1. Introduction
Over the past few decades, energy storage systems (ESSs) have been widely explored in search of higher energy and higher power density, long cycle life, and faster charging for electronic devices, including cell phones, laptops, and especially for all-electric vehicles [
1,
2,
3]. It is well documented that the energy storage capacity and electrochemical stability in conventional lithium-ion batteries have primarily relied on the quality of the cathode material and thus, it has been regarded as the ‘Holy Grail’ of batteries. Microporous polyolefin separators and polyvinylidene fluoride (PVDF) binders are inherently inert without any contribution to energy storage except for mechanical stability. Liquid electrolytes derived from organic electrolyte solvents such as ethylene carbonate (EC) and dimethyl carbonate (DMC) were customarily used in conventional batteries by virtue of their higher ionic conductivity, electrochemical stability, and low cost. However, these organic liquid electrolytes are highly flammable due to their high vapor pressure and low flash points coupled with lithium dendrite formation, resulting in explosion and catching fire. Consequently, many efforts have been directed to the development of solid-state electrolytes such as inorganic ceramic electrolytes (ICE), solid polymer electrolytes (SPE), and, recently, solid polymer electrolyte membranes (PEM) to overcome these drawbacks [
4,
5,
6,
7,
8,
9,
10,
11,
12].
Recently, an innovative approach was introduced for storing extra energy within a multifunctional polymer electrolyte membrane (PEM) beyond the storage capacity of the conventional metal oxide or phosphate cathode. This achievement was realized via ion-dipole interaction (or complexation) between the functional groups of the PEM network and dissociated lithium cations. Excess lithium ions can be supplied from the lithium metal anode to the cathode through the PEM networks, i.e., a process known as ‘in situ lithiation’, by deeply discharging the battery to a very low potential range of the lithium metal electrode (i.e., −0.5 V to 0.5 V) during the battery conditioning process. The impact of such in-situ lithiation on the PEM’s electrochemical performance was manifested in cyclic voltammetry (CV) and galvanostatic charge/discharge cycling, encompassing diverse potential ranges, including the PEM (0.01 to 3.5 V), nickel manganese cobalt oxide (NMC 622) cathode (2.5 to 5 V), and the full battery (0.01 to 5 V) [
11]. Notably, the lithiated polysulfide (PS)-co-polyoxide (PO) polymer network-based PEM exhibited a remarkably high ionic conductivity of 1.18 × 10
−3 S/cm at room temperature [
12]. Furthermore, the above polysulfide-co-polyoxide PEM displayed an ability to store additional energy while boasting a specific capacity of approximately 150 mAh/g at a 0.1 C-rate within the PEM voltage range (0.01 to 3.5 V) in addition to 165 mAh/g at 0.2 C of NMC622 cathode (i.e., 2.5 to 4.6 V). Moreover, given the broader potential window of energy storage in PEM plus cathode (i.e., 0.5~4.6 V), the discharge cycling time can be prolonged, thereby covering a longer range [
13,
14,
15,
16,
17,
18].
Lithium Titanate (LTO, Li
4Ti
5O
12) is primarily used as an active material for anodes by virtue of its minimal volumetric expansion, which virtually eliminates the need for electrode swelling during charge and discharge cycling processes. Additionally, LTO anode formulations, which include binders and conductive additives, have been shown to be compatible with three-dimensional (3D) printing alongside LFP (LiFePO
4) formulations for the cathode [
19,
20]. By optimizing the ratios of the components, these formulations can be tailored to achieve the rheological properties necessary for successful 3D printing. While this study does not specifically focus on 3D printing electrodes, the current research on the LTO anode lays the groundwork for future exploration into 3D printing entire battery components. 3D printing of all the active battery components offers impactful design customization and structural optimization, thereby enhancing battery performance [
21,
22]. Various 3D printing methods are employed in battery fabrication, such as Vat Photopolymerization (VPP) and Material Extrusion. The selection of 3D printable materials controlling melt viscosity, such as cross-linkable polysulfide and polyoxide, is of paramount importance. Additionally, it is essential to consider the commercial implications in terms of scalability and sustainability [
23].
The choice of LTO electrode is two-fold. First, the specific capacity of LTO electrode and LFP cathode are comparable in experiments (i.e., about 120~130 mAh/g) as well as theoretically (i.e., 170 mAh/g for LTO cathode and 175 mAh/g for LFP cathode). However, LTO can be operated in the potential range (0.2~2.5 V), which is comparable to the present polysulfide-co-polyoxide-based PEM but lower than the higher potential range LFP (2.5~4.2 V). Hence, in lieu of Li metal, it is meritorious to use LTO (Li4Ti5O12) as anode and also as a potential Li-ion source against the high potential LFP (LiFePO4) cathode, both of which can be fabricated by 3D printing while covering a wider voltage of 0.1 V~4.3 V. The present study of LTO electrode is a first step to compare with the electrochemical performance of 3D printed LTO electrode using polymer precursor binders to control the viscosity (or shear thinning) in 3D printing.
Another goal is aimed at the comparison of the role of a single network (i.e., TMPETA, polyoxide) versus a dual PEM network (TMPETA-co-polysulfide network) in the electrochemical performance of the solid-state battery in the lithium metal/PEM network/LTO cathode configuration. The complexation of Li cations with ether oxygen of polyoxide (TMPETA) and with thiols, ether, and disulfide bonds of polysulfide (Thioplast) is anticipated to slow down the lithium-ion transport, which is reminiscent of the temporary holding of Li ions within the PEM network. Such ion-dipole complexation mechanism provides extra Li-ion storage capacity within the polysulfide-co-polyoxide dual-network PEM in addition to those of the LTO anode and LFP cathode. It can be anticipated that the more functional groups in the matrix network for ion-dipole complexation, the larger the storage capacity, and thus, dual polysulfide-polyoxide networks are likely to outperform the single polyoxide network. Moreover, the Li-ion concentration in the vicinity of ether oxygen of TMPETA and thiols, disulfide, and ether groups of Thioplast would be different, which in turn gives rise to the local ion concentration gradients between the two polysulfide and polyoxide networks (note that ether oxygen can hold one Li-ion whereas one sulfur can hold six Li ions). Consequently, the electrical potential difference thus generated locally between two different networks, further enhances electrochemical performance in the dual PS-co-PO PEM networks. The ultimate goal is the 3D printability of LTO and LFP electrodes in drone applications while realizing extra energy storage capacity afforded by the PS-co-PO dual networks along with improved electrochemical stability and capacity retention in comparison with those of the single polyoxide network.
2. Experimental Section
2.1. Materials
Trimethylolpropane ethoxylate triacrylate (TMPETA; molecular weight~912) and 2,2-dimethoxy-2-phenylacetophenone (DMPA; photo-initiator, purity of 99%) were purchased from Sigma Aldrich, Co. St. Louis MO. USA. Succinonitrile (SCN), poly(vinylidene fluoride) (PVDF; molecular weight~534,000) and 1-methyl-2-pyrrolidinone (NMP) were acquired from Sigma Aldrich, Co. Polysulfide precursor, Thioplast G1, was purchased from Nouryon, Co. Lithium bis(trifluoromethane sulfonyl)imide (LiTFSI) was obtained from TCI America, Inc. Portland, OR. USA. The activated material, lithium titanium oxide (LTO), conductive carbon (Super-P), and lithium metal foils were purchased from MTI Corporation.
2.2. Preparation of Polysulfide-co-Polyoxide Polymer Electrolyte Membrane
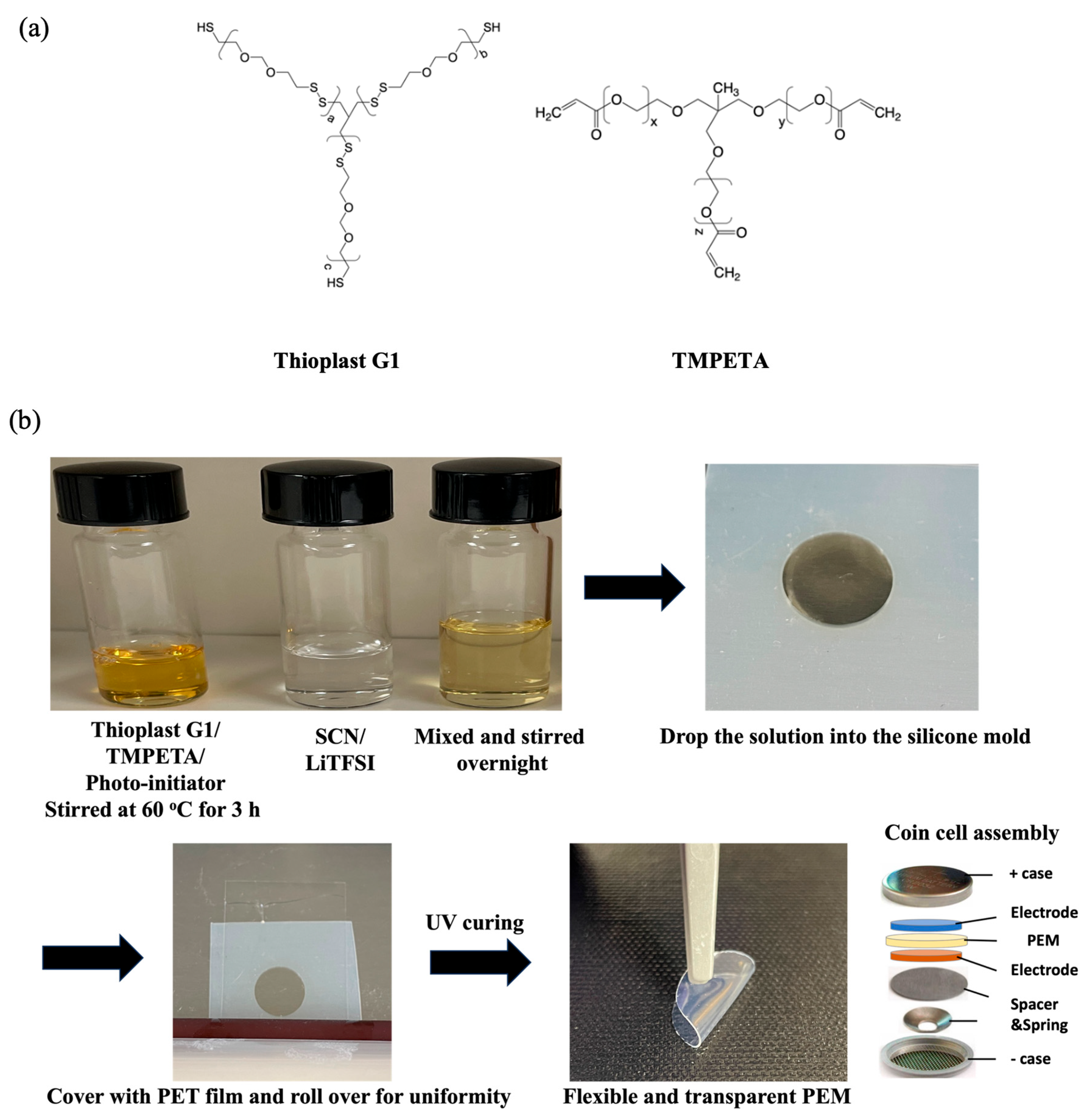
In the polymer electrolyte membrane preparation, polysulfide and polyoxide precursors were mixed in a weight ratio of 1:1 by rigorously stirring for 3 h until a homogeneous solution was obtained. A photo-initiator was subsequently added to the solution at a concentration of 3 wt% of the total polymer precursor weight.
The SCN plasticizer was first heated in a glovebox until it melted at a temperature of 50 °C and then LiTFSI salt was added and stirred until fully dissolved. Subsequently, polymer precursor was added to the transparent solution containing SCN plasticizer and LiTFSI salt and mixed thoroughly inside the glovebox. The solution was stirred using a magnetic stirrer for an additional 3 h until a homogeneous and transparent electrolyte mixture was obtained. The ratios of polymer precursor/SCN/LiTFSI salt in the solution were 30/35/35 by weight (%), respectively.
Silicon rubber sheets with a thickness of 300 μm were punched into a circular shape having a diameter of 15 mm. Subsequently, the polymer electrolyte solution was carefully poured into the prefabricated silicon molds and then exposed to UV light for 3 min to initiate a ‘thiol-ene’ click reaction between the thiol groups of polysulfide and the double bonds of TMPETA. This cross-linking reaction resulted in the formation of the polysulfide-co-polyoxide dual network structure. Interested readers are referred to the spectroscopic characterization of the detailed reaction mechanism and conversion in our previous paper [
11]. For the purpose of the comparison of the electrochemical performance of a dual network versus a single network, PEM containing TMPETA, SCN, and LiTFSI was fabricated using the same protocol. The resulting polysulfide-co-polyoxide/SCN/LiTFSI polymer electrolyte membranes remained homogeneous and exhibited a transparent appearance, suggesting miscibility among all components, although by no means a proof of complete miscibility.
2.3. Fabrication of Lithium Titanium Oxide (LTO) Cathode
Lithium titanium oxide (LTO), conductive carbon black (Super P), and poly(vinylidene fluoride) (PVDF) were dried in a vacuum oven at a temperature of 120 °C overnight to remove moisture from the materials. Subsequently, PVDF powder was fully dissolved in NMP solvent at a weight ratio of 10:90.
Next, LTO, carbon black (Super P), and PVDF were mixed together in a Thinky® mixer at the weight ratio was 80:10:10. The viscosity of the mixture was controlled by adjusting the amount of NMP solvent added to dissolve PVDF. The resulting cathode solution was then coated onto an aluminum foil using a coating machine equipped with a roller or coating bar. The coating speed was set at 3 mm/s, and a gap of 200 μm was maintained between the Al foil and the coating bar. After coating, the cathode was dried on a hot plate at 120 °C for 1 h. Subsequently, it was transferred to a vacuum oven chamber controlled at 100 °C under vacuum. This step was implemented to eliminate residual NMP solvent in the cathode, if any. The dried LTO cathode had a uniform thickness of about 65 μm and was assembled with PEM and lithium chips to fabricate coin cell batteries.
2.4. Electrochemical Characterization of PEM and Cathode
2.4.1. Ionic Conductivity Measurement
The prepared polymer electrolyte membrane (PEM) solution was poured into a homemade mold that was specifically designed for determining ionic conductivity. The dimensions of the mold were 10 mm × 10 mm × 1 mm (length × width × thickness). The PEM solution was then cured using a UV lamp, ensuring the solidification of the membrane. The cells were mounted on an AC impedance spectrometer (HP4192A, Hewlett-Packard) to measure the ionic conductivity of the PEMs. The temperature effect of the ionic conductivity was tested after placing the coin cells in a heating chamber, and the measurement was conducted by ramping the temperature from 25 °C to 100 °C at a rate of 1 °C/min. After reaching the target temperature, the cells were conditioned in the heating chamber for 15 min prior to running the experiment. By applying a constant voltage of 10 mV, the frequency scan was undertaken from 13 MHz to 5 Hz.
2.4.2. Cyclic Voltammetry (CV) and Charge/Discharge Cycle Measurement
CV tests were performed on the lithium metal/PEM/LTO coin cell batteries. Lithium metal batteries were assembled using a coin-cell (CR2032; MSE Supplies, Tucson, AZ, USA) configuration, including a spring for improved contact among all components. The PEM was sandwiched between a lithium metal anode and an LTO cathode. To ensure proper sealing and prevent current leakages from the edges, the PEM films were cut slightly larger, approximately 15 mm in diameter. The test was conducted at ambient conditions using a Multi Autolab-M204 potentiostat (Metrohm, Plainsboro, NJ, USA). CV scans were performed at a scan rate of 0.1 mV/s within a potential range of 0.3 to 3.2 V for lithium metal/PEM/LTO configuration and −0.5 to 4.0 V for lithium metal/PEM/stainless steel. This voltage range covered the oxidation and reduction peaks of both the disulfide and LTO cathode.
Galvanostatic charge and discharge (GCD) cycling tests were conducted in the lithium metal/PEM/LTO configuration. The charge/discharge cycling was performed at a rate of 0.1 C within a potential range from 0.3 to 3.2 V to determine the capacity retention.
3. Results and Discussion
In the present study, commercially available polysulfide (Thioplast G1) and polyoxide (TMPETA) with low T
g values of about −60 °C were chosen as polymer matrix precursors. As depicted in
Figure 1a, the chemical structure of polysulfide (Thioplast, PS) has one disulfide bond at each PEG arm near the core along with polyoxide (TMPETA, PO) network precursor. Solid polymer electrolyte based on polysulfide-co-polyoxide (PS-co-PO) dual network containing SCN plasticizer and LiTFSI salt was prepared inside the Argon glovebox by adding 3 wt% of photo-initiator. The thiol-ene click reaction was carried out by exposing the ternary mixture (i.e., polymer co-network/SCN/LiTFSI in the 30/35/35 weight ratio) to UV light at 365 nm for 60 s [
11]. For the purpose of comparison, a single polymer network was prepared in the same manner based on polyoxide (PO) precursor at the same weight ratio of 30/35/35. The prepared PEMs were transparent and flexible, as shown in
Figure 1b. The prepared polysulfide-co-polyoxide co-networks of polymer electrolyte membranes (PEMs) were assembled with a lithium metal anion and LTO cathode to verify their electrochemical properties in the next steps.
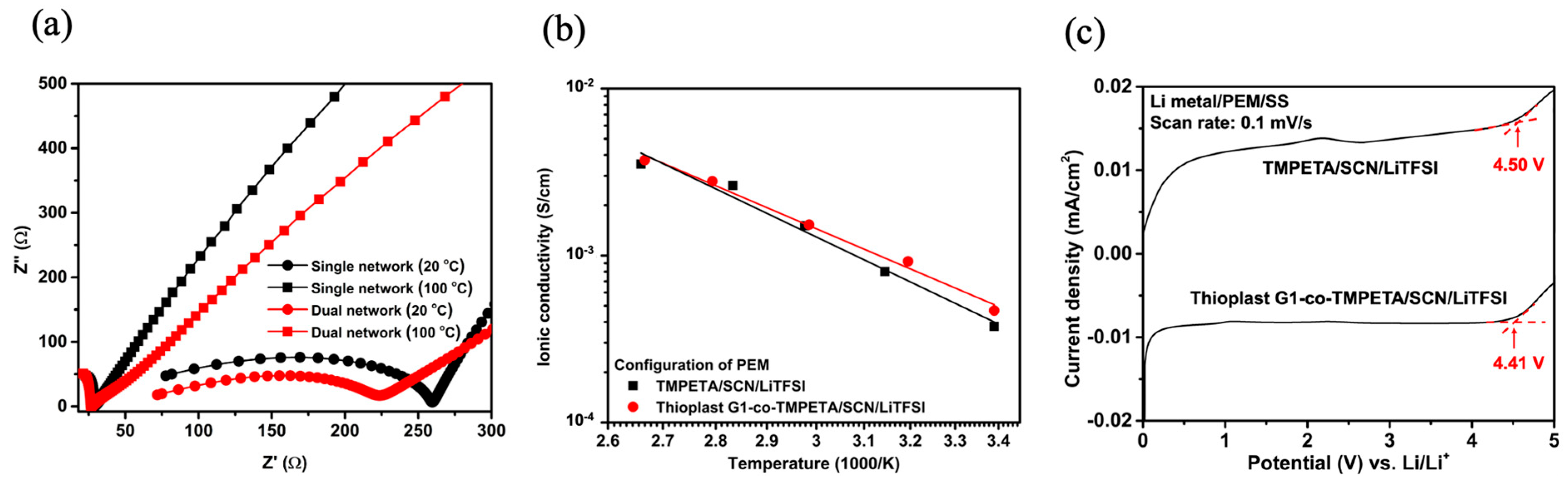
The ionic conductivity measurement conducted on the developed polysulfide-co-polyoxide dual network was conducted using an AC impedance spectrometer in comparison with a single polyoxide network containing succinonitrile (SCN) plasticizer and lithium bis(trifluoromethane)sulfonyl imide (LiTFSI) salt. The ionic conductivity for both PS-co-PO/SCN/LiTFSI and PO/SCN/LiTFSI at the ratio of 30/35/35 by weight are almost comparable, having reasonable conductivity values of about 4 × 10
−4 S/cm (
Figure 2a).
Based on these findings, the 30/35/35 polymer precursor/SCN plasticizer/LiTFSI salt-based PEM was chosen, having reasonably high conductivity levels of 4.67 × 10
−4 for the dual network PS-co-PO/SCN/LiTFSI and 3.76 × 10
−4 S/cm for the single network PO/SCN/LiTFSI at room temperature. As can be expected, these values increased to over 3.5 × 10
−3 S/cm at elevated temperatures of 90~100 °C. In the linear sweep voltammetry (LSV) scans, as indicated by the arrows at the onsets of the CV curves, PEMs appear stable against the stainless-steel (SS) electrode and comparable, i.e., 4.5 V for TMPETA/SCN/LiTFSI and 4.41 V for Thioplast G1-co-TMPETA/SCN/LiTFSI polymer electrolytes (
Figure 2b).
It is well known that the capacity of rechargeable batteries is primarily dominated by active materials in the cathodes. In order to enhance the capacity of battery systems, major efforts have been directed at modifying cathodes to improve their storage capacity, particularly in lithium–sulfur batteries. However, the present research takes a different approach by focusing on a recently discovered extra energy storage approach through the interaction of lithium ions with functional groups of a polymer electrolyte membrane (PEM). The present PEM is composed of polysulfide-co-polyoxide networks, LiTFSI salt, and SCN plasticizer. To comprehend the Li ion storage mechanism within the PEM, cyclic voltammetry (CV) and galvanostatic charge/discharge cyclic tests were conducted. These tests were performed in two configurations, i.e., lithium metal/PEM/LTO cathode (
Figure 3a,b) and lithium metal/PEM/stainless steel (
Figure 3c,d). The potential window ranged from 0.3 to 3.2 V for the former and from −0.5 to 4 V for the latter, with the inclusion of a single PO network and dual PS-co-PO network-based PEMs.
In
Figure 3a, the CV results at a 1 mV/s scan rate show clear oxidation and reduction peaks from the LTO cathode around 1.9 V and 1.2 V, respectively. These peaks originate from the battery cell containing the PEM configured with PO/SCN/LiTFSI at a 30/35/35 weight ratio. The peaks exhibit slight fluctuation in the initial cycles but stabilize after the third cycle. Conversely,
Figure 3b reveals data not only from the LTO oxidation and reduction peaks but also from a new oxidation peak around 2.9 V. This peak was absent in the 30/35/35 PO/SCN/LiTFSI cell configuration. A plausible explanation for this peak is due to the cleavage of disulfide bonds by dissociated Li cations forming sulfide radicals, which subsequently undergo complexation with Li ions.
Figure 3c,d illustrates the CV behavior within the combined potential range of the lithium metal and single PO network or dual PS-co-PO network-based PEMs (i.e., −0.5 to 4 V). In the lithium metal/PEM/stainless steel configuration, the oxidation and reduction processes primarily occur at the interface between the PEM and the lithium metal but less so at the interface of stainless steel. Notably,
Figure 3c shows no identifiable oxidation peak in the PO_PEM in the vicinity of 2.5 to 4 V, suggesting the lack of oxidation at the interface between the PEM and stainless-steel electrode. A very broad and weak peak appears around 1.5 V in the oxidation cycle. A minute reduction peak can be discerned at about 1.2 V, which may be ascribed to the reduction within the PO_PEM matrix. In contrast,
Figure 3d reveals stronger oxidation and reduction peaks when PS-co-PO_PEM is incorporated in the battery configuration. These peaks, located at around 2.5~3.5 V and 1.2 V, have approximately 10 times larger intensity relative to those in the single network PO-PEM. These peaks in PS-co-PO_PEM may be ascribed to chain scission of disulfide bonds in the PO-co-PO_PEM accompanied by the interaction between sulfur radicals and lithium ions undergoing oxidation and reduction at the PEM/SS electrode interface. More importantly, these peaks are clearly identifiable in
Figure 3b, in which LTO was employed as a cathode in its battery configuration. The oxidation peak from the PS-co-PO_ PEM located at around 2.9 V seemingly overlaps with the LTO oxidation peak at about 2~2.2 V. Of particular importance is that these CV results are reproducible, thereby further supporting their potential roles.
To substantiate the aforementioned argument regarding the extra energy storage capacity afforded by polysulfide constituents in the dual network of PS-co-PO_PEM, a series of charge/discharge cycling tests were conducted for 50 cycles at room temperature.
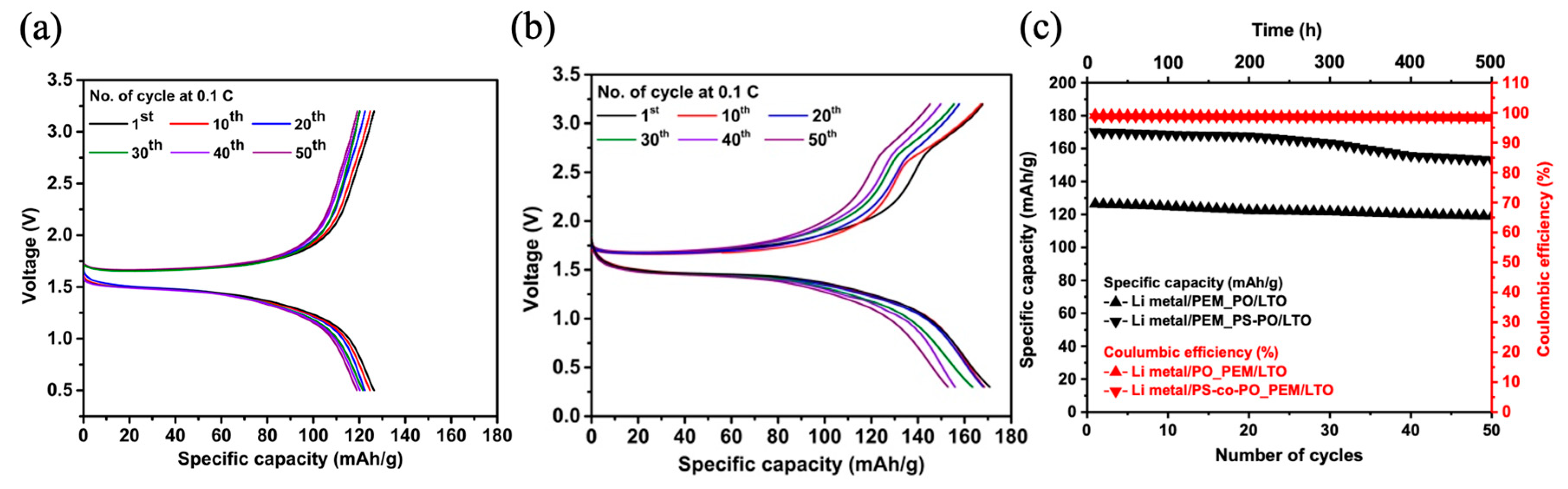
Figure 4a–c depicts the variations in specific capacity and Coulombic efficiency with the increasing number of cycles within a voltage range spanning from 0.3 to 3.2 V.
Initially, the specific capacity of the LTO cathode within the applied voltage window of 0.3 V to 3.2 V is approximately 126 mAh/g for the PO_PEM battery. This value slightly decreases to around 120 mAh/g after 50 cycles (i.e., about 95% capacity retention) with a Coulombic efficiency of approximately 99% (
Figure 4a,c). Notably, in the case of PS-PO-based PEM battery, the specific capacity of about 170 mAh/g, i.e., comparable to the theoretical value of LTO cathode, was obtained (
Figure 4b). Subsequently, the capacity settles around 153 mAh/g after 50 cycles, having capacity retention of approximately 90% with a Coulombic efficiency of 99% (
Figure 4b,c). The above-improved capacity can be undoubtedly attributed to the multifunctionality of the dual PS-co-PO networks. During the charging cycle, two discernible steps are observed at 1.7 V and 2.7 V, mirroring the oxidation peaks seen in CV results. Moreover, the discharge curve initiates at 1.7 V, stabilizes around 1.5 V, and then exhibits only a single step drop until reaching 0.3 V. This behavior aligns with the reduction reaction observed in the CV test using PS-co-PO-based PEM.
Of particular importance is the PEM co-networks incorporating disulfide groups within their backbone demonstrate an extra specific capacity, around 44 mAh/g initially and 33 mAh/g after 50th cycles, compared to those without such S–S groups. This enhancement may be attributed to the ability of disulfide bonds to interact with lithium cations, enabling oxidation and reduction processes facilitated by electron acceptance or release of the sulfur radicals at the PEM and electrode interfaces. These observations hold the promise of modifying the functional groups of polymeric matrix materials, which can contribute to higher battery storage capacities within the same volume and similar weight of the batteries. This phenomenon has also been noticed in the high nickel cobalt manganese cathodes (NCM622), whereby polysulfide (PS) containing disulfide bonds was utilized as a polymer binder in the cathode configuration instead of the traditional PVDF [
19]. In both cases, the incorporation of polysulfide-containing disulfide bonds in the dual network indeed affords additional energy storage relative to that of a single PO network. It is reasonable to consider polysulfide derivatives for use not only in polymer electrolyte matrix, but also as a polymer binder for cathode for promoting the extra storage capacity of solid-state batteries [
11].



{kind=link}
{kind=link}
{kind=link}
{kind=link}