1. Introduction
End-of-life batteries present a number of safety and logistics challenges due to the possibility of uncontrolled release of residual energy. These challenges are most apparent in high-energy-density lithium-ion batteries; however, any battery with sufficient energy is capable of creating an unsafe scenario when improperly stored or handled. A recent emphasis on battery recycling has also highlighted the need to safely transport end-of-life batteries toward a recycling destination, with two recent studies evaluating the dangers of lithium-ion batteries in municipal waste treatment facilities [
1,
2]. While the fires reported in waste treatment facilities have to date been caused by small batteries (e.g., batteries powering portable electronics devices or household electronics), the same concerns are amplified for end-of-life electric vehicle batteries [
3]. The U.S. Department of Transportation has issued safety advisory notices for the “Transportation of Lithium Batteries for Disposal or Recycling”, emphasizing the need to comply with the Hazardous Materials Regulations of the U.S. Code of Federal Regulations, which govern the requirement for shipping hazardous goods, including batteries [
4]. Due to the safety concerns presented by the transportation of batteries, the U.S. Code of Federal Regulations (49CFR§173.21 “Forbidden materials and packages”) states that the shipping of batteries and battery-powered devices is forbidden unless packaged in a manner that precludes the creation of sparks or the generation of dangerous amounts of heat. Damaged, defective, and recalled (DDR) batteries present even greater risks during transportation and are therefore subject to additional restrictions outlined in 49CFR§173.185 “Lithium cells and batteries”, which specifies that each cell or battery must be placed in individual, non-metallic inner packaging that is then surrounded by non-combustible, non-conductive, and absorbent cushioning material and then individually placed in appropriately marked outer packaging. While these guidelines exist to minimize issues occurring during the transportation of batteries, several recent examples of significant fires involving end-of-life batteries, including at least two fires at warehouses that became EPA superfund sites (April 2011 fire in Cartersville, GA, and June 2021 fire in Morris, IL, superfund site IDs GAR000035162 and ILN000520733, respectively), highlight that risks remain in the handling of end-of-life batteries. While current federal guidance focuses on packaging, labeling, and the education of potential shippers, a complimentary approach would be to reduce or completely prevent the possibility of energetic battery failure. One effective option to achieve this is by lowering the state-of-charge (SOC) of cells or batteries prior to transportation. Lithium-ion cells at a lower SOC are less likely to undergo thermal runaway in response to abuse, which has been widely reported by authors studying a variety of cell form factors and chemistries [
5,
6,
7,
8,
9,
10,
11]. In many cases, cells reduced to 0% SOC are inert to abuse and will not undergo thermal runaway. Therefore, reducing the SOC of end-of-life cells prior to transportation, storage, or recycling would be an ideal approach to mitigating the effects of accidental abuse.
In a detailed review on battery pretreatment from Yu et al., deactivation methodologies were categorized into four broad methods: (1) discharge through an electrical resistor, (2) discharge via electrolysis in a salt water solution, (3) disassembly under cryogenic conditions, and (4) thermal deactivation (i.e., controlled thermal abuse) [
12]. All of these methods have limitations and tradeoffs, and generally none are appropriate for use at the point of collection, implying that batteries must be stored or transported containing residual energy prior to deactivation. Mikita et al. presented a novel methodology for battery deactivation by introducing a redox shuttle, which resolves issues presented by cells with electrodes disconnected from their terminals; however, this method requires the deactivation solution to be injected into the spent battery via a small hole [
13]. Similar limitations apply to deactivation methods involving the extraction of electrolytes via pressurized or supercritical CO
2, in which cells must be breached or otherwise disassembled to enable electrolyte extraction [
14,
15,
16].
In this work, we demonstrate an electrical discharge methodology that is generically applicable to cells and batteries of any form factor or chemistry and is sufficiently low cost and provides a simple means to enable deactivation at the point of collection. Discharge through some method frequently precedes other materials recovery methods; however, electrical discharge (i.e., connection of battery terminals to a resistive load) is reported as potentially hazardous due to the generation of heat [
12,
17]. Therefore, it is critical to appropriately adjust the resistive load to the batteries being deactivated. Rather than rely on a conventional electrical resistor, which would need to be soldered or welded to the battery, we instead employ a polymer–carbon composite gel using water-soluble polyvinyl alcohol (PVA). By cross-linking PVA with sodium tetraborate (borax), a gel of adjustable rheology can be formed. Our approach was informed by previous reports of PVA–carbon composites for flexible electronics, which show the expected percolation behavior of electrical conductivity when increasing the carbon content in the PVA–carbon composite [
18,
19,
20,
21]. Borax cross-linked PVA has many interesting properties, notably its “self-healing” ability, which in the present application aids in forming a continuous network of gel between battery terminals [
22,
23]. Both borax and PVA are also relatively non-toxic and low cost, and for this reason have been used as an educational demonstration for many years, making them ideal as a potentially scaled up deactivation methodology [
24]. PVA–carbon gels were successfully used to completely deactivate several cell types, rendering them inert to abuse and therefore much safer for storage and transportation.
3. Results
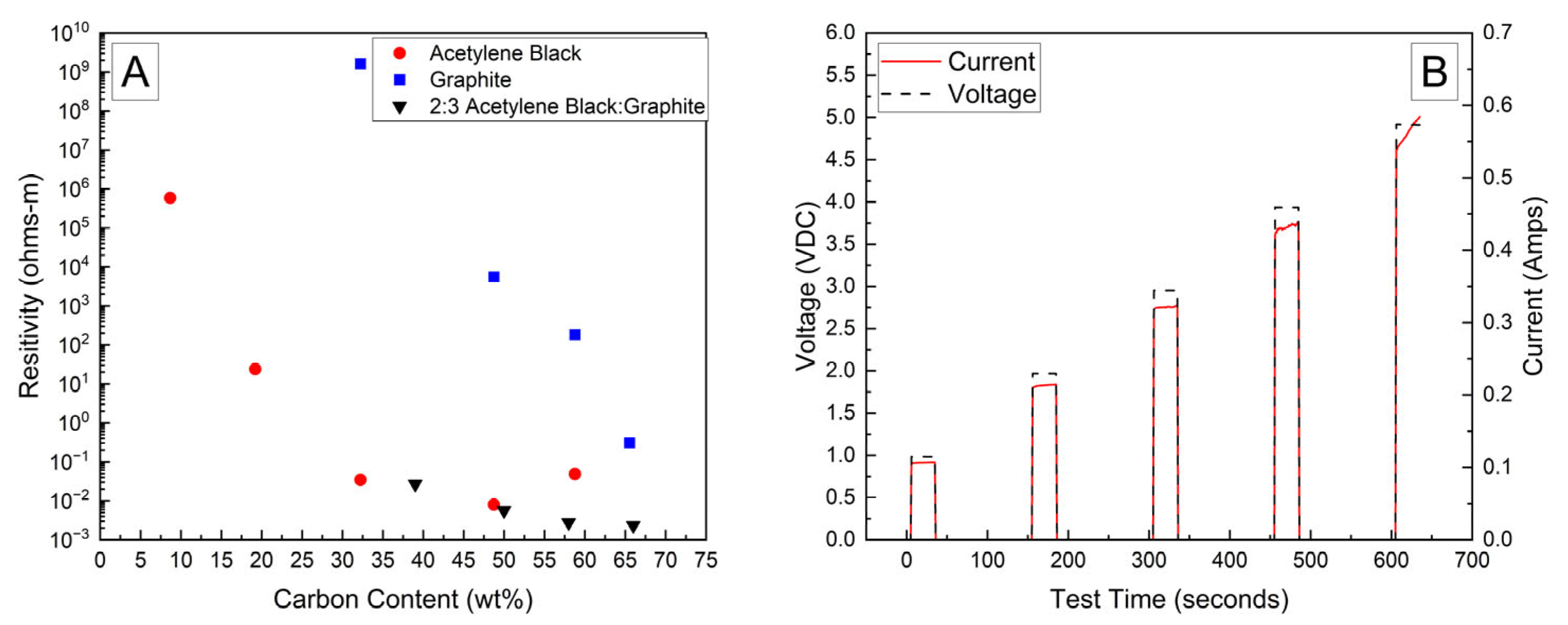
Bulk resistivities for a range of carbon contents and compositions are presented in
Figure 1A. The bulk resistivity of the fully dried samples of the PVA–carbon composite loaded on filter paper containing only acetylene black or graphite were observed to decrease by several orders of magnitude with increasing carbon content; however, the resistivity of the acetylene black-containing strips was much lower than those containing only graphite due to the lower volumetric mass density of acetylene black, which corresponds to a higher volume fraction of conductive carbon at a given mass fraction. Above about 50 wt% of acetylene black, however, the resistivity was observed to increase, which was attributed to the inability of these samples to maintain structural integrity when drying. Large cracks formed in the samples containing a high weight fraction of acetylene black, preventing the formation of an electrically conductive pathway. No cracking was observed for any weight fraction of the PVA–carbon composite containing only graphite, which is attributed to the much lower tap density of the graphite powder. Higher weight fractions of acetylene black led to a greater volume fraction of carbon in the PVA–carbon composite, which above approximately 50 wt% led to cracking. A mixture of acetylene black and graphite was found to resolve the cracking issue for a composite wt% of greater than 50% carbon, resulting in the lowest observed bulk resistivity for any sample composition tested. A ratio of 2:3 acetylene black to graphite by weight was found to produce the best consistency in the prepared gel, so this ratio was applied across a range of total carbon content compositions. Bulk resistivities of 2 × 10
−3 Ω-m were measured for strips of dried PVA–carbon gel containing 66 wt% of total carbon, which is consistent with the value expected for amorphous carbon and lower than the resistivity reported for a similar weight fraction of carbon nanotubes in PVA by Shaffer and Windle, suggesting that the dried composite is well mixed and the PVA does not significantly impede current flow [
18].
A representative current response for a dried sample containing 58 wt% of carbon black and graphite in a 2:3 ratio is shown in
Figure 1B. At voltages of 2 V and above, the current is observed to increase as a function of time, which becomes more prominent with increasing voltage. This behavior is attributed to a decrease in bulk resistivity due to resistive heating of the PVA–carbon composite, which is expected for both graphite and carbon black [
25]. Resistive heating of the lowest bulk resistivity samples was a potential issue when applying these formulations to high-voltage and or high-power-capable batterie; therefore, lower carbon content samples were selected in these cases to achieve a safe rate of discharge. Unless otherwise indicated, the 58 wt% carbon gel composition corresponding to a bulk resistivity of 2.8 × 10
−3 Ω-m was used for all deactivation experiments.
The voltage and temperature vs. time for a LiPo pouch cell deactivated by PVA–carbon gel is shown in
Figure 2. The cell voltage is seen to drop to <0% SOC (2.5 V) after approximately 19 h. At approximately 4 h, the voltage begins to vary unrelated to the deactivation process due to the placement of the voltage sense wires, which were likely influenced by the coincident placement of the PVA–carbon gel. Minimal heating was observed by a thermocouple applied to the surface of the cell. Temperature vs. time plots of the 1.2 Ah LiPo pouch cells in response to nail penetration at a 100% SOC and after deactivation are shown in
Figure 3. The deactivated cell voltage is below 2.5 V (0% SOC), which reflects a rebound from the ~0 V measured during the deactivation process shown in
Figure 2. At the moment of nail penetration, both the charged and deactivated cells experience an immediate drop in cell potential to 0 V; however, only the 100% SOC cell shows a rapid rise in temperature. While the maximum temperature seen for the nail penetration of the 100% SOC LiPo cell is fairly mild at only 88 °C, the maximum heating rate exceeded 1300 °C/min, far above the rule-of-thumb heating rate of 5 °C/min attributed to thermal runaway by Doughty and Roth [
26].
Cylindrical 18,650 cells were deactivated by first removing a portion of the shrink wrap covering the majority of the cell surface. Because 18,650 cells are can-negative, removing the shrink wrap significantly reduces the distance between the positively grounded header and negatively grounded can, which aids in creating the electrical connection between them. The voltage and temperature of an 18,650 LIB cell deactivated by the PVA–carbon gel is shown in
Figure 4, which shows that the 18,650 cell dropped below 0% SOC (<2.5 V) within about 15 h. The same 58 wt% carbon gel composition used on the LiPo was used on the 18,650 cell, and similar to the LiPo pouch cell, only a modest temperature rise was observed. Despite using an identical gel composition and starting cell OCV, the implied current for the 18,650 discharge rate (2.5 Ah over 15 h) is much higher than that of the LiPo cell (1.2 Ah over 20 h), which may be due to variations in the cell construction and resulting internal impedance or the effective resistance of the dried gel due to the difference in electrode spacing between the two cell types. An evaluation of the 18,650 cells before and after deactivation was conducted by external heating of the cells inside of a pressure vessel. The cells were heated at a rate of 1–2 °C/min to a maximum of 180 °C. The bomb was continuously purged with nitrogen, which was connected directly to the heated sampling lines of the FTIR gas analyzer. The voltage and temperature response of the 18,650 cells inside of the pressure vessel, along with a pressure transducer reading of the pressure inside of the vessel, are presented in
Figure 5A. Heating the 100% SOC 18,650 cell leads to a voltage loss attributed to activation of a current interrupt device (CID) at 130 °C. Further heating of the cell leads to an increase in measured pressure and a slight decrease in cell temperature attributed to cell venting at 157 °C. Once the cell reaches 199 °C, a rapid increase in temperature and pressure was observed, with the cell temperature reaching a maximum value of 475 °C. An identical heating profile was applied to an 18,650 cell deactivated by the PVA–carbon gel, which resulted in no signatures of thermal runaway, as shown in
Figure 5B. For the deactivated cell, a small increase in pressure was observed at approximately 120 °C; however, no rapid increase in pressure or temperature followed up to the maximum experimental temperature of 180 °C. A corresponding plot of carbon dioxide (CO
2), carbon monoxide (CO), methane (CH4), ethyl methyl carbonate (EMC), and hydrofluoric acid (HF), as measured by FTIR, is shown in
Figure 6 for both the 100% SOC cell (
Figure 6A) and PVA–carbon deactivated cell (
Figure 6B). For the 100% SOC cell, an initial spike in gas concentration is observed at approximately 123 min, which corresponds with the venting observed in the pressure and temperature traces shown in
Figure 5A. A much larger increase in concentration is observed for the 100% SOC cell at 160 min, which is correlated to the large increase in temperature and pressure from thermal runaway. Meanwhile, an increase in CO
2 and CO gas concentrations is seen around 70 min for the 18,650 cell deactivated by the PVA–carbon gel. This is attributed to the forced venting of the electrolyte from the cell due to external heating; however CH4 and HF remain near the detection limit throughout the test. Interestingly, the presence of EMC is detected from the deactivated cell after only 60 min of heating, corresponding to a temperature of only 106 °C. This suggests that the header of the deactivated cell may have been compromised. The gas composition of the deactivated cell compared to the 100% SOC cell shows that the concentration of CO
2 was reduced from 90,000 ppm to 4300 ppm, CO was reduced from 230,000 ppm to 200 ppm, CH4 was reduced from 35,000 ppm to 12 ppm, and HF was reduced from 200 ppm to 16 ppm. Concentrations of CO
2, CO, CH4, and HF first appear after the cell vents (noted by a decrease in temperature and an increase in measured pressure for the 100% SOC cell). Gas concentrations in the 100% SOC cell reach a maximum value commensurate with thermal runaway, while the decomposition gases emitted from the deactivated cell rapidly drop off after venting. While CO
2, CO, CH4, and HF are attributed to thermal decomposition of the electrolyte, the concentration of ethyl methyl carbonate (EMC), which is an assumed electrolyte solvent, was relatively consistent at 500 ppm from the 100% SOC cell and 280 ppm from the deactivated cell. For the 100% SOC cell, the EMC signal slowly increases and then drops off during thermal runaway, while for the deactivated cell, the EMC signal is fairly constant until the cell reaches 180 °C (the limit of the external heaters for this experiment) and begins to cool. This suggests that the EMC content is due to evaporation of the electrolyte solvent from the vented cell without significant thermal decomposition, which is also supported by the significantly lower concentrations of CO
2, CO, CH4, and HF.
The voltage and temperature response for a 6 V alkaline battery deactivated by the PVA–carbon gel is shown in
Figure 7. Both the voltage and capacity of the 6 V alkaline cell are significantly larger than the single cell shown in
Figure 2 and
Figure 4, and a correspondingly a larger temperature response is observed for the 6 V battery despite a relatively slower discharge time of approximately 25 h to reach 0% SOC (3 V lower voltage cutoff when under load). While faster discharge times are certainly possible based on the bulk resistivities demonstrated in
Figure 1, an even greater temperature rise would result. When the PVA–carbon gel was removed, the voltage of the alkaline batteries rebounded to approximately 5 V; however, this corresponds to a per-cell voltage of about 1.2 V (i.e., four cells connected in series), which is near 10% SOC according to the manufacturer specifications. As alkaline cells do not undergo thermal runaway, deactivation of the 6 V alkaline battery was demonstrated via an external short circuit. Resistive heating from a short circuit can lead to fires in packaging or other combustible materials even for batteries that do not undergo thermal runaway; therefore, even relatively benign battery chemistries still present some transportation hazards when improperly handled. As-received and post-deactivation alkaline batteries were shorted through a 333 µΩ shunt to determine the short-circuit current and temperature rise in response to electrical abuse. Temperature and voltage response for the alkaline batteries subjected to external short circuit is shown in
Figure 8. For both tests, the short-circuit condition was applied at an approximately 10 min test time, resulting in an immediate drop in voltage to near zero. For the as-received battery, a short-circuit current of over 2 A was briefly measured, and a corresponding rise in temperatures measured across the battery surface was observed. Thermocouples placed on the positive terminal recorded temperatures of over 200 °C, and the temperatures and short circuit current fell over approximately 2 h. For the post-deactivation battery, the current and temperature response were significantly reduced, with the short-circuit current reaching a maximum value of approximately 150 mA and the temperature increasing to only 30 °C. Like with the lithium-ion cells, reducing the state of charge of the alkaline battery has a clear beneficial effect on abuse tolerance.
A final demonstration of the PVA–carbon gel was conducted using a small power-tool battery containing three 18,650 cells connected in series. Due to the higher voltage and power capability, a relatively low carbon content of 39 wt% was used in the gel, corresponding to a bulk resistivity of 2.68 × 10
−2 Ω-m. The power-tool battery includes simple control circuitry between the external terminals and cells, which prevents operation of the battery in response to excessive temperatures and maintains appropriate cell voltages; however, no indication that this control circuit was engaged was observed, possibly because the control circuit only serves to provide a signal to a charger or tool. The temperature and voltage response of the 12 V power-tool battery is shown in
Figure 9, which resulted in a complete discharge to 0% SOC (7.5 V) after approximately 35 h. Both the voltage decrease and temperature increase are low for the first 20 h of the deactivation, which is attributed first to the time required to dry the residual water content of the gel and later to the decreasing resistance with temperature shown in
Figure 1 at higher voltages. Note that while the LiPo pouch cell, 18,650 lithium-ion cell, and 6 V alkaline battery deactivations were conducted in a fume hood with a high volume of air flow, the 12 V power-tool battery deactivation was conducted inside of the ARC enclosure used for abusive testing out of an abundance of caution. The ARC enclosure is sealed with very little air flow, which likely delayed the onset of significant current flow for the power-tool battery. After 25 h, the temperature recorded at the top of the power-tool battery using a thermocouple placed between the battery terminals began to rise rapidly, reaching a maximum temperature of approximately 80 °C, while a thermocouple placed on the front of the battery casing reached a maximum of about 35 °C. An infrared camera was used to confirm that heating was localized to the PVA–carbon material, while the majority of the battery remained relatively cool.
Following deactivation by the PVA–carbon gel, the power-tool batteries were dissembled, and the three 18,650 cells were removed for abusive testing via nail penetration. Upon disassembly, the voltage of the cells in the deactivated pack rebounded slightly and had voltages ranging from 1 to 2 V, similar to the value observed for the LiPO pouch cell. For comparison purposes, an untested battery was also disassembled, and individual cells were charged to 100% SOC prior to abuse testing. The voltage and temperature response of cells recovered from the 12 V power-tool battery is shown in
Figure 10. Like with the previous abuse tests of 100% lithium-ion cells, a rapid increase in temperature is observed at the moment of nail penetration occurring at 10 min of test time, while the deactivated cell shows a slight decrease in temperature attributed to the nitrogen overflow from the pneumatic nail driver.
The gas analysis conducted by FTIR is shown for the recovered power-tool cells in
Figure 11. Like with the 18,650 cells heated in a pressure vessel shown in
Figure 6, the cells recovered from the 12 V power-tool battery generated no detectable concentrations of any of the measured gases after deactivation. CO
2, CO, CH4, and DMC dropped from 45,000 ppm, 23,000 ppm, 2700 ppm, and 500 ppm to ~0 ppm after deactivation. HF was not observed for either case, presumably due to the highly reactive nature of the acid gas, which precluded detection when tested in open air, as required for the nail penetration apparatus. During nail penetration, the walls of the ARC chamber were not heated, and the gas samples were collected from the open chamber rather than directly from the sealed pressure vessel, both factors likely contributing to the undetectable HF and lower overall gas concentrations.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}