
Comparing the SEI Formation on Copper and Amorphous Carbon: A Study with Combined Operando Methods

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. The Electrochemical Cell, Materials, and Equipment
2.2. The Formation Procedure and Experimental Details
3. Results and Discussion
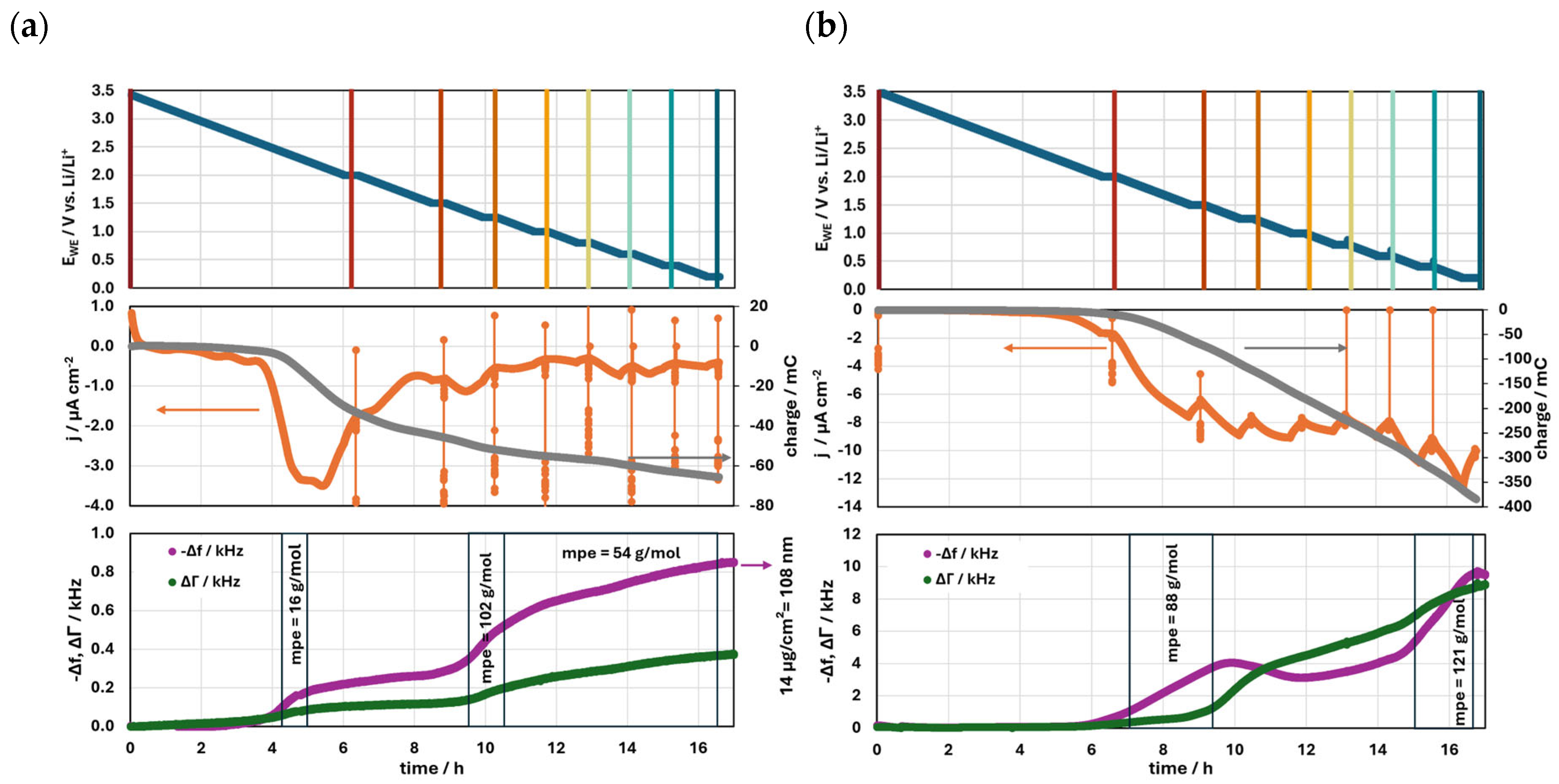
3.1. EQCM Results
3.1.1. Slow Formation Procedure
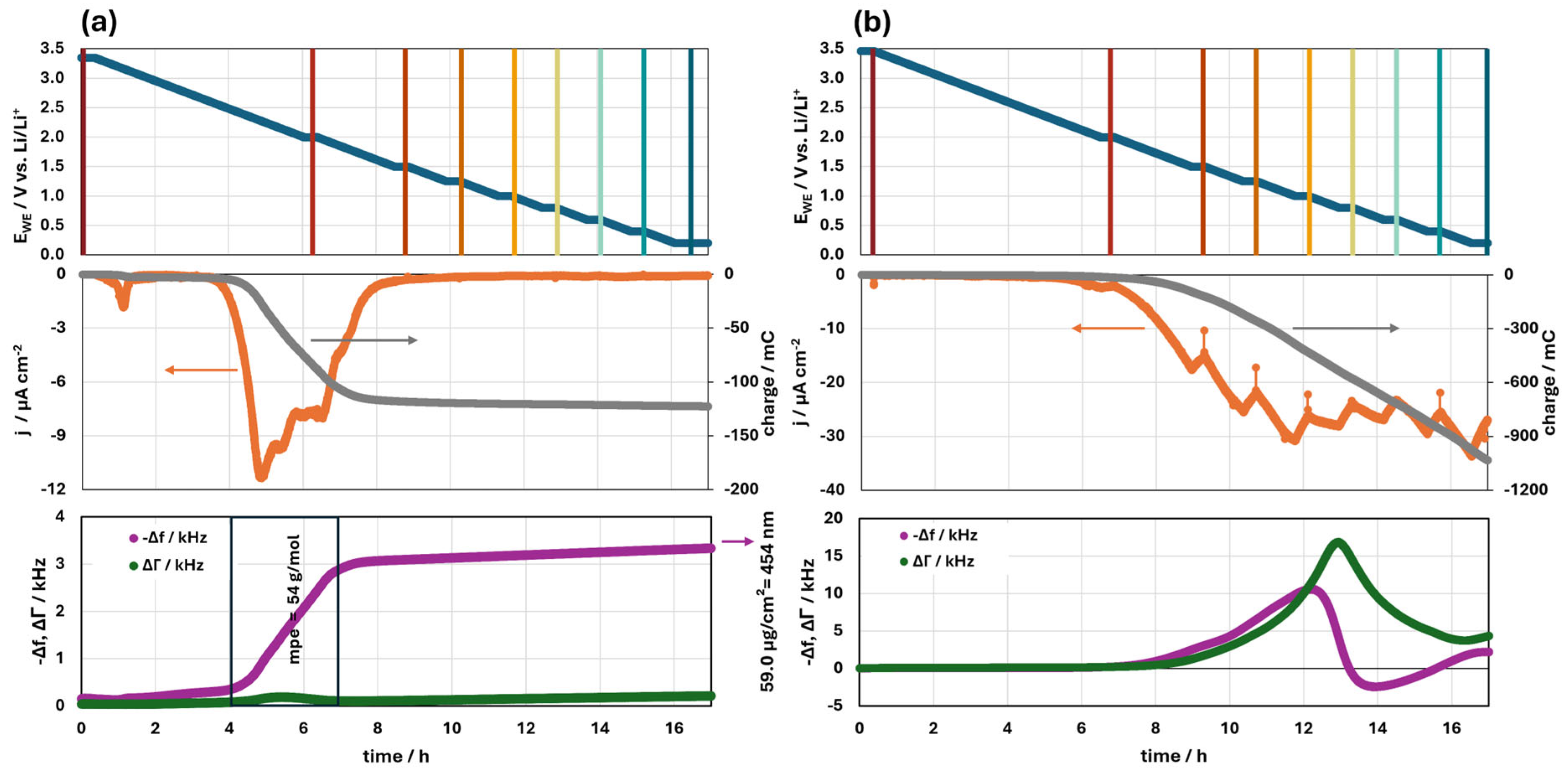
3.1.2. Fast Formation Procedure
3.1.3. The Effect of the VC Additive
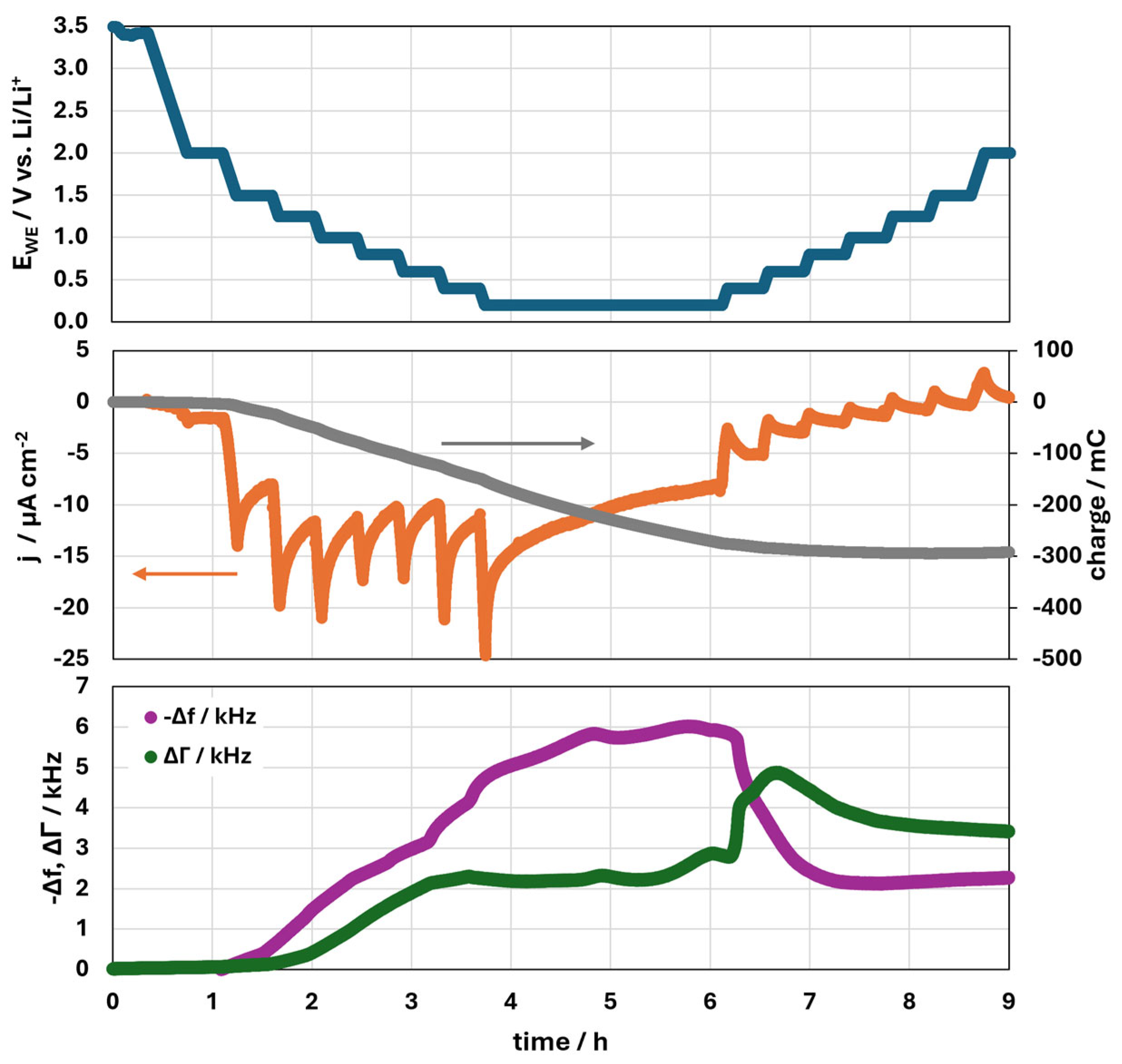
3.1.4. Continued SEI Formation and Potential Increase
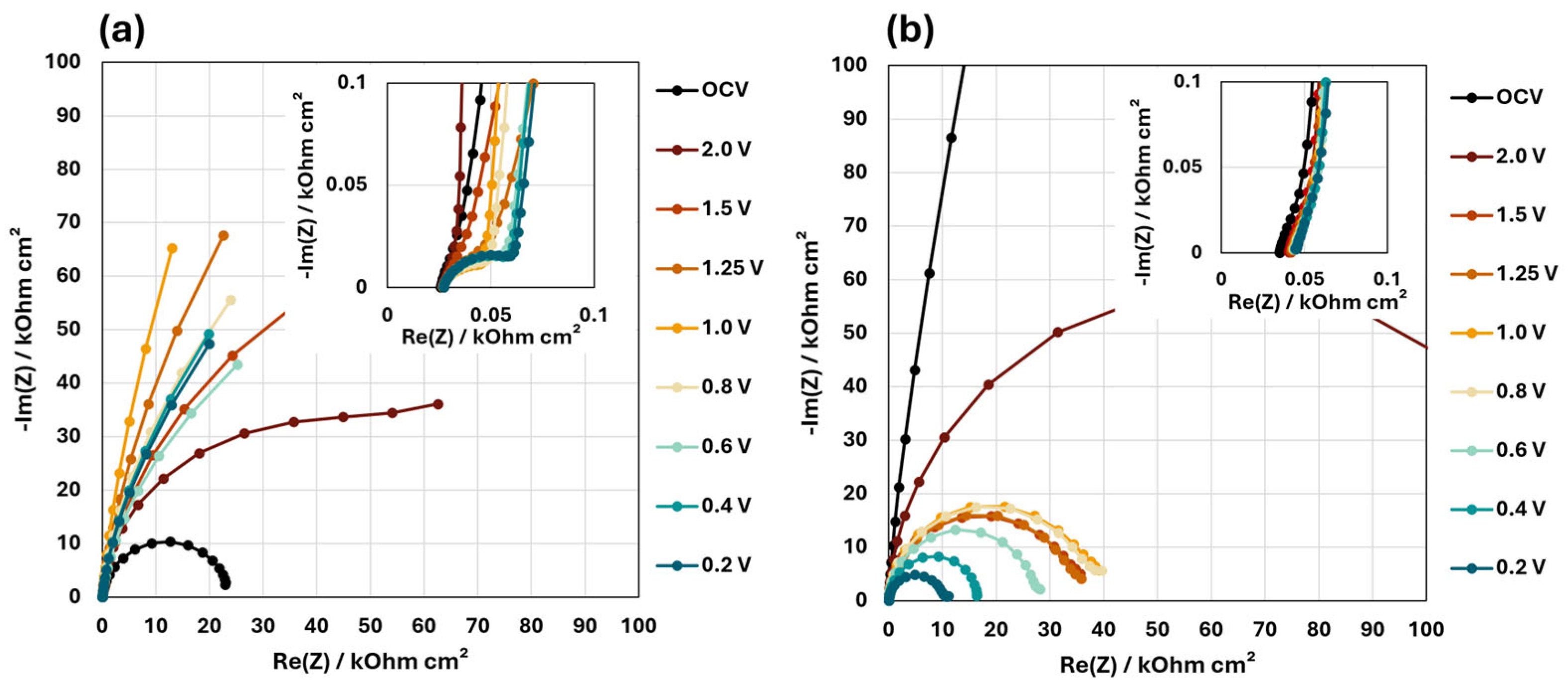
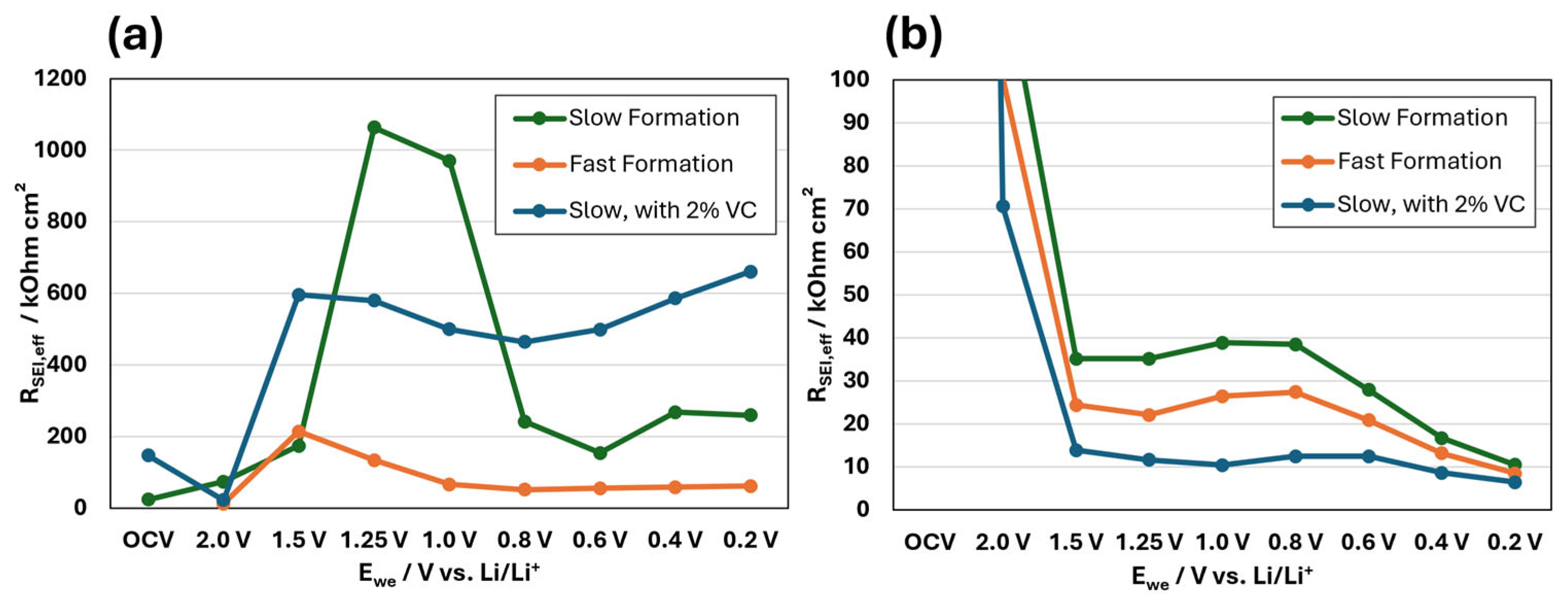
3.2. EIS Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Peled, E. The Electrochemical Behavior of Alkali and Alkaline Earth Metals in Nonaqueous Battery Systems—The Solid Electrolyte Interphase Model. J. Electrochem. Soc. 1979, 126, 2047. [Google Scholar] [CrossRef]
- Winter, M. The Solid Electrolyte Interphase—The Most Important and the Least Understood Solid Electrolyte in Rechargeable Li Batteries. Z. Physiol. Chem. 2009, 223, 1395–1406. [Google Scholar] [CrossRef]
- Jia, T.; Zhong, G.; Lv, Y.; Li, N.; Liu, Y.; Yu, X.; Zou, J.; Chen, Z.; Peng, L.; Kang, F.; et al. Prelithiation Strategies for Silicon-Based Anode in High Energy Density Lithium-Ion Battery. Green Energy Environ. 2023, 8, 1325–1340. [Google Scholar] [CrossRef]
- Li, H.; Zhou, H. Enhancing the Performances of Li-Ion Batteries by Carbon-Coating: Present and Future. Chem. Commun. 2012, 48, 1201–1217. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, C.; Fujimoto, H.; Tokumitsu, K.; Mabuchi, A.; Kasuh, T. Reduction of the Irreversible Capacity of a Graphite Anode by the CVD Process. Carbon 2001, 39, 1409–1413. [Google Scholar] [CrossRef]
- Zhu, P.; Gastol, D.; Marshall, J.; Sommerville, R.; Goodship, V.; Kendrick, E. A Review of Current Collectors for Lithium-Ion Batteries. J. Power Sources 2021, 485, 229321. [Google Scholar] [CrossRef]
- Matsumoto, F.; Fukunishi, M. Review of Current Collector-, Binder-, Conductive Additive-Free, and Freestanding Electrodes in Lithium and Related Batteries. Batteries 2024, 10, 330. [Google Scholar] [CrossRef]
- Wu, B.; Chen, C.; Raijmakers, L.H.J.; Liu, J.; Danilov, D.L.; Eichel, R.-A.; Notten, P.H.L. Li-Growth and SEI Engineering for Anode-Free Li-Metal Rechargeable Batteries: A Review of Current Advances. Energy Storage Mater. 2023, 57, 508–539. [Google Scholar] [CrossRef]
- Menkin, S.; O’Keefe, C.A.; Gunnarsdóttir, A.B.; Dey, S.; Pesci, F.M.; Shen, Z.; Aguadero, A.; Grey, C.P. Toward an Understanding of SEI Formation and Lithium Plating on Copper in Anode-Free Batteries. J. Phys. Chem. C 2021, 125, 16719–16732. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.; Pasta, M. Solid Electrolyte Interphases in Lithium Metal Batteries. Joule 2023, 7, 2228–2244. [Google Scholar] [CrossRef]
- Edström, K.; Herstedt, M.; Abraham, D.P. A New Look at the Solid Electrolyte Interphase on Graphite Anodes in Li-Ion Batteries. J. Power Sources 2006, 153, 380–384. [Google Scholar] [CrossRef]
- Schroder, K.W.; Celio, H.; Webb, L.J.; Stevenson, K.J. Examining Solid Electrolyte Interphase Formation on Crystalline Silicon Electrodes: Influence of Electrochemical Preparation and Ambient Exposure Conditions. J. Phys. Chem. C 2012, 116, 19737–19747. [Google Scholar] [CrossRef]
- Solchenbach, S.; Huang, X.; Pritzl, D.; Landesfeind, J.; Gasteiger, H.A. Monitoring SEI Formation on Graphite Electrodes in Lithium-Ion Cells by Impedance Spectroscopy. J. Electrochem. Soc. 2021, 168, 110503. [Google Scholar] [CrossRef]
- Steinhauer, M.; Risse, S.; Wagner, N.; Friedrich, K.A. Investigation of the Solid Electrolyte Interphase Formation at Graphite Anodes in Lithium-Ion Batteries with Electrochemical Impedance Spectroscopy. Electrochim. Acta 2017, 228, 652–658. [Google Scholar] [CrossRef]
- Iurilli, P.; Brivio, C.; Wood, V. On the Use of Electrochemical Impedance Spectroscopy to Characterize and Model the Aging Phenomena of Lithium-Ion Batteries: A Critical Review. J. Power Sources 2021, 505, 229860. [Google Scholar] [CrossRef]
- Kiani, S.; Gharibi, H.; Javadian, S.; Zhiani, M.; Kashani, H. The Effect of the SEI Layer on the Electrochemical Impedance in the Graphite/Li[Ni0.5Mn0.3Co0.2]O2 Lithium-Ion Full Cells. Appl. Surf. Sci. 2023, 633, 157638. [Google Scholar] [CrossRef]
- Luchkin, S.Y.; Lipovskikh, S.A.; Katorova, N.S.; Savina, A.A.; Abakumov, A.M.; Stevenson, K.J. Solid-Electrolyte Interphase Nucleation and Growth on Carbonaceous Negative Electrodes for Li-Ion Batteries Visualized with in Situ Atomic Force Microscopy. Sci. Rep. 2020, 10, 8550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Smith, K.; Jervis, R.; Shearing, P.R.; Miller, T.S.; Brett, D.J.L. Operando Electrochemical Atomic Force Microscopy of Solid–Electrolyte Interphase Formation on Graphite Anodes: The Evolution of SEI Morphology and Mechanical Properties. ACS Appl. Mater. Interfaces 2020, 12, 35132–35141. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Li, W.; Chen, Y.; Huang, F.; Jie, Y.; Li, X.; Cao, R.; Jiao, S. Nanoscale Characterization of the Solid Electrolyte Interphase and Lithium Growth by Atomic Force Microscopy. Battery Energy 2024, 3, 20230045. [Google Scholar] [CrossRef]
- Kawaura, H.; Harada, M.; Kondo, Y.; Kondo, H.; Suganuma, Y.; Takahashi, N.; Sugiyama, J.; Seno, Y.; Yamada, N.L. Operando Measurement of Solid Electrolyte Interphase Formation at Working Electrode of Li-Ion Battery by Time-Slicing Neutron Reflectometry. ACS Appl. Mater. Interfaces 2016, 8, 9540–9544. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, M.; Stich, M.; Kurniawan, M.; Seidlhofer, B.-K.; Trapp, M.; Bund, A.; Wagner, N.; Friedrich, K.A. In Situ Studies of Solid Electrolyte Interphase (SEI) Formation on Crystalline Carbon Surfaces by Neutron Reflectometry and Atomic Force Microscopy. ACS Appl. Mater. Interfaces 2017, 9, 35794–35801. [Google Scholar] [CrossRef] [PubMed]
- Kitz, P.G.; Lacey, M.J.; Novák, P.; Berg, E.J. Operando EQCM-D with Simultaneous in Situ EIS: New Insights into Interphase Formation in Li Ion Batteries. Anal. Chem. 2019, 91, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Kitz, P.G.; Novák, P.; Berg, E.J. Influence of Water Contamination on the SEI Formation in Li-Ion Cells: An Operando EQCM-D Study. ACS Appl. Mater. Interfaces 2020, 12, 15934–15942. [Google Scholar] [CrossRef] [PubMed]
- Kitz, P.G.; Lacey, M.J.; Novák, P.; Berg, E.J. Operando Investigation of the Solid Electrolyte Interphase Mechanical and Transport Properties Formed from Vinylene Carbonate and Fluoroethylene Carbonate. J. Power Sources 2020, 477, 228567. [Google Scholar] [CrossRef]
- Ivanov, S.; Mai, S.; Himmerlich, M.; Dimitrova, A.; Krischok, S.; Bund, A. Microgravimetric and Spectroscopic Analysis of Solid−Electrolyte Interphase Formation in Presence of Additives. ChemPhysChem 2019, 20, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Capone, F.G.; Sottmann, J.; Meunier, V.; Ramírez, L.P.; Grimaud, A.; Iadecola, A.; Scardamaglia, M.; Rueff, J.-P.; Dedryvère, R. Operando Observation of the Dynamic SEI Formation on a Carbonaceous Electrode by Near-Ambient Pressure XPS. Energy Environ. Sci. 2024, 17, 1509–1519. [Google Scholar] [CrossRef]
- Bommier, C.; Chang, W.; Li, J.; Biswas, S.; Davies, G.; Nanda, J.; Steingart, D. Operando Acoustic Monitoring of SEI Formation and Long-Term Cycling in NMC/SiGr Composite Pouch Cells. J. Electrochem. Soc. 2020, 167, 020517. [Google Scholar] [CrossRef]
- Stich, M.; Valdes Landa, J.E.; Pantenburg, I.; Krauss, F.T.; Baumer, C.; Roling, B.; Bund, A. Combining Operando Techniques for an Accurate Depiction of the SEI Formation in Lithium-Ion Batteries. Batteries 2025, 11, 117. [Google Scholar] [CrossRef]
- Johannsmann, D.; Langhoff, A.; Leppin, C. Studying Soft Interfaces with Shear Waves: Principles and Applications of the Quartz Crystal Microbalance (QCM). Sensors 2021, 21, 3490. [Google Scholar] [CrossRef] [PubMed]
- Sauerbrey, G. Verwendung von Schwingquarzen zur Wägung dünner Schichten und zur Mikrowägung. Z. Phys. 1959, 155, 206–222. [Google Scholar] [CrossRef]
- Wan, T.H.; Saccoccio, M.; Chen, C.; Ciucci, F. Influence of the Discretization Methods on the Distribution of Relaxation Times Deconvolution: Implementing Radial Basis Functions with DRTtools. Electrochim. Acta 2015, 184, 483–499. [Google Scholar] [CrossRef]
- Maradesa, A.; Py, B.; Wan, T.H.; Effat, M.B.; Ciucci, F. Selecting the Regularization Parameter in the Distribution of Relaxation Times. J. Electrochem. Soc. 2023, 170, 030502. [Google Scholar] [CrossRef]
- Peled, E.; Menkin, S. Review—SEI: Past, Present and Future. J. Electrochem. Soc. 2017, 164, A1703. [Google Scholar] [CrossRef]
- Song, H.; Gong, Y.; Su, J.; Li, Y.; Li, Y.; Gu, L.; Wang, C. Surfaces/Interfaces Modification for Vacancies Enhancing Lithium Storage Capability of Cu2O Ultrasmall Nanocrystals. ACS Appl. Mater. Interfaces 2018, 10, 35137–35144. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.; Strehle, B.; Solchenbach, S.; Gasteiger, H.A. Hydrolysis of Ethylene Carbonate with Water and Hydroxide under Battery Operating Conditions. J. Electrochem. Soc. 2016, 163, A1219. [Google Scholar] [CrossRef]
- Kwon, K.; Kong, F.; McLarnon, F.; Evans, J.W. Characterization of the SEI on a Carbon Film Electrode by Combined EQCM and Spectroscopic Ellipsometry. J. Electrochem. Soc. 2003, 150, A229. [Google Scholar] [CrossRef]
- Thaman, H.L.; Li, M.; Rose, J.A.; Narasimhan, S.; Xu, X.; Yeh, C.-N.; Jin, N.; Akbashev, A.; Davidoff, I.; Bazant, M.Z.; et al. Two-Stage Growth of Solid Electrolyte Interphase on Copper: Imaging and Quantification by Operando Atomic Force Microscopy. ACS Nano 2025, 19, 11949–11960. [Google Scholar] [CrossRef] [PubMed]
- Putra, R.P.; Matsushita, K.; Ohnishi, T.; Masuda, T. Operando Nanomechanical Mapping of Amorphous Silicon Thin Film Electrodes in All-Solid-State Lithium-Ion Battery Configuration during Electrochemical Lithiation and Delithiation. J. Phys. Chem. Lett. 2024, 15, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Thornton, D.B.; Koronfel, M.A.; Stephens, I.E.L.; Ryan, M.P. Degradation in Lithium Ion Battery Current Collectors. J. Phys. Energy 2021, 3, 032015. [Google Scholar] [CrossRef]
- Lee, H.; Cho, J.-J.; Kim, J.; Kim, H.-J. Comparison of Voltammetric Responses over the Cathodic Region in LiPF6 and LiBETI with and without HF. J. Electrochem. Soc. 2005, 152, A1193. [Google Scholar] [CrossRef]
- Soto, F.A.; Ma, Y.; Martinez de la Hoz, J.M.; Seminario, J.M.; Balbuena, P.B. Formation and Growth Mechanisms of Solid-Electrolyte Interphase Layers in Rechargeable Batteries. Chem. Mater. 2015, 27, 7990–8000. [Google Scholar] [CrossRef]
- Domack, A.; Prucker, O.; Rühe, J.; Johannsmann, D. Swelling of a Polymer Brush Probed with a Quartz Crystal Resonator. Phys. Rev. E 1997, 56, 680–689. [Google Scholar] [CrossRef]
- Johannsmann, D.; Petri, J.; Leppin, C.; Langhoff, A.; Ibrahim, H. Particle Fouling at Hot Reactor Walls Monitored In Situ with a QCM-D and Modeled with the Frequency-Domain Lattice Boltzmann Method. Results Phys. 2023, 45, 106219. [Google Scholar] [CrossRef]
- Plank, C.; Rüther, T.; Jahn, L.; Schamel, M.; Schmidt, J.P.; Ciucci, F.; Danzer, M.A. A Review on the Distribution of Relaxation Times Analysis: A Powerful Tool for Process Identification of Electrochemical Systems. J. Power Sources 2024, 594, 233845. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stich, M.; Leppin, C.; Krauss, F.T.; Valdes Landa, J.E.; Pantenburg, I.; Roling, B.; Bund, A. Comparing the SEI Formation on Copper and Amorphous Carbon: A Study with Combined Operando Methods. Batteries 2025, 11, 273. https://doi.org/10.3390/batteries11070273
Stich M, Leppin C, Krauss FT, Valdes Landa JE, Pantenburg I, Roling B, Bund A. Comparing the SEI Formation on Copper and Amorphous Carbon: A Study with Combined Operando Methods. Batteries. 2025; 11(7):273. https://doi.org/10.3390/batteries11070273
Chicago/Turabian StyleStich, Michael, Christian Leppin, Falk Thorsten Krauss, Jesus Eduardo Valdes Landa, Isabel Pantenburg, Bernhard Roling, and Andreas Bund. 2025. "Comparing the SEI Formation on Copper and Amorphous Carbon: A Study with Combined Operando Methods" Batteries 11, no. 7: 273. https://doi.org/10.3390/batteries11070273
APA StyleStich, M., Leppin, C., Krauss, F. T., Valdes Landa, J. E., Pantenburg, I., Roling, B., & Bund, A. (2025). Comparing the SEI Formation on Copper and Amorphous Carbon: A Study with Combined Operando Methods. Batteries, 11(7), 273. https://doi.org/10.3390/batteries11070273