
Humidity-Induced Degradation of Lithium-Stabilized Sodium-Beta Alumina Solid Electrolytes


Abstract
:
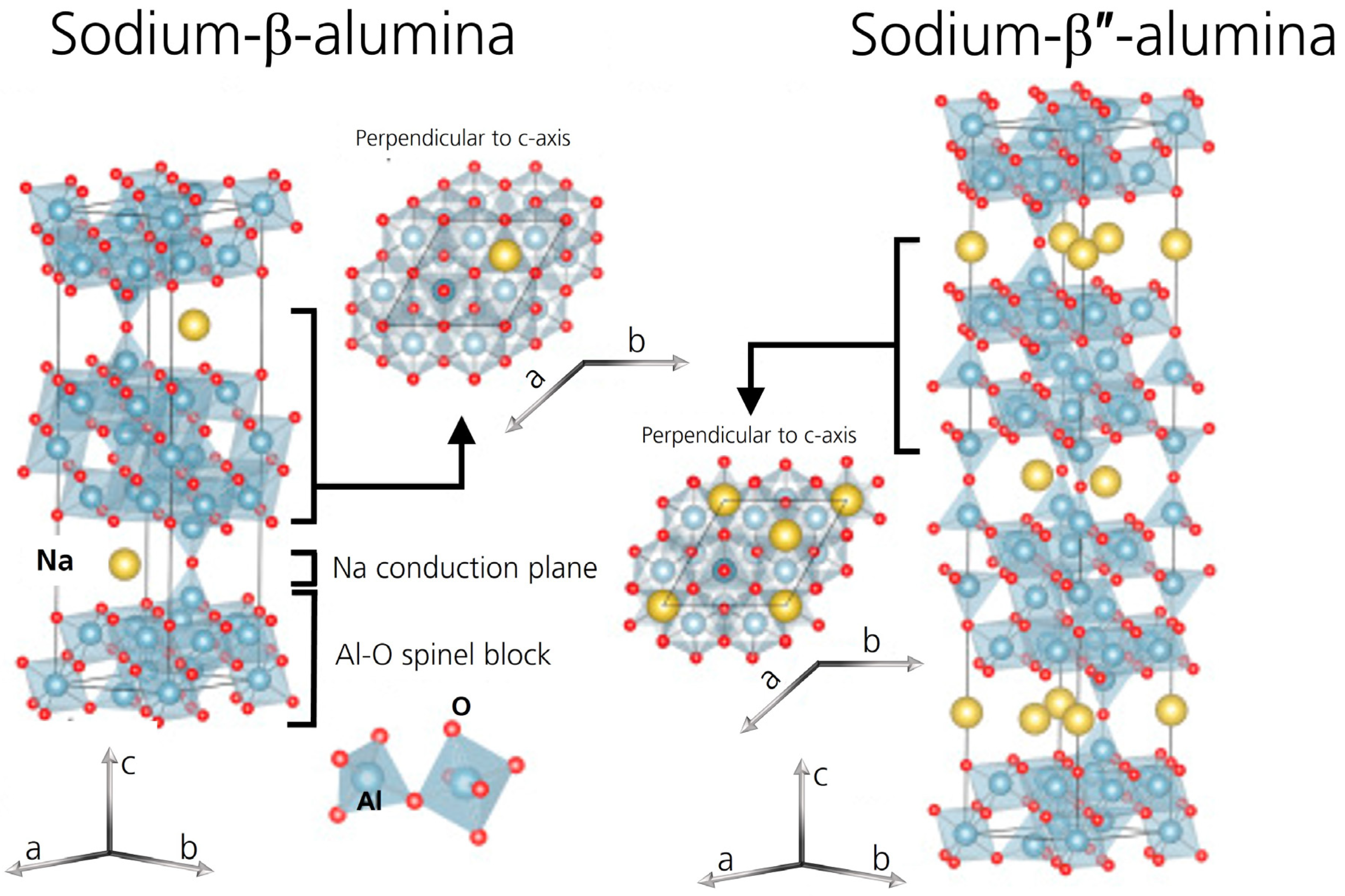
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Sample Characterization
3. Results
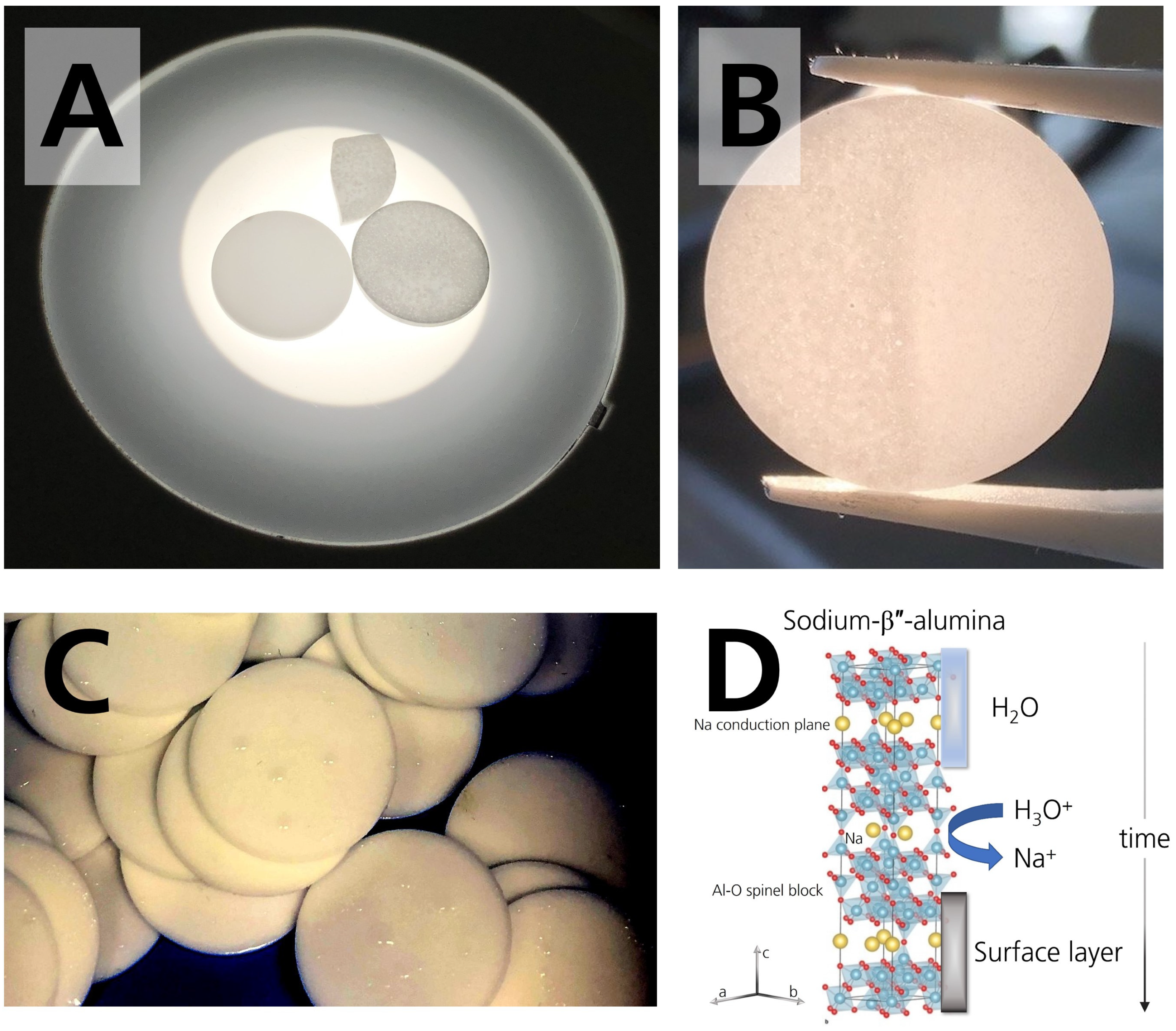
3.1. Density and Microstructure Analysis
3.2. Chemical Characterization and Additional Observations
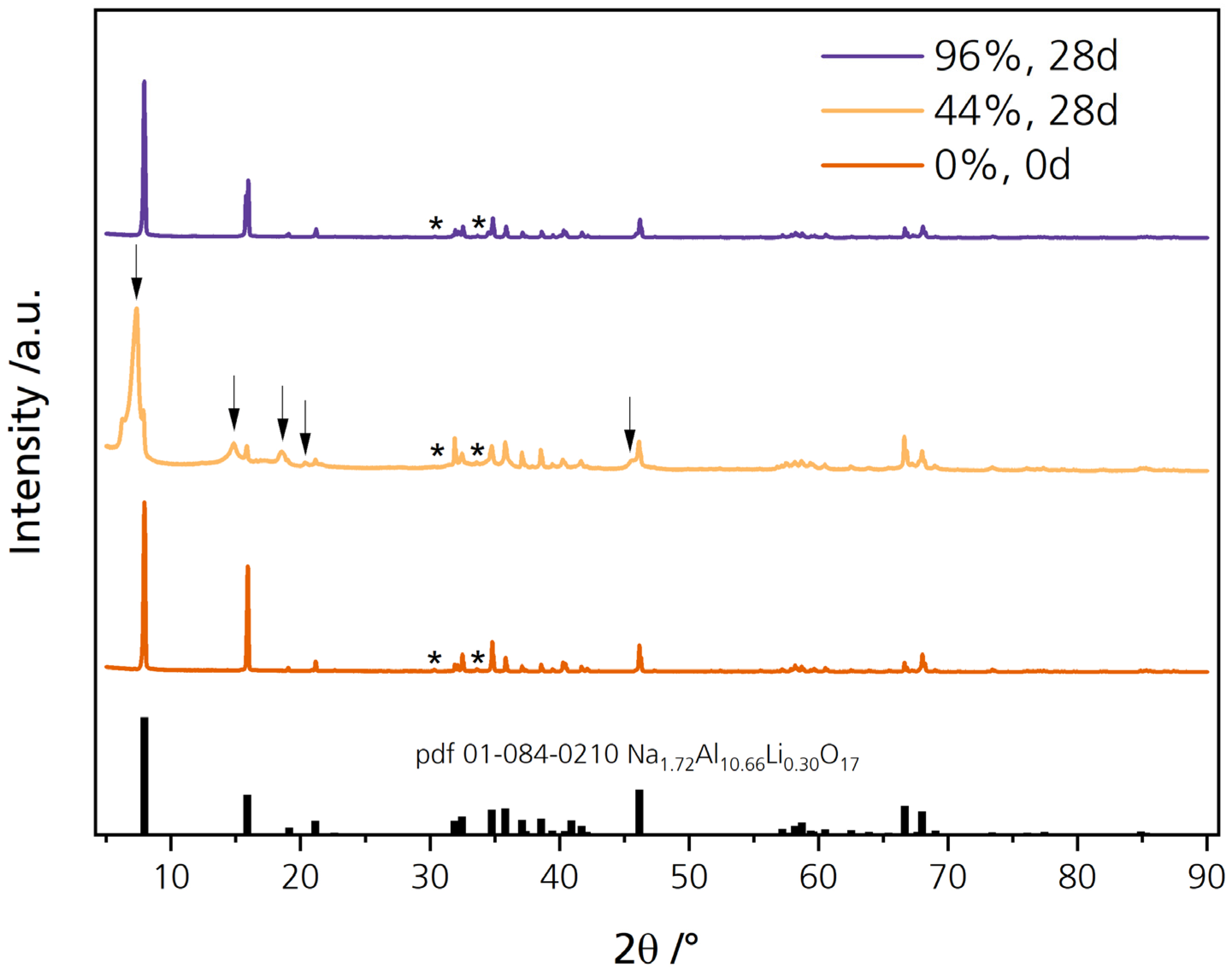
3.3. XRD Analysis
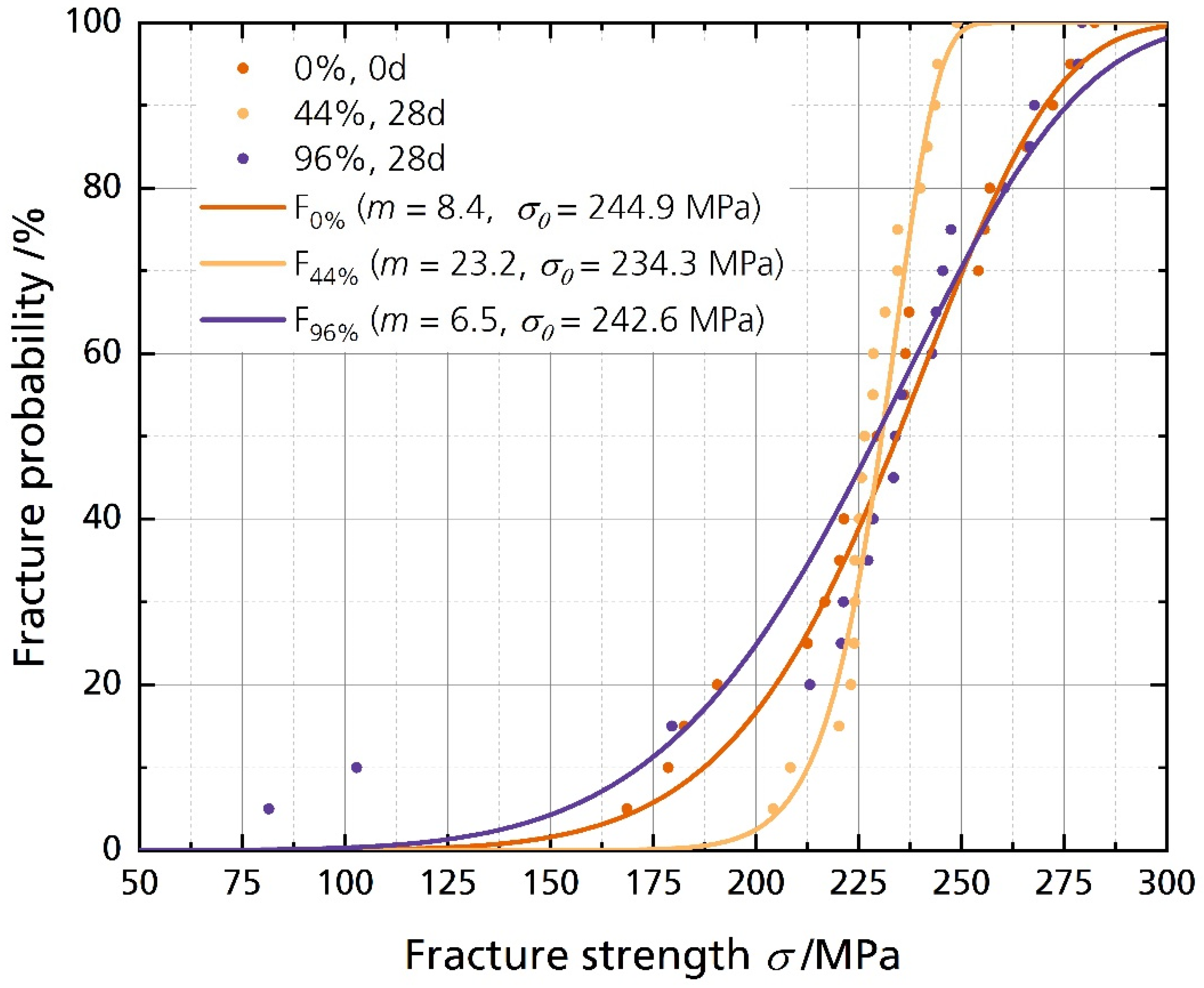
3.4. Fracture Strength Analysis
3.5. Conductivity Analysis
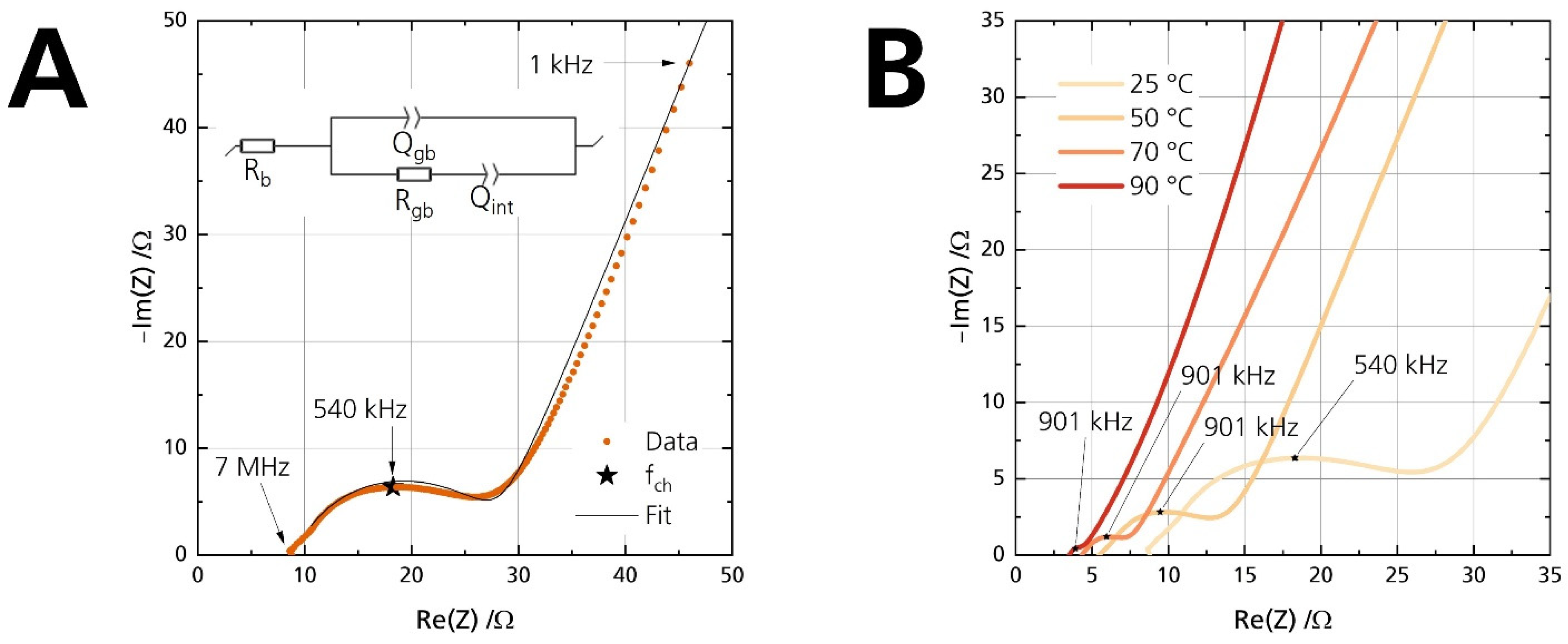
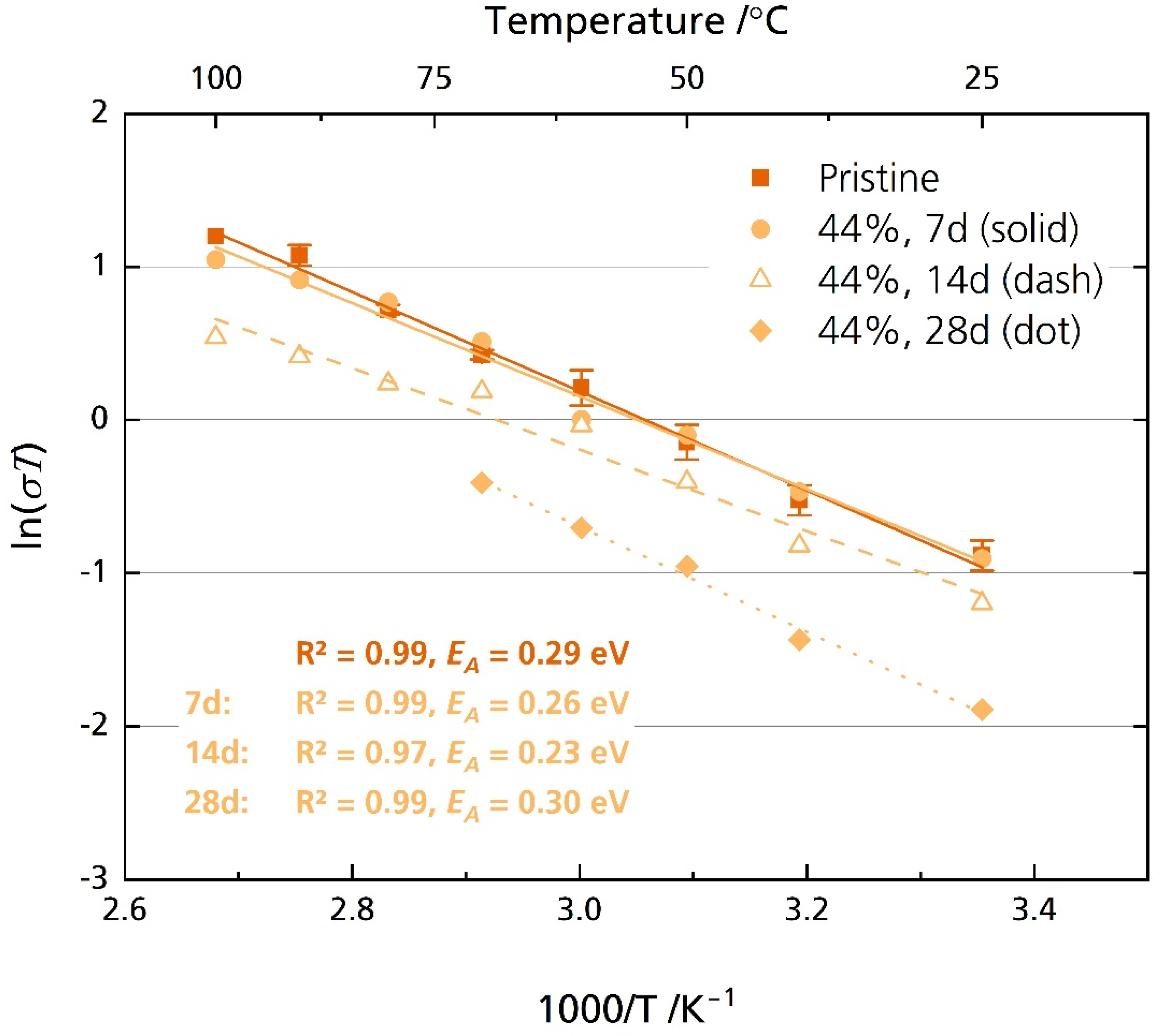
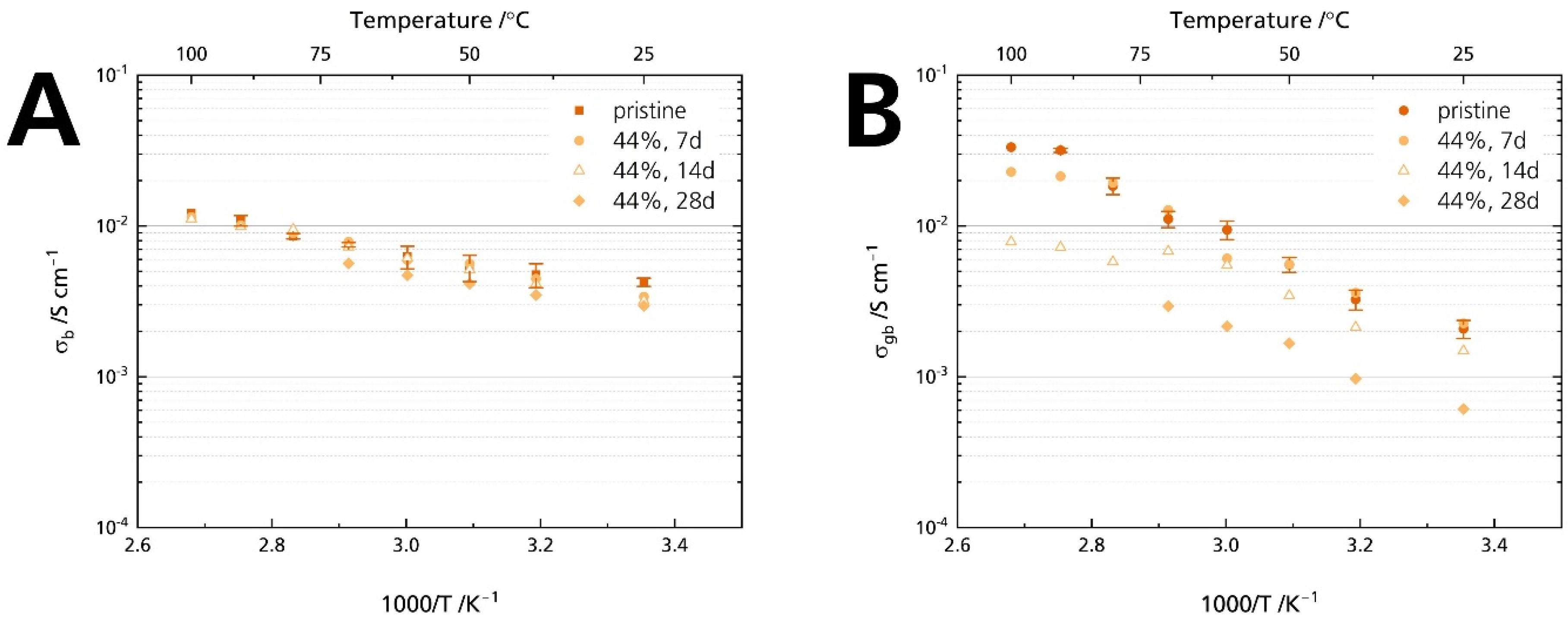
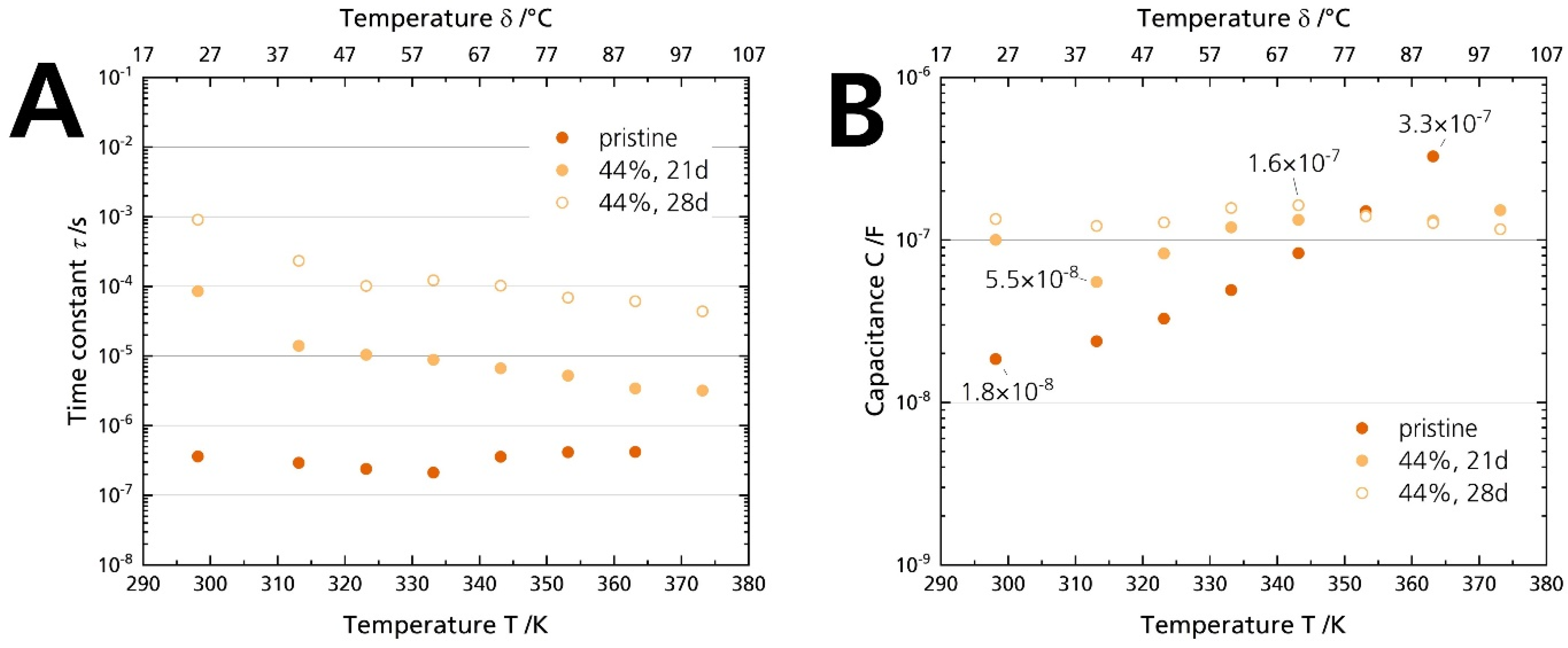
3.5.1. Ionic Conductivity
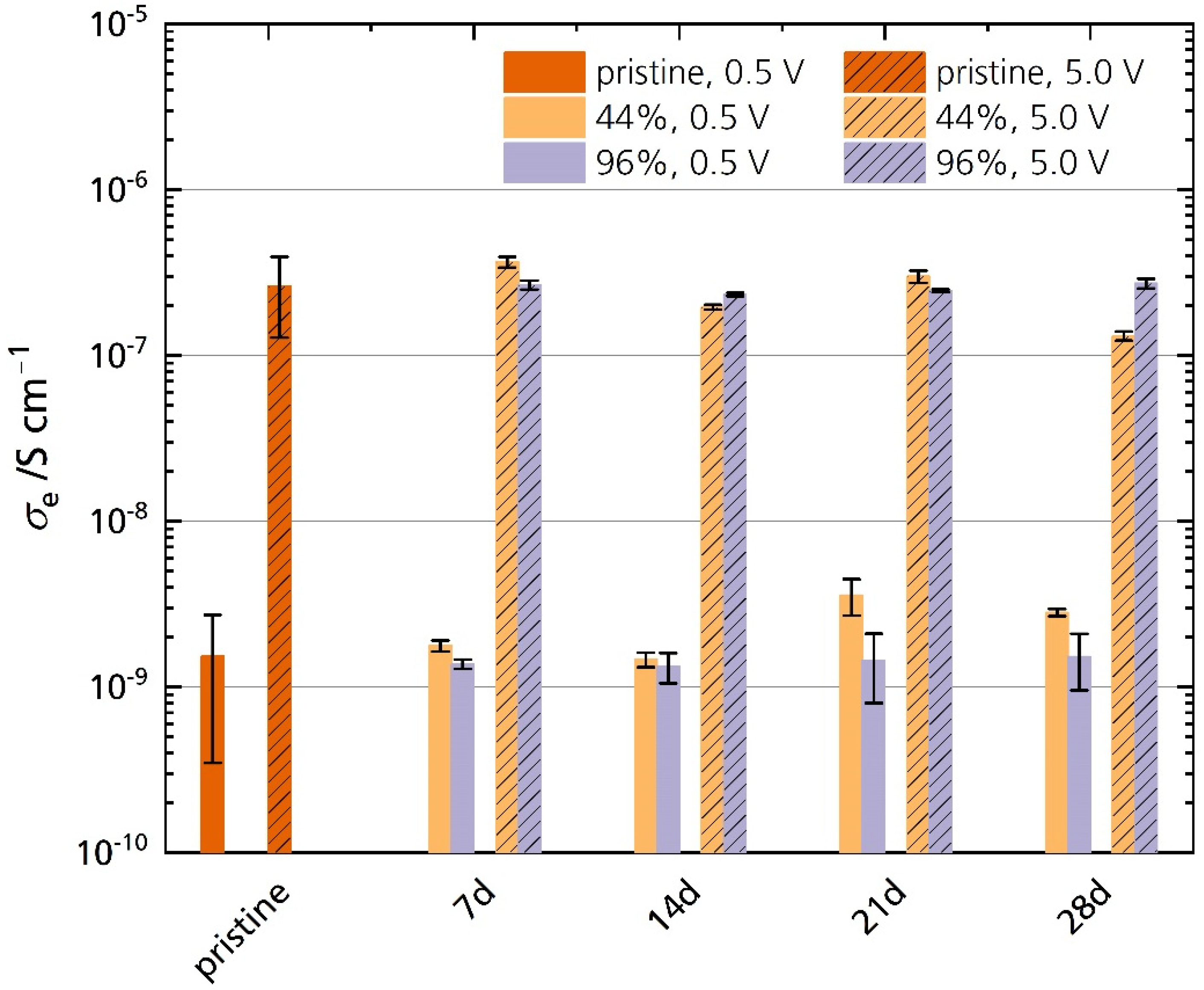
3.5.2. Electronic Conductivity
3.6. Critical Current Density
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, X.; Xia, G.; Lemmon, J.P.; Yang, Z. Advanced materials for sodium-beta alumina batteries: Status, challenges and perspectives. J. Power Sources 2010, 195, 2431–2442. [Google Scholar] [CrossRef]
- Wenzel, S.; Leichtweiss, T.; Weber, D.A.; Sann, J.; Zeier, W.G.; Janek, J. Interfacial Reactivity Benchmarking of the Sodium Ion Conductors Na3PS4 and Sodium β-Alumina for Protected Sodium Metal Anodes and Sodium All-Solid-State Batteries. ACS Appl. Mater. Interfaces 2016, 8, 28216–28224. [Google Scholar] [CrossRef]
- Pujar, P.; Gupta, B.; Sengupta, P.; Gupta, D.; Mandal, S. Sodium ion incorporated alumina—A versatile anisotropic ceramic. J. Eur. Ceram. Soc. 2019, 39, 4473–4486. [Google Scholar] [CrossRef]
- Hueso, K.B.; Palomares, V.; Armand, M.; Rojo, T. Challenges and perspectives on high and intermediate-temperature sodium batteries. Nano Res. 2017, 255, 410. [Google Scholar] [CrossRef]
- Li, G.; Lu, X.; Kim, J.Y.; Lemmon, J.P.; Sprenkle, V.L. Cell degradation of a Na–NiCl2 (ZEBRA) battery. J. Mater. Chem. A 2013, 1, 14935–14942. [Google Scholar] [CrossRef]
- Fertig, M.P.; Skadell, K.; Schulz, M.; Dirksen, C.; Adelhelm, P.; Stelter, M. From High- to Low-Temperature: The Revival of Sodium-Beta Alumina for Sodium Solid-State Batteries. Batter. Supercaps 2022, 5, e202100131. [Google Scholar] [CrossRef]
- Deng, T.; Ji, X.; Zou, L.; Chiekezi, O.; Cao, L.; Fan, X.; Adebisi, T.R.; Chang, H.-J.; Wang, H.; Li, B.; et al. Interfacial-engineering-enabled practical low-temperature sodium metal battery. Nat. Nanotechnol. 2022, 17, 269–277. [Google Scholar] [CrossRef]
- Hasa, I.; Mariyappan, S.; Saurel, D.; Adelhelm, P.; Koposov, A.Y.; Masquelier, C.; Croguennec, L.; Casas-Cabanas, M. Challenges of today for Na-based batteries of the future: From materials to cell metrics. J. Power Sources 2021, 482, 228872. [Google Scholar] [CrossRef]
- Vaalma, C.; Buchholz, D.; Weil, M.; Passerini, S. A cost and resource analysis of sodium-ion batteries. Nat. Rev. Mater. 2018, 3, 652. [Google Scholar] [CrossRef]
- Beevers, C.A.; Ross, Μ.A.S. The Crystal Structure of “Beta Alumina” Na2O·11Al2O3. Z. Krist. Cryst. Mater. 1937, 97, 59–66. [Google Scholar] [CrossRef]
- Vries, R.C.; Roth, W.L. Critical Evaluation of the Literature Data on Beta Alumina and Related Phases: I, Phase Equilibria and Characterization of Beta Alumina Phases. J. Am. Ceram. Soc. 1969, 52, 364–369. [Google Scholar] [CrossRef]
- Sudworth, J.L.; Barrow, P.; Dong, W.; Dunn, B.; Farrington, G.C.; Thomas, J.O. Toward Commercialization of the Beta-Alumina Family of Ionic Conductors. MRS Bull. 2000, 25, 22–26. [Google Scholar] [CrossRef]
- Bragg, W.L.; Gottfried, C.; West, J. The Structure of β Alumina. Z. Krist. Cryst. Mater. 1931, 77, 255–274. [Google Scholar] [CrossRef]
- Dell, R.M.; Moseley, P.T. Beta-alumina electrolyte for use in sodium/sulphur batteries: Part I. Fundamental properties. J. Power Sources 1981, 6, 143–160. [Google Scholar] [CrossRef]
- Yamaguchi, G.; Suzuki, K. On the Structures of Alkali Polyaluminates. Bull. Chem. Soc. Jpn. 1968, 41, 93–99. [Google Scholar] [CrossRef]
- Dirksen, C.L.; Skadell, K.; Schulz, M.; Fertig, M.P.; Stelter, M. Influence of 3d Transition Metal Doping on Lithium Stabilized Na-β″-Alumina Solid Electrolytes. Materials 2021, 14, 5389. [Google Scholar] [CrossRef]
- Dunn, B. Effect of Air Exposure on the Resistivity of Sodium Beta and Beta Aluminas. J. Am. Ceram. Soc. 1981, 64, 125–128. [Google Scholar] [CrossRef]
- Chi, C.; Katsui, H.; Goto, T. Effect of Li addition on the formation of Na-β/βʹʹ-alumina film by laser chemical vapor deposition. Ceram. Int. 2017, 43, 1278–1283. [Google Scholar] [CrossRef]
- Armstrong, R.D.; Sellick, D.P. A. c. impedance studies on sodium β-alumina tubes for use in sodium-sulphur cells. J. Appl. Electrochem. 1979, 9, 623–628. [Google Scholar] [CrossRef]
- Armstrong, R.D.; Sellick, D.P. A Study into the Effect of Water Vapour on Sodium β-Aluminas. Electrochim. Acta 1980, 25, 1199–1204. [Google Scholar] [CrossRef]
- Will, F.G. Effect of Water on Beta Alumina Conductivity. J. Electrochem. Soc 1976, 123, 834–836. [Google Scholar] [CrossRef]
- Harbach, F. Further experiments concerning the water take-up of fully and partially stabilized beta″ alumina ceramics. Solid State Ion. 1983, 9–10 Pt 1, 231–236. [Google Scholar] [CrossRef]
- Heavens, S.N. Surface layers in polycrystalline sodium\-alumina. J. Mater. Sci. 1982, 17, 965–969. [Google Scholar] [CrossRef]
- Flor, G.; Marini, A.; Massarotti, V.; Villa, M. Reactivity of β-Aluminas with Water. Solid State Ion. 1981, 2, 195–204. [Google Scholar] [CrossRef]
- Viswanathan, L.; Virkar, A.V. Wetting characteristics of sodium on β-alumina and on nasicon. J. Mater. Sci. 1982, 17, 753–759. [Google Scholar] [CrossRef]
- Kawamura, T.; Okada, S.; Yamaki, J. Decomposition reaction of LiPF6-based electrolytes for lithium ion cells. J. Power Sources 2006, 156, 547–554. [Google Scholar] [CrossRef]
- Lux, S.F.; Lucas, I.T.; Pollak, E.; Passerini, S.; Winter, M.; Kostecki, R. The mechanism of HF formation in LiPF6 based organic carbonate electrolytes. Electrochem. Commun. 2012, 14, 47–50. [Google Scholar] [CrossRef]
- Sharafi, A.; Meyer, H.M.; Nanda, J.; Wolfenstine, J.; Sakamoto, J. Characterizing the Li–Li7La3Zr2O12 interface stability and kinetics as a function of temperature and current density. J. Power Sources 2016, 302, 135–139. [Google Scholar] [CrossRef]
- Dirksen, C.L.; Skadell, K.; Schulz, M.; Stelter, M. Effects of TiO2 doping on Li+-stabilized Na-β″-alumina for energy storage applications. Sep. Purif. Technol. 2019, 213, 88–92. [Google Scholar] [CrossRef]
- De Jonghe, L.C.; Buechele, A. Chemical colouration of sodium beta-aluminas. J. Mater. Sci. 1982, 17, 885–892. [Google Scholar] [CrossRef]
- Bay, M.-C.; Heinz, M.V.; Danilewsky, A.N.; Battaglia, C.; Vogt, U.F. Analysis of c-lattice parameters to evaluate Na2O loss from and Na2O content in β″-alumina ceramics. Ceram. Int. 2021, 47, 13402–13408. [Google Scholar] [CrossRef]
- Sharafi, A.; Yu, S.; Naguib, M.; Lee, M.; Ma, C.; Meyer, H.M.; Nanda, J.; Chi, M.; Siegel, D.J.; Sakamoto, J. Impact of air exposure and surface chemistry on Li–Li7La3Zr2O12 interfacial resistance. J. Mater. Chem. A 2017, 5, 13475–13487. [Google Scholar] [CrossRef]
- Garbarczyk, J.E.; Jakubowski, W.; Wasiucionek, M. Effect of selected mobile ions on moisture uptake by beta″ alumina. Solid State Ion. 1983, 9–10 Pt 1, 249–253. [Google Scholar] [CrossRef]
- Bay, M.-C.; Wang, M.; Grissa, R.; Heinz, M.V.F.; Sakamoto, J.; Battaglia, C. Sodium Plating from Na-β″-Alumina Ceramics at Room Temperature, Paving the Way for Fast-Charging All-Solid-State Batteries. Adv. Energy Mater. 2020, 10, 1902899. [Google Scholar] [CrossRef]
- Spencer Jolly, D.; Ning, Z.; Darnbrough, J.E.; Kasemchainan, J.; Hartley, G.O.; Adamson, P.; Armstrong, D.E.J.; Marrow, J.; Bruce, P.G. Sodium/Na β″ Alumina Interface: Effect of Pressure on Voids. ACS Appl. Mater. Interfaces 2020, 12, 678–685. [Google Scholar] [CrossRef]
- Lu, X.; Chang, H.J.; Bonnett, J.F.; Canfield, N.L.; Jung, K.; Sprenkle, V.L.; Li, G. Effect of cathode thickness on the performance of planar Na-NiCl2 battery. J. Power Sources 2017, 365, 456–462. [Google Scholar] [CrossRef]
- Heinz, M.V.; Graeber, G.; Landmann, D.; Battaglia, C. Pressure management and cell design in solid-electrolyte batteries, at the example of a sodium-nickel chloride battery. J. Power Sources 2020, 465, 228268. [Google Scholar] [CrossRef]
- Huggins, R.A. Simple method to determine electronic and ionic components of the conductivity in mixed conductors a review. Ionics 2002, 8, 300–313. [Google Scholar] [CrossRef]
- Armstrong, R.D.; Burnham, R.A. The effect of roughness on the impedance of the interface between a solid electrolyte and a blocking electrode. J. Electroanal. Chem. Interfacial Electrochem. 1976, 72, 257–266. [Google Scholar] [CrossRef]
- Irvine, J.T.S.; Sinclair, D.C.; West, A.R. Electroceramics: Characterization by Impedance Spectroscopy. Adv. Mater. 1990, 2, 132–138. [Google Scholar] [CrossRef]
- Hüttl, J.; Seidl, C.; Auer, H.; Nikolowski, K.; Görne, A.L.; Arnold, M.; Heubner, C.; Wolter, M.; Michaelis, A. Ultra-low LPS/LLZO interfacial resistance—Towards stable hybrid solid-state batteries with Li-metal anodes. Energy Storage Mater. 2021, 40, 259–267. [Google Scholar] [CrossRef]
- Han, F.; Westover, A.S.; Yue, J.; Fan, X.; Wang, F.; Chi, M.; Leonard, D.N.; Dudney, N.J.; Wang, H.; Wang, C. High electronic conductivity as the origin of lithium dendrite formation within solid electrolytes. Nat. Energy 2019, 4, 187–196. [Google Scholar] [CrossRef]
- Nazri, G.-A. (Ed.) Lithium Batteries: Science and Technology; Springer: Boston, MA, USA, 2003; ISBN 978-0-387-92674-2. [Google Scholar]
- Lu, Y.; Zhao, C.-Z.; Yuan, H.; Cheng, X.-B.; Huang, J.-Q.; Zhang, Q. Critical Current Density in Solid-State Lithium Metal Batteries: Mechanism, Influences, and Strategies. Adv. Funct. Mater. 2021, 31, 2009925. [Google Scholar] [CrossRef]
- Park, R.J.-Y.; Eschler, C.M.; Fincher, C.D.; Badel, A.F.; Guan, P.; Pharr, M.; Sheldon, B.W.; Carter, W.C.; Viswanathan, V.; Chiang, Y.-M. Semi-solid alkali metal electrodes enabling high critical current densities in solid electrolyte batteries. Nat. Energy 2021, 6, 314–322. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dopant | β″ [%] | r.h. [%] | t [d] | R | σmechanical | c0 |
|---|---|---|---|---|---|---|
| Li [19] | 85 | n/a | n/a | n/a | n/a | n/a |
| Li, Mg [19] | 50 | 100 | 9 | o | n/a | n/a |
| Li [20] | 80 | <15 | 30 | o | o | n/a |
| Mg [20] | 55 | 30–70 | 40 | o | n/a | n/a |
| Li, Mg [20] | 55 | 30–70 | 40 | o | n/a | n/a |
| Li [20] | 80 | 30–70 | 40 | + | − | n/a |
| Mg [20] | 55 | 100 | 40 | o | o | n/a |
| Li, Mg [20] | 55 | 100 | 40 | o | o | n/a |
| Li [20] | 70 | 100 | 40 | − | − | n/a |
| Mg, Y [21] | n/a | 30 mbar * | 14 | + | n/a | n/a |
| Mg [22] | n/a | 100 | >100 | n/a | o | + |
| n/a [23] | n/a | air | n/a | n/a | n/a | + |
| Li (this work) | 95 | 0 | 28 | o | o | n/a |
| Li (this work) | 95 | 44 | 28 | + | o | n/a |
| Li (this work) | 95 | 96 | 28 | n/a | o | n/a |
| Sample | σ0/MPa | σ0.001/MPa | m | σavg/MPa | σσ/MPa |
|---|---|---|---|---|---|
| pristine | 244.9 | 108.0 | 8.4 | 239.9 | 30.4 |
| 44%, 28 d | 234.3 | 174.0 | 23.2 | 233.3 | 8.2 |
| 96%, 28 d | 242.6 | 84.4 | 6.5 | 224.7 | 54.8 |
| Grain Boundary Capacitance Cgb /F | Interfacial Capacitance | |
|---|---|---|
| Derived from Figure 7 | 0.5 × 10−8 | Qint = 2.5 × 10−5 sn−1/Ω |
| Irvine et al. [40] | 10−11–10−8 | Cint = 10−7–10−5 s/Ω |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fertig, M.P.; Dirksen, C.; Schulz, M.; Stelter, M. Humidity-Induced Degradation of Lithium-Stabilized Sodium-Beta Alumina Solid Electrolytes. Batteries 2022, 8, 103. https://doi.org/10.3390/batteries8090103
Fertig MP, Dirksen C, Schulz M, Stelter M. Humidity-Induced Degradation of Lithium-Stabilized Sodium-Beta Alumina Solid Electrolytes. Batteries. 2022; 8(9):103. https://doi.org/10.3390/batteries8090103
Chicago/Turabian StyleFertig, Micha P., Cornelius Dirksen, Matthias Schulz, and Michael Stelter. 2022. "Humidity-Induced Degradation of Lithium-Stabilized Sodium-Beta Alumina Solid Electrolytes" Batteries 8, no. 9: 103. https://doi.org/10.3390/batteries8090103
APA StyleFertig, M. P., Dirksen, C., Schulz, M., & Stelter, M. (2022). Humidity-Induced Degradation of Lithium-Stabilized Sodium-Beta Alumina Solid Electrolytes. Batteries, 8(9), 103. https://doi.org/10.3390/batteries8090103