430. [Google Scholar] [CrossRef]
- Zhao, X.; Cheruvally, G.; Kim, C.; Cho, K.K.; Ahn, H.J.; Kim, K.W.; Ahn, J.H. Lithium/sulfur secondary batteries: A review. J. Electrochem. Sci. Technol. 2016, 7, 97–114. [Google Scholar] [CrossRef]
- Thomitzek, M.; Cerdas, F.; Thiede, S.; Herrmann, C. Cradle-to-gate analysis of the embodied energy in lithium ion batteries. Procedia CIRP 2019, 80, 304–309. [Google Scholar] [CrossRef]
- Kwade, A.; Haselrieder, W.; Leithoff, R.; Modlinger, A.; Dietrich, F.; Droeder, K. Current status and challenges for automotive battery production technologies. Nat. Energy 2018, 3, 290–300. [Google Scholar] [CrossRef]
- Hawley, W.B.; Li, J. Electrode manufacturing for lithium-ion batteries—Analysis of current and next generation processing. J. Energy Storage 2019, 25, 100862. [Google Scholar] [CrossRef]
- Chen, C.H.; Kelder, E.M.; Jak, M.J.; Schoonman, J. Electrostatic spray deposition of thin layers of cathode materials for lithium battery. Solid State Ionics 1996, 86–88, 1301–1306. [Google Scholar] [CrossRef]
- Yoon, W.S.; Ban, S.H.; Lee, K.K.; Kim, K.B.; Kim, M.G.; Lee, J.M. Electrochemical characterization of layered LiCoO2 films prepared by electrostatic spray deposition. J. Power Sources 2001, 97, 282–286. [Google Scholar] [CrossRef]
- Hiroya, A.; Akira, K.; Makio, N.; Masayuki, Y. Electrostatic Spray Deposition for Fabrication of Li-ion Batteries. Trans. JWRI 2015, 44, 9. [Google Scholar]
- Koike, S.; Tatsumi, K. Preparation and performances of highly porous layered LiCoO2 films for lithium batteries. J. Power Sources 2007, 174, 976–980. [Google Scholar] [CrossRef]
- Orlenius, J.; Lyckfeldt, O.; Kasvayee, K.A.; Johander, P. Water based processing of LiFePO 4/C cathode material for Li-ion batteries utilizing freeze granulation. J. Power Sources 2012, 213, 119–127. [Google Scholar] [CrossRef]
- Schälicke, G.; Landwehr, I.; Dinter, A.; Pettinger, K.H.; Haselrieder, W.; Kwade, A. Solvent-Free Manufacturing of Electrodes for Lithium-Ion Batteries via Electrostatic Coating. Energy Technol. 2020, 8, 1900309. [Google Scholar] [CrossRef]
- Shin, J.; Yudi, Y.; Magsino, P.; Wong, W.; Duong, H. Dry Processed Nickel-Rich Layered Transition Metal Oxide Cathode Electrode. Meet. Abstr. 2019, 2, 317. [Google Scholar] [CrossRef]
- Al-Shroofy, M.; Zhang, Q.; Xu, J.; Chen, T.; Kaur, A.P.; Cheng, Y.T. Solvent-free dry powder coating process for low-cost manufacturing of LiNi1/3Mn1/3Co1/3O2 cathodes in lithium-ion batteries. J. Power Sources 2017, 352, 187–193. [Google Scholar] [CrossRef]
- Wang, M.; Hu, J.; Wang, Y.; Cheng, Y.T. The influence of polyvinylidene fluoride (PVDF) binder properties on LiNi0.33Co0.33Mn0.33O2 (NMC) electrodes made by a dry-powder-coating process. J. Electrochem. Soc. 2019, 166, A2151–A2157. [Google Scholar] [CrossRef]
- Rollag, K.; Juarez-Robles, D.; Du, Z.; Wood, D.L.; Mukherjee, P.P. Drying Temperature and Capillarity-Driven Crack Formation in Aqueous Processing of Li-Ion Battery Electrodes. ACS Appl. Energy Mater. 2019, 2, 4464–4476. [Google Scholar] [CrossRef]
- Lim, S.; Kim, S.; Ahn, K.H.; Lee, S.J. Stress development of Li-ion battery anode slurries during the drying process. Ind. Eng. Chem. Res. 2015, 54, 6146–6155. [Google Scholar] [CrossRef]
- Westphal, B.; Bockholt, H.; Gunther, T.; Haselrieder, W.; Kwade, A. Influence of Convective Drying Parameters on Electrode Performance and Physical Electrode Properties. ECS Trans. 2015, 64, 57. [Google Scholar] [CrossRef]
- Jaiser, S.; Kumberg, J.; Klaver, J.; Urai, J.L.; Schabel, W.; Schmatz, J.; Scharfer, P. Microstructure formation of lithium-ion battery electrodes during drying—An ex-situ study using cryogenic broad ion beam slope-cutting and scanning electron microscopy (Cryo-BIB-SEM). J. Power Sources 2017, 345, 97–107. [Google Scholar] [CrossRef]
- Font, F.; Protas, B.; Richardson, G.; Foster, J.M. Binder migration during drying of lithium-ion battery electrodes: Modelling and comparison to experiment. J. Power Sources 2018, 393, 177–185. [Google Scholar] [CrossRef]
- Du, Z.; Rollag, K.M.; Li, J.; An, S.J.; Wood, M.; Sheng, Y.; Mukherjee, P.P.; Daniel, C.; Wood, D.L. Enabling aqueous processing for crack-free thick electrodes. J. Power Sources 2017, 354, 200–206. [Google Scholar] [CrossRef]
- Westphal, B.G.; Kwade, A. Critical electrode properties and drying conditions causing component segregation in graphitic anodes for lithium-ion batteries. J. Energy Storage 2018, 18, 509–517. [Google Scholar] [CrossRef]
- Jaiser, S.; Sanchez Salach, N.; Baunach, M.; Scharfer, P.; Schabel, W. Impact of drying conditions and wet film properties on adhesion and film solidification of lithium-ion battery anodes. Dry. Technol. 2017, 35, 1807–1817. [Google Scholar] [CrossRef]
- Jaiser, S.; Friske, A.; Baunach, M.; Scharfer, P.; Schabel, W. Development of a three-stage drying profile based on characteristic drying stages for lithium-ion battery anodes. Dry. Technol. 2017, 35, 1266–1275. [Google Scholar] [CrossRef]
- Jaiser, S.; Müller, M.; Baunach, M.; Bauer, W.; Scharfer, P.; Schabel, W. Investigation of film solidification and binder migration during drying of Li-Ion battery anodes. J. Power Sources 2016, 318, 210–219. [Google Scholar] [CrossRef]
- Oppegård, E.; Jinasena, A.; Hammer Strømman, A.; Are Suul, J.; Stokke Burheim, O. Study of an Industrial Electrode Dryer of a Lithium-Ion Battery Manufacturing Plant: Dynamic Modeling. In Proceedings of the 61st SIMS Conference on Simulation and Modelling, Virtual Conference, Finland, 22–24 September 2020; Linköping University Electronic Press: Linköping, Sweden, 2020; Volume 176, pp. 77–84. [Google Scholar] [CrossRef]
- Kumberg, J.; Müller, M.; Diehm, R.; Spiegel, S.; Wachsmann, C.; Bauer, W.; Scharfer, P.; Schabel, W. Drying of Lithium-Ion Battery Anodes for Use in High-Energy Cells: Influence of Electrode Thickness on Drying Time, Adhesion, and Crack Formation. Energy Technol. 2019, 7, 1900722. [Google Scholar] [CrossRef]
- Richardson, J.F.; Harker, J.H.; Backhurst, J.R. Chemical Engineering: Particle Technology and Separation Processes, 5th ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2003; Volume 2, pp. 1–1156. [Google Scholar] [CrossRef]
- Bhadra, R.; Muthukumarappan, K.; Rosentrater, K.; Kannadhason, S. Drying Kinetics of Distillers Wet Grains (DWG) Under Varying Condensed Distillers Solubles (CDS) and Temperature Levels. Cereal Chem. 2011, 88, 451–458. [Google Scholar] [CrossRef]
- Ghiaasiaan, S. Convective Heat and Mass Transfer; Cambridge University Press: Cambridge, UK, 2011; p. 548. [Google Scholar]
- Susarla, N.; Ahmed, S.; Dees, D.W. Modeling and analysis of solvent removal during Li-ion battery electrode drying. J. Power Sources 2018, 378, 660–670. [Google Scholar] [CrossRef]
- Müller, M.; Pfaffmann, L.; Jaiser, S.; Baunach, M.; Trouillet, V.; Scheiba, F.; Scharfer, P.; Schabel, W.; Bauer, W. Investigation of binder distribution in graphite anodes for lithium-ion batteries. J. Power Sources 2017, 340, 1–5. [Google Scholar] [CrossRef]
- Kim, J.y.; Jeong, Y.W.; Cho, H.Y.; Chang, H.J. Alternative Sample Preparation Method for Large-Area Cross-Section View Observation of Lithium Ion Battery. Appl. Microsc. 2017, 47, 77–83. [Google Scholar] [CrossRef]
- Edge, J.S.; O’Kane, S.; Prosser, R.; Kirkaldy, N.D.; Patel, A.N.; Hales, A.; Ghosh, A.; Ai, W.; Chen, J.; Yang, J.; et al. Lithium ion battery degradation: What you need to know. Phys. Chem. Chem. Phys. 2021, 23, 8200–8221. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, G.; Song, X.; Ridgway, P.; Xun, S.; Battaglia, V.S. Cathode Performance as a Function of Inactive Material and Void Fractions. J. Electrochem. Soc. 2010, 157, A1060. [Google Scholar] [CrossRef]
- Ahmad, I.; Zhang, P. Advanced Drying Process for Lower Manufacturing Cost of Electrodes; Technical report; National Energy Technology Laboratory: Pittsburgh, PA, USA; Morgantown, WV, USA, 2016. [Google Scholar] [CrossRef]
- Xu, R.; Sun, H.; de Vasconcelos, L.S.; Zhao, K. Mechanical and Structural Degradation of LiNixMnyCozO2 Cathode in Li-Ion Batteries: An Experimental Study. J. Electrochem. Soc. 2017, 164, A3333. [Google Scholar] [CrossRef]
- Bryntesen, S.N.; Tolstorebrov, I.; Svensson, A.M.; Shearing, P.; Lamb, J.J.; Burheim, O.S. Introducing Lignin as a Binder Material for the Aqueous Production of NMC Cathodes for Li-ion Batteries. Mater. Adv. 2023, 4, 523–541. [Google Scholar] [CrossRef]
- Bresser, D.; Buchholz, D.; Moretti, A.; Varzi, A.; Passerini, S. Alternative binders for sustainable electrochemical energy storage-the transition to aqueous electrode processing and bio-derived polymers. Energy Environ. Sci. 2018, 11, 3096–3127. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).