Abstract
Mulberry (Morus alba L.) is a perennial woody plant with significant economic benefits and ecological value. The floral character of mulberry has an important impact on the yield and quality to its fruits and leaves. However, little is known about the molecular mechanism of mulberry floral differentiation still now. The transcriptome data were obtained via Illumina HiSeq high-throughput sequencing from male and female inflorescences of the monoecious mulberry. A total of 26.21 Gb clean data were obtained, and as many as 100,177 unigenes with an average length of 821.66 bp were successfully assembled. In comparative-omics analysis, 1717 differentially expressed genes (DEGs) were identified between male and female flowers and only a quarter of the DEGs were highly expressed in female flowers. The KEGG pathway enrichment analysis revealed that DEGs were involved in glucose and lipid metabolism, hormone signal transduction, and the regulation of related transcription factors. In addition, many DEGs related to flower development and plant sex differentiation have also been detected, such as PMADS1/2, AGAMOUS, FLOWERING LOCUS T (FT), APETALA 2 (AP2), TASSELSEED2 (TS2), and ARABIDOPSIS RESPONSE REGULATOR 17 (ARR17). Finally, the expression patterns of selected 20 DEGs were validated by q-PCR and the results showed that the transcriptome data were highly reliable. This study shows that the differentiation of male and female flowers of mulberry is affected and regulated by multiple factors, with transcription factors and hormone signals playing a key role. Briefly, the current data provide comprehensive insights into the mulberry tree’s floral differentiation as well as a bioinformatics framework for the development of molecular breeding of mulberry.
1. Introduction
Mulberry (Morus alba. L.) is an ideal plant for sustainable development for its rapid regeneration ability after cutting and it is distributed broadly across the world and has multiple uses. It has a positive impact on environment protection approaches such as water conservation, prevention of soil erosion, and improvement of air quality via carbon sequestration. It is widely recognized for its economic importance in producing Mori silk through the feeding of the leaves to silkworm (Bombyx mori) larvae [1]. In recent years, this plant species has been well-regarded as a multipurpose plant due to recognition of its role in the environmental safety approach. It has been exploited as every part of mulberry is utilized in the preparation of various products in the pharmaceutical, food, cosmetic, and health care industries [2]. Mulberry leaves and fruits possess health-promoting essential phytonutrient compounds like minerals, amino acids, fatty acids, sugars, phenolics, flavonoids, vitamins, and others [3,4]. Mulberry leaves are also consumed as an anti-diabetic medicine for diabetic patients as they contain anti-diabetic molecules such as 1-deoxynojirimycin (1-DNJ), isobavachalcone, morachalcon, fagomine, quercetin, and so on [5]. It has been widely used to treat various diseases such as hypertension, hyperglycemia, inflammation, fever, cough, and cancer for hundreds of years in China [6,7]. Mulberry fruits provide potential health benefits to humans, for example, delaying the aging process, protecting from cardiovascular diseases, antidiabetic activity, and reducing the cancer risk [7]. Mulberry leaves and fruits, therefore, are both agronomically and economically significant.
The majority of mulberry varieties are dioecious. Mulberry plants with male flowers cannot bear berries (mulberry fruit). Only the plants with whole female flowers can bear berries and not all of the female plants can produce mulberry fruit because there are few flowers on most mulberry plants. The mulberry floral character is, therefore, an important factor in determining the use and economic yield [8].
From the perspective of evolutionary history, dioecious plants are more advanced than monoecious plants, and gender differentiation of plants is a strategy to enhance the adaptation to environmental changes [9]. Currently, it is generally believed that genetic factors determine sex differentiation in plants. Hormones and the external environment also have a significant influence on plant sex determination [10]. Presently, about 70 plant sex-determining genes have been discovered. However, with the advancement of biotechnology, other sex-determining genes have been identified in several plants like grape (APRT3), persimmon (MeGI), black poplar (FERR-R and MSL), and kiwifruit (SyGI and FrBy) [11,12,13,14,15]. In the classic ABCDE model for flower development, relevant genes that regulate flower development in plants are classified into five groups, which are typically expressed in five types of floral organs (sepals, petals, stamens, carpels, and ovules). Furthermore, many studies have demonstrated that flowering is regulated by key genes related to six regulatory pathways including photoperiod, vernalization, autonomy, temperature, gibberellin, and aging [16]. In addition, hormone signals also play important roles in the transition from vegetative to reproductive growth [17]. Many genes are involved in the flowering mechanism and act directly or indirectly with each other, forming complex regulatory networks.
Transcriptome sequencing is well-established and widely used in screening candidate functional genes. In 2017, Shang et al. used Illumina RNA-Seq to sequence the transcriptomes of mulberry female flower buds at six developmental stages and found many DEGs, and concluded that ethylene signal is significant in the development of mulberry female flowers [18]. However, there is limited information about the DEGs between male and female flowers and during the development of floral organs in mulberry. In order to address this lack of knowledge, this study sequenced the transcriptomes of fully developed female and male flowers of two monoecious mulberry varieties and obtained a large number of DEGs via bioinformatic analysis. In addition, the expression of some candidate genes was quantitatively analyzed at different stages of male and female flower development, providing significant information for the floral differentiation and organ development of male and female flowers in mulberry. This study aimed to develop a scientific basis for the regulation of mulberry flowers for plant breeding.
2. Materials & Methods
2.1. Plant Materials
The mulberry (Morus alba. L.) plants were grown in a field at the Southwest University Mulberry Garden (Chongqing, China, 29°48′56′′ N, 106°24′49′′ E). After observation, it was previously found that only two mulberry trees showed a sex transition from completely female to monoecious among over 600 mulberry varieties. These two trees belong to different mulberry varieties, namely JiaLing40 and HongGuo1. Therefore, each of the two monoecious plants was sampled twice, male and female flowers in stage II, in total four sample pools (Figure 1a), and a single sample pool contained at least five inflorescences with consistent attributes. Half of the plant material in each sample pool was used for sequencing, and the other was used for q-PCR verification. Furthermore, due to the rarity of inflorescence material from monoecious mulberries, samples from four developmental stages (Figure 1d) of inflorescences of two completely dioecious mulberries were selected to characterize the expression patterns of MADS-box genes, each stage containing three biological replicates (Figure 1b,c). All the tissue samples were immediately frozen in liquid nitrogen and stored at −80 °C until further use.

Figure 1.
The flower pictures of mulberry. (a) Monoecious buds of JiaLing40 (one of the sequencing samples); (b,c) Male and female flowers in two completely dioecious mulberry varieties. (d) Four development stages of mulberry flowers. I: Flower buds stage; II: Bud-breaking stage; III: Swelling stage; IV: mature stage. ♂ represents male flowers, ♀ represents female flowers.
2.2. RNA Extraction and Sequencing
Total RNA was extracted separately from the two varieties and different sex of mulberry flowers with the TRIZOL Kit (Takara, Otsu, Japan). The purity, concentration, and integrity of the RNA samples were detected using a Nanodrop 2000 instrument (Thermo Scientific, Wilmington, DE, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). All indicators met the sequencing requirements of subsequent experiments. Then, approximately 3 μg of RNA per sample was used to construct the sequencing library by the NEBNext® Ultra™ Directional RNA Library Prep Kit (NEB, Ipswich, MA, USA). Based on SBS (Sequencing By Sequencing) technology, the Illumina Solexa HiSeq 2000 high-throughput sequencing platform (Biomarker Technologies Co., Beijing, China) was used to sequence the cDNA library with pair-end. After sequencing, the raw reads were filtered by removing adaptor sequences, contamination, and low-quality reads.
2.3. De Novo Assembly and Functional Annotation
After obtaining high-quality sequencing data, the filtered reads were assessed with FASTQC (Babraham Bioinformatics—FastQC A Quality Control tool for High Throughput Sequence Data), and the third-party software (Trinity V2.0.6, Arlington, TX, USA) with the default parameters was used to perform de novo transcriptome assembly. To investigate the function, the assembled unigenes were aligned in seven bioinformatics databases using the BLAST2.0 alignment algorithm with an E-value threshold of 1e-5. These databases included the Nr (http://www.ncbi.nlm.nih.gov, accessed on 5 June 2019), COG (http://www.ncbi.nlm.nih.gov/COG, accessed on 5 June 2019), KOG (http://genome.jgi-psf.org/help/kogbrowser.jsf, accessed on 5 June 2019), eggNOG (http://eggnog5.embl.de/#/app/home, accessed on 5 June 2019), SWISS-PROT (http://www.expasy.ch/sprot, accessed on 5 June 2019), and KEGG pathway database (http://www.genome.jp/kegg, accessed on 5 June 2019). Blast2GO was used for the GO (http://geneontology.org/, accessed on 5 June 2019) annotation results for the unigenes. The functional classification of unigenes was performed using the WEGO2.0 software (http://wego.genomics.org.cn/cgibin/wego/index.pl, accessed on 5 June 2019).
2.4. DEG Analysis
The Bowtie software (Bowtie: Manual (https://sourceforge.net), accessed on 8 June 2019) was used to compare the reads obtained by sequencing with the unigene library and combined with RSEM (RNA-Seq by Expectation-Maximization) to estimate the expression level based on the comparison results. The FPKM (Fragments Per Kilobase of transcript per Million mapped reads) value was used to indicate the expression abundance of the corresponding unigenes. In the process of differential expression analysis, the standard and effective Benjamini–Hochberg method was used to correct the p-value of the original hypothesis test. Finally, the corrected p-value, namely FDR (False Discovery Rate), as a key indicator for DEG screening was used, to reduce false positives caused by independent statistical hypothesis tests on the expression values of a large number of genes. In addition, FC (Fold Change), indicating the ratio of expression levels between two samples, was also taken into consideration. Then, the DEGs were screened by setting the threshold parameters in EBSeq software (Bioconductor—EBSeq v1.26.0, Buffalo, NY, USA) to FDR ≤ 0.001 and |Log2FC| ≥ 2. The DEGs were further used for GO and KEGG enrichment analyses, and combined with the gene functions reported in related literature to perform heatmap analysis by TBtools [19]. In addition, we uploaded the DEGs obtained from the screening to the PlantTFDB (PlantTFDB—Plant Transcription Factor Database @ CBI, PKU (gao-lab.org), accessed on 14 December 2020) to obtain the information of the differentially enriched transcription factor family types between male and female inflorescences.
2.5. q-PCR Validation
In order to verify the reliability of our Illumina sequencing data, 20 significantly DEGs were selected for q-PCR. The RNA samples used for q-PCR were the same as the sequencing samples. Besides, four unigenes that related to plant flower development were selected for q-PCR, using the RNA samples of flowers from the various stages of male and female plants, which were different from the sequencing samples. All samples conducted three biological replicates, and each biological replicate includes three technical replicates. The gene β-actin (LOC21394037) from Morus notabilis was used as an internal reference gene for q-PCR [20]. All the specific primers were designed by Primer Premier 6 software (PREMIER Biosoft, San Francisco, CA, USA) and are summarized in the Supplementary Table S1. Quantitative Real-time PCR was carried out using the ABI Step One with TB GreenTM Premix Ex TaqTM II kit (Takara, Otsu, Japan) and the 2−ΔΔCT and 2−ΔCT methods were used to measure the relative expression levels of unigenes [21]. And the data in this study were analyzed by SPSS software (IBM SPSS Statistics, Chicago, IL, USA) with general parameters, and a one-way ANOVA and Duncan’s multiple range test with biological statistical significance when p < 0.05. In addition, the correlation between RNA-Seq and q-PCR was analyzed using the Pearson correlation coefficient method with SPSS software.
2.6. Phylogenetic Analyses
In Supplementary Figures S4 and S5, the phylogenetic trees of two types of genes, MDAS-box and ARRs, were constructed, respectively. The partial gene sequences of the two types of genes in poplar and Arabidopsis were downloaded from the NCBI (National Center for Biotechnology Information (ncbi.nlm.nih.gov), accessed on 18 December 2020) website, and the sequences of the two types of genes in mulberry were provided by transcriptome data. The obtained genes of each category were aligned using MUSCLE. Then, two unrooted NJ trees were generated using MEGA-X (Home (megasoftware.net), accessed on 20 December 2020), ascertained with 1000 bootstrap.
3. Results
3.1. Sequencing, Assembly, and Annotation of the Mulberry Flower Reference Transcriptome
To obtain a reference transcriptome for mulberry flowers, RNA-seq libraries were constructed using RNA samples from male and female flowers and 27.06 Gb of raw data were obtained after sequencing. After filtering the raw data by removing the linker sequences and low-quality reads, 26.21 Gb of high-quality clean data were obtained. The clean data of each sample reached 6.36 Gb, with Q30 base percentage of 94.56% and greater, and with GC content of around 46% (Table 1).

Table 1.
Summary of sequencing data generated and analyzed in this study.
Trinity software (Arlington, TX, USA) was used for de novo assembly and a total of 100,177 unigenes were obtained, of which 23,246 were more than 1 kb in length. The total length of all assembled unigenes were 82,311,264 bp, the average length was 821.66 bp, and the N50 was 1555 bp (Table S2). The online BLAST tool was used to compare unigenes with public databases and perform functional annotations. A total of 55,288 (55.19%) unigenes have been annotated (Table S3). Of these, the NR database has the most annotations with 49,553 unigenes accounting for 49.46% of the total unigenes. Alternatively, the COG database had the least annotations, with only 16,356 unigenes accounting for 16.33% of the total unigenes (Figure 2a).
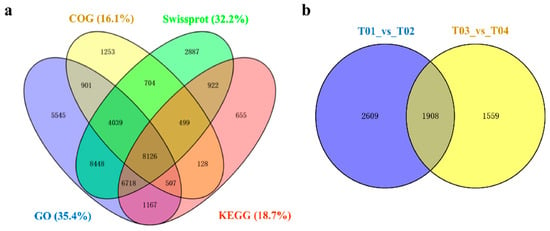
Figure 2.
Venn diagrams shared with all Unigenes and DEGs. (a) Venn diagram of all Unigenes in four annotation databases. (b) Venn diagram of total DEGs in different species.
3.2. Cluster Analysis of DEGs between the Male and Female Flowers in Mulberry
There were 4517 DEGs from group 1 (T01/T02) and 3467 DEGs from group 2 (T03/T04). There were 1593 up-regulated and 2924 down-regulated genes in male flowers compared to female flowers from group 1. There were 1090 up-regulated and 2377 down-regulated genes in male flowers compared to female flowers from group 2. Furthermore, 1908 genes appeared in common in the two groups (Figure 2b) and 1717 DEGs showed consistency in expression levels between males and females in group 1 and group 2 (Figure 3). Among these, female flowers (T02, T04) contained 351 up-regulated and 1366 down-regulated genes compared to male flowers (T01, T03) (Table 2 and Table S4). These 1717 DEGs were used for subsequent bioinformatics analysis.
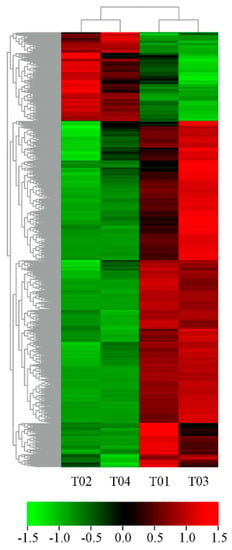
Figure 3.
Heat map for cluster analysis of the 1717 DEGs by K−means method. Red indicates up-regulated genes and green indicates down-regulated genes.
Table 2.
Three groups of DEGs.
3.3. GO Enrichment Analysis
The GO function enrichment analysis was performed on the DEGs in samples of different sexes of mulberry plants. GO terms are divided into three categories including molecular functions, cellular components, and biological processes. All the molecular functions and the cellular components were divided into 15 subcategories and the biological processes were divided into 22 subcategories, for a total of 52 functional subcategories (Figure 4). In all DEGs, 325 GO terms were annotated into the molecular function category with DNA binding (GO: 0003677, 86 unigenes), metal ion binding (GO: 0046872, 61 unigenes), and protein serine/threonine kinase activity (GO: 0004674, 50 unigenes) as the three groups with the largest number of DEGs. Among the cellular component, 95 GO terms were annotated to an integral component of membrane (GO: 0016021, 73 unigenes), cell wall (GO: 0005618, 45 unigenes), and membrane (GO: 0016020, 45 unigenes) in three groups that have the largest number of DEGs. In the biological process, 411 GO terms annotated to protein phosphorylation (GO: 0006468, 86 unigenes), regulation of transcription and DNA-templated (GO: 0006355, 70 unigenes), and oxidation-reduction process (GO: 0055114, 69 unigenes) were classified into three groups containing the largest number of DEGs.
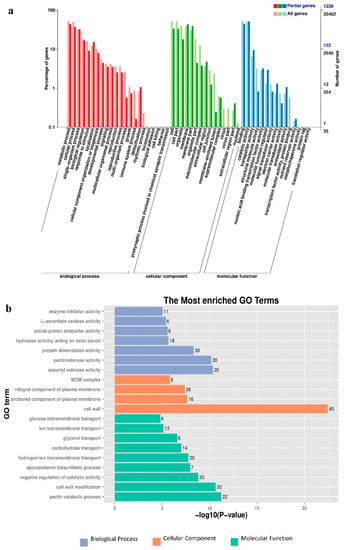
Figure 4.
Annotation analysis of GO functions of all DEGs. (a) The GO functional classification of co-expression DEGs. Deep colors represent all expressed Unigenes, and light colors represent DEGs. (b) The most enriched GO terms.
3.4. KEGG Pathway Analysis
In order to further explore the function of the identified DEGs, a KEGG pathway enrichment analysis was performed. A total of 505 DEGs were compared and annotated to 80 different pathways (Table S5). These pathways were divided into five categories: organic systems, genetic information processing, metabolism, environmental information processing, and cellular processes (Figure 5a). The three metabolic pathways with the largest number of annotated DEGs were starch and sucrose metabolism (37 unigenes), pentose and glucuronate interconversions (22 unigenes), and plant hormone signal transduction (17 unigenes). Interestingly, the first and second pathways mentioned above were also considered to be the most credible pathways for enrichment significance (Figure 5b) with 19 DEGs shared by the two pathways. Among the 19 DEGs in the two KEGG pathways, Unigene_009630 and Unigene_093301 simultaneously encode the pectate lyase synthesis, while Unigene_097983 encodes aldehyde dehydrogenase synthesis. These three DEGs were highly expressed in male flowers. In addition, the starch and sucrose metabolism pathway also displayed the most significant difference in the expression of DEGs between male and female flowers. Among these, there were seven DEGs with a Log2FC value of more than 10, that were also highly expressed in male flowers. These DEGs encode exopolygalacturonase (Unigene_007785, Unigene_013639, and Unigene_098084), pectinesterase (Unigene_004587, Unigene_007147, and Unigene_010800), and UDP-glucuronic acid decarboxylase 2 (UXS2, Unigene_009084), suggesting that carbohydrates play an important role in the development of male and female flowers in mulberry. In the plant hormone signal transduction pathway, only three DEGs were highly expressed in female flowers and the remaining 14 DEGs were highly expressed in male flowers (Figure S1). Moreover, the three downstream response factors IAA, AUXIN RESISTANT 1 (AUX1), SMALL AUXIN UPREGULATED RNA (SAUR) related to auxin all showed a trend of high expression in male flowers and low expression in female flowers. The regulatory factors PROTEIN PHOSPHATASE 2C (PP2C), SUCROSE NON-FERMENTED RELATED PROTEIN KINASE 2 (SnRK2), and ABA-RESPONSIVE ELEMENT BINDING FACTORS (ABF) that respond to abscisic acid showed the same expression pattern as the auxin (IAA, AUX1, SAUR) and had higher expression levels in the male flowers. This indicates that both auxin and abscisic acid have specific regulatory effects on the development of mulberry male flowers.
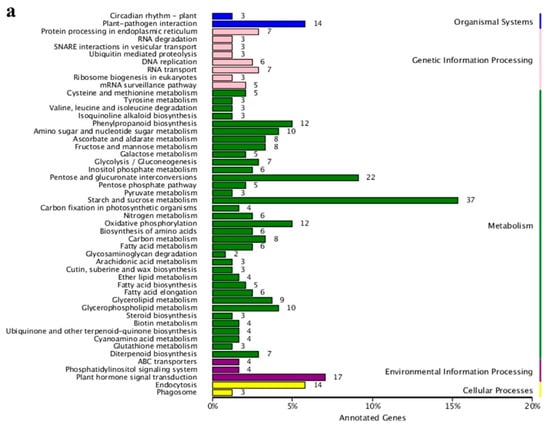
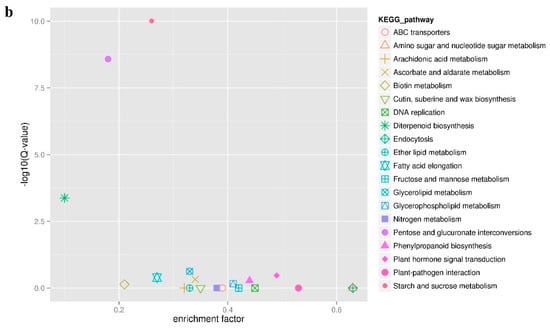
Figure 5.
Annotation analysis of KEGG pathway of all DEGs. (a) Statistic analysis of co-expression DEGs in KEGG pathways, DEGs were grouped into five major pathway categories as mentioned in the right panel. (b) Scatter plot of KEGG pathway enrichment for all DEGs. Each small shape in the figure represents a KEGG path, and the path names are shown in the legend on the right. The horizontal coordinate is the enrichment factor, the smaller the enrichment factor, the more significant the enrichment level of DEGs in the pathway. The ordinate is -log10 (Q-value), the larger the ordinate, the more reliable the enrichment of DEGs in the pathway.
3.5. Classification of GO and KEGG Terms of Male and Female Flowers
Furthermore, we also performed both GO and KEGG enrichment analysis on DEGs unique to female and male flowers, respectively. The results showed that cell killing (GO: 0001906) and nutrient reservoir activity (GO: 0045735) were unique to female flowers in GO classification, while the GO term of the membrane-enclosed lumen (GO: 0031974) was unique to male flowers. In all DEGs, 325 and 699 GO entries were identified in female and male flowers, respectively. Comparing the two groups of enriched GO terms, DEGs of female flowers were primarily enriched in molecular functional subclasses, while the DEGs of male flowers were primarily enriched in biological process subclasses. In addition, the three most abundant GO terms in female flowers were DNA binding (GO: 0003677), protein phosphorylation (GO: 0006468) and transcriptional regulation, and DNA templating (GO: 0006355). For male flowers, most of the GO terms were related to the membrane system (Figure S2). Further comparison of the KEGG enrichment analysis of the two DEGs groups identified that several metabolic pathways such as starch and sucrose metabolism, pentose and glucuronic acid mutual conversion, and plant hormone signal transduction showed high levels of enrichment in both male and female groups, indicating that these pathways may be essential for the development of plant floral organs. Interestingly, in the genetic information processing subcategory unit of the KEGG metabolic pathway, the DEGs of female flowers were mostly related to DNA replication, while the DEGs of male flowers were more abundant at the transcription level and post-transcriptional protein expression level (Figure S3). This suggests that the male and female flowers of mulberry have a different destiny at the end of their development, with male flowers beginning to wither and fall off after pollen ejection, while female flowers gradually expand after fertilization and accumulate nutrients stored in fruits.
3.6. Candidate DEGs Related to Flower Development
Based on the NR database annotation on the NCBI website, among the 1717 DEGs, 26 were highly homologous with other genes reported in the literature related to flower organ development (Figure 6a). Some of these homologous genes are attributed to several classic flowering regulation pathways [22].
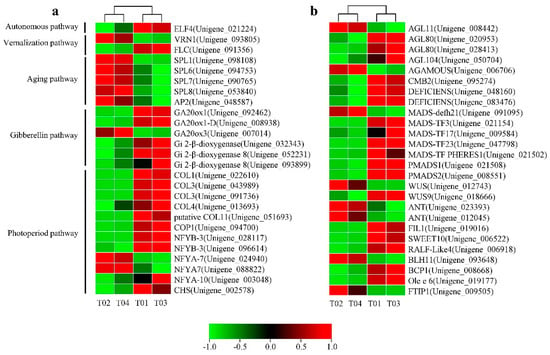
Figure 6.
Heat map diagram of expression patterns for candidate DEGs between the four samples. (a) The floral transition and flower development-associated genes. (b) Some other important target genes involved in the development of floral organs.
In the vernalization pathway, there were two DEGs homologous to the reported flower regulation genes VERNALIZATION 1 (VRN1, 1 unigene) and FLOWERING LOCUS C (FLC, 1 unigene). The autonomous pathway had only one DEG EARLY FLOWERING 4 (ELF4, 1 unigene). SQUAMOSA PROMOTER-BINDING-LIKE PROTEIN (SPL9, 4 unigenes) and APETELA2 (AP2, 1 unigene) were identified in the aging pathway and all highly expressed in female flowers. In addition, six DEGs were annotated as GIBBERELLIC ACID INSENSITIVE (GAI, 6 unigenes) belonging to the gibberellin pathway. In the photoperiod pathway, 12 DEGs matched the flower homologous genes reported in other plants, including CONSTANS (CO, 5 unigenes), NUCLEAR FACTOR Y (NF-Y, 5 unigenes), CONSTITUTIVELY PHOTOMORPHOGENIC 1 (COP1, 1 unigene), and CHALONE SYNTHASE (CHS, 1 unigene).
In addition, some other unclassified floral organ development regulatory genes were identified (Figure 6b). These genes included WUSCHEL (WUS), AP2-LIKE ETHYLENE-RESPONSIVE TRANSCRIPTION FACTOR ANT (ANT-Like), RAPID ALKALINIZATION FACTOR (RALF-Like), FT-INTERACTING PROTEIN 1 (FTIP1), and several unigenes containing the MADS-box domain and other homologous DEGs (Table S6).
Considering the enrichment of plant hormone signal transduction pathways (ko: 04075) in KEGG and the extensive involvement of phytohormones in the regulation of plant floral development, we searched the genes directly involved in hormone regulation in the DEGs set and retrieved a total of 53 DEGs (Figure 7 and Table S7). The number of DEGs contained in ethylene was up to 15, followed by 11 in cytokinin, and the least in jasmonic acid, which contained only 1 DEG.
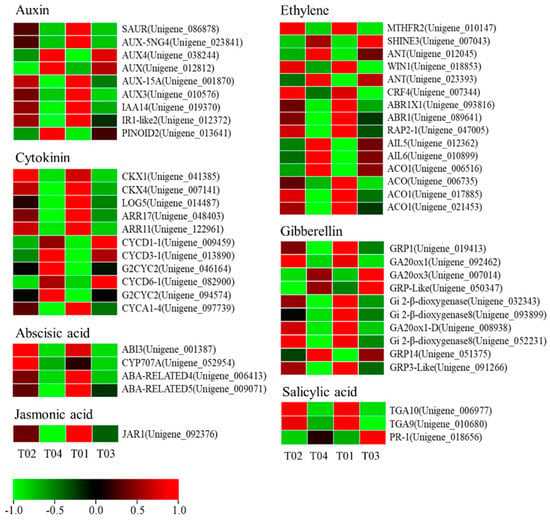
Figure 7.
Differentially expressed phytohormone-related unigenes between male and female mulberry flower materials.
3.7. Differential Expression of TFs in Male and Female Flowers
Across the two male and female datasets, a total of 124 TFs were identified as differentially expressed (Table S8). Among these, 39 differentially expressed TFs were highly expressed in female flowers and 85 were highly expressed in male flowers. Figure 8 shows the differential expression profiles of TFs in different samples. In the differentially expressed TFs, there were 17 C2H2, 15 bHLH, 13 MYB, and some other common TF families including MADS, ERF, LBD, NAC, and others and several other small TF family members.
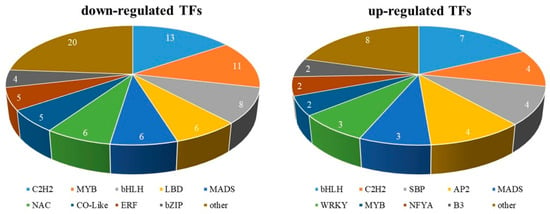
Figure 8.
Transcription factors associated with flower development in Morus alba L.
3.8. Q-PCR Validation of DEGs
In order to verify the reliability of the sequencing data, 20 DEGs were selected for q-PCR verification. Among them, eight DEGs were highly expressed in female flowers and 12 were highly expressed in male flowers. The sequencing and quantitative data of T01_vs_T02 and T03_vs_T04 showed a high correlation with R2 of 0.8512 and 0.975 in the two groups, respectively (Figure 9). In addition, we also selected four DEGs with MADS-box domains for their expression pattern analysis at different developmental stages of male and female mulberry flowers. The expression levels of these DEGs showed a trend of continuous up-regulation with the gradual maturation of flowers, and all had very low expression levels in the flower bud stage. In addition, the two DEGs of AGL11 and PMADS2 both showed the highest expression levels during flower enlargement, then decreased significantly thereafter (Figure 10).
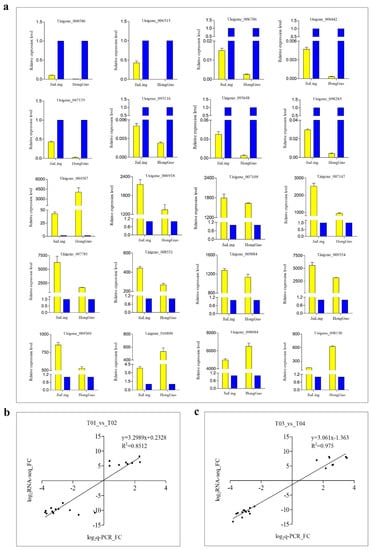
Figure 9.
(a) Q-PCR validation of DEGs randomly selected between male and female mulberry flower buds. The yellow color represents male flowers, and the blue color represents female flowers. (b,c) Correlation analysis between the transcriptome data and quantitative data of JiaLing 40 and HongGuo 1, respectively.
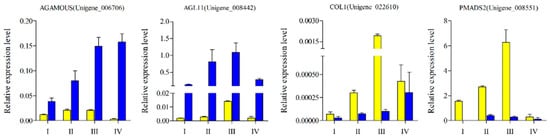
Figure 10.
Q-PCR of four candidate DEGs between male and female mulberry flower buds of four different developmental stages. The yellow color represents male flowers, and the blue color represents female flowers.
4. Discussion
At present, most of the understanding of mulberry sex differentiation is still at the basic morphological observation level with few literature reports on its specific sex differentiation regulation mechanism and genes related to the flower development process. Currently, with the popularity and importance of sequencing technology in various fields of biology, some researchers have used this technology to identify candidate genes for flower development in woody plants such as sugar apple (Annona squamosa L.), Chinese chinquapin (Castanea henryi), and jatropha (Jatropha curcas L) [23,24,25]. In this study, male and female flowers of two varieties of mulberry were sequenced and some regulatory genes and transcription factors putatively involved in the development of mulberry floral sexual differentiation were identified.
The classic flower development ABC model proposed by Coen and Meyerowitz (1991) was gradually perfected by other scholars to the ABCDE model [26,27]. Studies have shown that all functional genes related to flower organ development in the ABCDE model are transcription factors, contain one highly conserved DNA binding domain, and belong to the MADS-box family, except for AP2 [28]. In our data, a total of 61 unigenes containing MADS domains were annotated, of which 12 unigenes were differentially expressed. Then, we constructed a phylogenetic tree using the Neighbor-Joining method for the 12 DEGs and some homologous genes in Arabidopsis and poplar. Among these DEGs, the two class B unigenes (PMADS1 and PMADS2) had the highest FPKM value in male flowers and their log2FC value was more than 5 compared to female flowers. The PMADS1 in mulberry had a higher similarity with the APETALA3 (AP3) gene in Arabidopsis, while PMADS2 had a higher similarity with the homologous genes of poplar, also showing high homology with the Arabidopsis PISTILLATA (PI) gene (Figure S4). In model plants such as Arabidopsis and snapdragon, AP3/DEF and PI/GLO are specifically expressed in petals and stamens and play leading roles in determining the characteristics of petals and stamens [29,30]. In the mutant aps of the quasi-model plant petunia, PMADS1/PhGLO2 and PMADS2/PhDEF are only expressed in the petals and stamens, indicating their function in the petalization of the calyx and the dehiscence of the corolla, as well as participate in the development of stamens [31]. In addition, the AP3 gene in Pyrola alba is located on the Y chromosome and is only expressed in male flower buds [32]. It is speculated that this gene may also have a sex-determining function.
The data presented in this study demonstrate that three DEGs were highly expressed in female flowers, namely AGL11, AGAMOUS, and DEFH21. Among these, the expression abundance of AGL11 and AGAMOUS was relatively higher and the Log2FC value of the difference between male and female samples of the two varieties was more than 5. In Arabidopsis, AGAMOUS is the only gene belonging to the C class and initiates expression when the sepal primordium bulges in the early stage of anthesis. The carpel primordium then appears in the later stages and the AG expression level reaches a peak [33]. This is consistent with the expression trend of AGAMOUS in different stages of mulberry female flowers (Figure 9). The D class gene AGL11/STK was primarily expressed in the middle stage of female flower development of mulberry, which corresponds to the most vigorous growth period of the mulberry fruit. It has recently been discovered that AGL11 can also regulate fruit growth by regulating the level of cytokinin and the content of fructose [34]. AGL11 also has a direct regulatory effect on the seedless phenotype in grapes [35]. From the perspective of evolutionary branches, AGL104, which regulates pollen development and late growth of pollen tubes, and AGL80, which cooperates with AGL61 to regulate central cell differentiation, both are relatively conserved genes [36,37]. In our study, MnAGL104 had the highest expression level at the fully mature stage of male flowers, and the pollen of male flowers in this period was basically fully mature and in a state of ejection and flight. It is, therefore, hypothesized that these genes may have similar functions in the development of mulberry flower organs.
Many studies have shown that the sex expression of higher plants is polymorphic and moldable, and phytohormones play a key role in the process of plant bisexual differentiation and flower development. [17,38]. Moreover, different hormones have different effects on the sex expression in plants and the same type of hormones have an opposite regulation of sex differentiation in different plants. For example, ethylene has a female-promoting effect on cucumbers, while gibberellin can promote the differentiation of cucumber male flowers [39,40]. Interestingly, applying gibberellin to bitter melon can increase the proportion of female flowers [41]. Shang et al. (2017) used transcriptomics to identify that the 1-AMINOCYCLOPROPANE-1-CARBOXYLIC ACID SYNTHASE (ACS) gene expression in the ethylene biosynthesis pathway was significantly different in the six developmental stages of mulberry female flower buds. The expression of the ACS gene is highest in the second stage and then rapidly declines [18].
In our data, some plant hormone-related DEGs have been identified (Figure 10). For example, one ACC synthase gene (ACS) and four ACC oxidase genes (ACO) in the ethylene biosynthetic pathway were identified, but the expression of the ACS gene was not consistent in the two sequenced varieties. This inconsistency in expression patterns may be caused by different genetic backgrounds but requires further research. In addition, some genetic elements that respond to ethylene signals have been annotated. In the plant hormone signaling pathway, several key genes in the auxin regulatory pathway, including SAUR, LAX3, and IAA14 were highly expressed in male flowers while the auxin transporter PINOID2 was one of the few DEGs highly expressed in female flowers. In Arabidopsis, the two homologous genes of PINOID (PINOID2/WGA1 and WGA2) have a negative regulatory relationship to the formation of cotyledons [42]. Recent studies have found that in rice, mutations in the PINOID gene can lead to the production of stigma-free mutants [43]. Further studies have shown that the PINOID gene can also interact with MADS16 or LAX1 in rice to regulate the development of floral organs [44]. In addition to auxin and ethylene, a large number of gibberellin-related genes have been annotated in mulberry including GA20ox1/1-D/3, GIBBERELLIN-REGULATED PROTEIN1/3/14, and GIBBERELLIN 2-BETA-DIOXYGENASE. Transient overexpression of PbGA20ox2 in tomatoes and pears, a homologous gene of mulberry GA20ox3 in pears, promotes fruit development, delays shedding of unpollinated fruits, and increases GA(4) content [45].
TFs are key regulatory proteins that respond to changes in the external environment during plant growth and development. They mediate a variety of regulatory pathways during the transformation of plants from vegetative to reproductive growth. In the ABCDE model, most of the genes belong to the MADS-box transcription factor family. Such as the SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) and AGAMOUS-LIKE24 (AGL24) genes that promote flowering during the regulation of plant flowering, as well as the FLOWERING LOCUS C (FLC) and SHORT VEGETATIVE PHASE (SVP) genes that inhibit flowering [46], these genes all contain a typical MADS-box domain and encode MIKC type protein. In this study, six MYB family TFs were identified (MYB35/61/62/78/97/101). Studies in Arabidopsis have found that down-regulation of MYB80 (MYB103) gene expression leads to a degradation of the tapetum, making the pollen unable to fully mature, and knocking out MYB80 will result in complete male sterility [47]. In addition, a single MYB33 or MYB65 mutant can form normally functional pollen, but the pollen is severely aborted in the double mutant [48]. The myb97myb101myb120 triple mutation displays non-defective pollen growth and development; however, the pollen tube shows uncontrolled growth and the elimination of sperm cells is blocked [49]. It is speculated that the MYB TF may have a specific regulatory effect on plant pollen. In addition, five transcription factors of the NFY family that specifically bind to CCAAT-box have also been identified in this study. Many subgenes of this family are involved in the regulation of plant flowering. For example, AtNF-YB2/3 interact with AtNF-YC3/4/9 and then through the photoperiod pathway regulate the flowering time of Arabidopsis [50]. Overexpression of AtNF-YC2/3 can promote plant flowering and also increase the transcription level of FT [51]. Moreover, we also annotated two TF members of the bZIP family (TGA9 and TGA10). In Arabidopsis, the development of anthers is affected in the homologous deletion mutant tga9tga10 and normal pollen grains cannot develop [52].
Furthermore, some specific DEGs that have a significant impact on plant sex regulation and flower development have also been discovered. Many articles on the sequencing of plant male and female flower buds show that both sugar and lipid metabolism are pathways in which DEGs are more enriched [53,54,55]. A large number of genes related to glycolipid synthesis have also been found to be involved in the regulation of plant sex and the development of floral organs. Among all DEGs, 18 SUGAR TRANSPORT PROTEINS (STPs) and a large number of UDP-glycosyltransferase (UGT) superfamily members have been annotated. During floral transition of Arabidopsis, SWEET10 acts downstream of FT and ectopic expression leads to early flowering [56]. The ectopic overexpression of CsUGT85A53 in Arabidopsis leads to a significant decrease in the accumulation of transcripts of the flowering suppressor genes FLC and ABI5, leading to earlier flowering [57]. Nineteen DEGs encoding GDSL (Gly-Asp-Ser-Leu domain proteins) lipase have been annotated and GDSL lipase is known to be involved in multiple life activities and morphogenesis of plants [58]. Recent studies have found that GDSL family members also play an important role in the reproductive development of plants. For example, AtEXL6, ZmMs30, and OsRMS2 are essential for the formation of the exine, and the aliphatic metabolic pathways required for the development of anther epidermis [59,60,61]. In addition, four DEGs encoding short peptides of RAPID ALKALIZATION FACTORS have been annotated. These are small peptide signals that have an important function in the fertilization processes in plants [62]. We also identified two DEGs belonging to the short-chain dehydrogenase family. Among these, Unigene_007952 was only expressed in male flowers and not in female flowers. The Log2FC value of expression difference between the two groups was greater than eight. Moreover, the Unigene_007952 was compared with homologous genes of other plants and it was found that this unigene has more than 60% homology with the sex-determining gene TS2 in maize [63]. In a recent study, Muller et al. (2020) used CRISPR-Cas9-induced mutations to show that PtARR17 functions as a sex switch in poplar [64]. In our data, three homologous DEGs of ARR were annotated and one of them (MnARR17) showed a close relationship with PtARR17 via evolutionary tree analysis (Figure S5). We hypothesize that these genes may be involved in the regulation of mulberry sex differentiation, but further research is necessary.
In summary, four high-quality sequencing libraries of male and female mulberry flowers were constructed. After integration and comparison, it was found that a large number of genes were differentially expressed in male or female mulberry flowers. Through NR database annotation and GO/KEGG enrichment analysis, the expression patterns of DEGs involved in the development of mulberry male and female flowers were analyzed. Some candidate genes for signal substances such as glucose and lipid metabolism, hormone signal transduction, transcription factor regulation, and short peptides have been screened. These genes may be involved in the sexual differentiation and flower development regulatory network of mulberry. Q-PCR verified the accuracy and reliability of the transcriptome data and analysis results, and further elaborated the expression patterns of some floral organ regulatory genes at different flower developmental stages. Our transcriptome data provide new insights into the study of mulberry sex regulation and floral organ development and lay a foundation for future in-depth analyses of the complex sex mechanism of mulberry.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/horticulturae8070625/s1, Table S1. Primers of random transcripts used in this study. Table S2. Summary of de novo transcriptome assembly. Table S3. All unigenes assembled and annotated based on transcripotome sequencing data of mulberry flower materials. Table S4. All DEGs of co-expressed between male and female flowers with two mulberry varieties (1717). Table S5. 80 different enriched KEGG pathways. Table S6. Differentially expressed unigenes which were annotated as flower-related genes. Table S7. Differentially expressed phytohormone-related unigenes between male and female mulberry flowers. Table S8. Differentially expressed transcription element unigenes between male and female Mulberry flower buds. Figure S1. Plant hormone signal transduction. Figure S2. Annotation analysis of GO functions of female and male flowers’ DEGs, respectively. Figure S3. Annotation analysis of KEGG pathway of female and male flowers’ DEGs, respectively. Figure S4. Phylogenetic analysis of MADS-box genes by Neighbor-Joining in Morus alba L. Figure S5. Phylogenetic analysis of ARR genes by Neighbor-Joining in Morus alba L.
Author Contributions
D.X. and Z.H. analyzed the transcriptome data and drafted the manuscript. D.X., N.D. and Y.L. conceived research and designed experiments; D.X., L.Y. and S.L. performed experiments; X.W. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by “Fundamental Research Funds for the Central Universities” (Project No. SWU120025), and by ChongQing Municipal Commission of Commerce Special Fund (Project No. 20210611150932818), and by China Agriculture Research System of MOF and MARA.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets used during the current study had been uploaded to NCBI SRA database (https://dataview.ncbi.nlm.nih.gov/object/PRJNA782379?reviewer=vmt143ocs8iv9nh1bpoovrbr8k, accessed on 22 November 2021). The plant materials are available from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vijayan, K.; Chakraborti, S.P.; Ghosh, P.D. Screening of mulberry (Morus spp.) for salinity tolerance through in vitro seed germination. Indian J. Biotechnol. 2004, 3, 47–51. [Google Scholar]
- Yang, X.; Yang, L.; Zheng, H. Hypolipidemic and antioxidant effects of mulberry (Morus alba L.) fruit in hyperlipidaemia rats. Food Chem. Toxicol. 2010, 48, 2374–2379. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiang, L.; Wang, C.; Tang, C.; He, X. Antidiabetic and antioxidant effects and phytochemicals of mulberry fruit (Morus alba L.) polyphenol enhanced extract. PLoS ONE 2013, 8, e71144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, Z.F.; Luo, X.; Li, X. Effects of Mulberry Fruit (Morus alba L.) Consumption on Health Outcomes: A Mini-Review. Antioxidants 2018, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Thakur, K.; Zhang, Y.-Y.; Mocan, A.; Zhang, F.; Zhang, J.-G.; Wei, Z.-J. 1-Deoxynojirimycin, its potential for management of non-communicable metabolic diseases. Trends Food Sci. Technol. 2019, 89, 88–99. [Google Scholar] [CrossRef]
- Butt, M.S.; Nazir, A.; Sultan, M.T.; Schroën, K. Morus alba L. nature’s functional tonic. Trends Food Sci. Technol. 2008, 19, 505–512. [Google Scholar] [CrossRef]
- Mnaa, S.; Aniess, W.; Olwy, Y.; Shaker, E. Antioxidant Activity of White (Morus alba L.) and Black (Morus nigra L.) Berries against CCl4 Hepatotoxic Agent. Adv. Tech. Biol. Med. 2015, 3, 1–7. [Google Scholar]
- Clement, W.L.; Weiblen, G.D. Morphological Evolution in the Mulberry Family (Moraceae). Syst. Bot. 2009, 34, 530–552. [Google Scholar] [CrossRef]
- Juvany, M.; Munne-Bosch, S. Sex-related differences in stress tolerance in dioecious plants: A critical appraisal in a physiological context. J. Exp. Bot. 2015, 66, 6083–6092. [Google Scholar] [CrossRef]
- Wessinger, C.A.; Hileman, L.C. Parallelism in Flower Evolution and Development. Annu. Rev. Ecol. Evol. Syst. 2020, 51, 387–408. [Google Scholar] [CrossRef]
- Akagi, T.; Henry, I.M.; Tao, R.; Comai, L. A Y-chromosome-encoded small RNA acts as a sex determinant in persimmons. Science 2014, 346, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Fechter, I.; Hausmann, L.; Daum, M.; Sorensen, T.R.; Viehover, P.; Weisshaar, B.; Topfer, R. Candidate genes within a 143 kb region of the flower sex locus in Vitis. Mol. Genet. Genom. 2012, 287, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Wu, H.; Chen, Y.; Li, X.; Hou, J.; Lu, J.; Wei, S.; Dai, X.; Olson, M.S.; Liu, J.; et al. Two antagonistic effect genes mediate separation of sexes in a fully dioecious plant. bioRxiv 2020. [Google Scholar] [CrossRef]
- Akagi, T.; Pilkington, S.M.; Varkonyi-Gasic, E.; Henry, I.M.; Sugano, S.S.; Sonoda, M.; Firl, A.; McNeilage, M.A.; Douglas, M.J.; Wang, T.; et al. Two Y-chromosome-encoded genes determine sex in kiwifruit. Nat. Plants 2019, 5, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Henry, I.M.; Ohtani, H.; Morimoto, T.; Beppu, K.; Kataoka, I.; Tao, R. A Y-Encoded Suppressor of Feminization Arose via Lineage-Specific Duplication of a Cytokinin Response Regulator in Kiwifruit. Plant Cell 2018, 30, 780–795. [Google Scholar] [CrossRef]
- Srikanth, A.; Schmid, M. Regulation of flowering time: All roads lead to Rome. Cell Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef] [PubMed]
- Golenberg, E.M.; West, N.W. Hormonal interactions and gene regulation can link monoecy and environmental plasticity to the evolution of dioecy in plants. Am. J. Bot. 2013, 100, 1022–1037. [Google Scholar] [CrossRef]
- Shang, J.; Liang, J.; Xiang, Z.; He, N. Anatomical and transcriptional dynamics of early floral development of mulberry (Morus alba). Tree Genet. Genom. 2017, 13, 1–14. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- He, N.J.; Zhang, C.; Qi, X.W.; Zhao, S.; Tao, Y.; Yang, G.; Lee, T.H.; Wang, X.; Cai, Q.; Li, D.; et al. Draft genome sequence of the mulberry tree Morus notabilis. Nat. Commun. 2013, 4, 2445. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Komeda, Y. Genetic regulation of time to flower in Arabidopsis thaliana. Annu. Rev. Plant Biol. 2004, 55, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Feng, S.; Pan, Y.; Zhong, J.; Chen, Y.; Yuan, C.; Li, H. Transcriptome Analysis and Identification of Genes Associated with Floral Transition and Flower Development in Sugar Apple (Annona squamosa L.). Front. Plant Sci. 2016, 7, 1695. [Google Scholar] [CrossRef]
- Fan, X.; Yuan, D.; Tian, X.; Zhu, Z.; Liu, M.; Cao, H. Comprehensive Transcriptome Analysis of Phytohormone Biosynthesis and Signaling Genes in the Flowers of Chinese Chinquapin (Castanea henryi). J. Agric. Food Chem. 2017, 65, 10332–10349. [Google Scholar] [CrossRef]
- Hui, W.; Yang, Y.; Wu, G.; Peng, C.; Chen, X.; Zayed, M.Z. Transcriptome profile analysis reveals the regulation mechanism of floral sex differentiation in Jatropha curcas L. Sci. Rep. 2017, 7, 16421. [Google Scholar] [CrossRef]
- Coen, E.S.; Meyerowitz, E.M. The war of the whorls-genetic interactions controlling flower development. Nature 1991, 353, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kater, M.M.; Dreni, L.; Colombo, L. Functional conservation of MADS-box factors controlling floral organ identity in rice and Arabidopsis. J. Exp. Bot. 2006, 57, 3433–3444. [Google Scholar] [CrossRef]
- Theissen, G.; Becker, A.; Di Rosa, A.; Kanno, A.; Kim, J.T.; Munster, T.; Winter, K.U.; Saedler, H. A short history of MADS-box genes in plants. Plant Mol. Biol. 2000, 42, 115–149. [Google Scholar] [CrossRef]
- Whipple, C.J.; Zanis, M.J.; Kellogg, E.A.; Schmidt, R.J. Conservation of B class gene expression in the second whorl of a basal grass and outgroups links the origin of lodicules and petals. Proc. Natl. Acad. Sci. USA 2007, 104, 1081–1086. [Google Scholar] [CrossRef]
- Irish, V.F. Evolution of petal identity. J. Exp. Bot. 2009, 60, 2517–2527. [Google Scholar] [CrossRef]
- Zhou, Q.; Lu, R.; Zhang, S.; Bao, M.; Liu, G. Phenotype characterization and genetic analysis of a floral mutant aps in petunia. Acta Hortic. Sin. 2019, 46, 317–329. [Google Scholar]
- Matsunaga, S.; Isono, E.; Kejnovsky, E.; Vyskot, B.; Dolezel, J.; Kawano, S.; Charlesworth, D. Duplicative transfer of a MADS box gene to a plant Y chromosome. Mol. Biol. Evol. 2003, 20, 1062–1069. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sieburth, L.E.; Meyerowitz, E.M. Molecular dissection of the AGAMOUS control region shows that cis elements for spatial regulation are located intragenically. Plant Cell 1997, 9, 355–365. [Google Scholar] [PubMed]
- Di Marzo, M.; Herrera-Ubaldo, H.; Caporali, E.; Novák, O.; Strnad, M.; Balanzà, V.; Ezquer, I.; Mendes, M.A.; de Folter, S.; Colombo, L. SEEDSTICK Controls Arabidopsis Fruit Size by Regulating Cytokinin Levels and FRUITFULL. Cell Rep. 2020, 30, 2846–2857. [Google Scholar] [CrossRef]
- Ocarez, N.; Mejia, N. Suppression of the D-class MADS-box AGL11 gene triggers seedlessness in fleshy fruits. Plant Cell Rep. 2016, 35, 239–254. [Google Scholar] [CrossRef]
- Bemer, M.; Wolters-Arts, M.; Grossniklaus, U.; Angenent, G.C. The MADS domain protein DIANA acts together with AGAMOUS-LIKE80 to specify the central cell in Arabidopsis ovules. Plant Cell 2008, 20, 2088–2101. [Google Scholar] [CrossRef]
- Xia, C.; Wang, Y.J.; Liang, Y.; Niu, Q.K.; Tan, X.Y.; Chu, L.C.; Chen, L.Q.; Zhang, X.Q.; Ye, D. The ARID-HMG DNA-binding protein AtHMGB15 is required for pollen tube growth in Arabidopsis thaliana. Plant J. 2014, 79, 741–756. [Google Scholar] [CrossRef]
- Khryanin, V.N. Evolution of the pathways of sex differentiation in plants. Russ. J. Plant Physiol. 2007, 54, 845–852. [Google Scholar] [CrossRef]
- Yamasaki, S.; Fujii, N.; Takahashi, H. The ethylene-regulated expression of CS-ETR2 and CS-ERS genes in cucumber plants and their possible involvement with sex expression in flowers. Plant Cell Physiol. 2000, 41, 608–616. [Google Scholar] [CrossRef]
- Peterson, C.E.; Anhder, L.D. Induction of staminate flowers on gynoecious cucumbers with Gibberellin-a3. Science 1960, 131, 1673–1674. [Google Scholar] [CrossRef]
- Thomas, T.D. The effect of in vivo and in vitro applications of ethrel and GA3 on sex expression in bitter melon (Momordica charantia L.). Euphytica 2008, 164, 317–323. [Google Scholar] [CrossRef]
- Cheng, Y.F.; Qin, G.J.; Dai, X.H.; Zhao, Y.D. NPY genes and AGC kinases define two key steps in auxin-mediated organogenesis in Arabidopsis. Proc. Natl. Acad. Sci. USA 2008, 105, 21017–21022. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tang, D.; Cheng, X.; Zhang, J.; Tang, Y.; Tao, Q.; Shi, W.; You, A.; Gu, M.; Cheng, Z.; et al. OsPINOID Regulates Stigma and Ovule Initiation through Maintenance of the Floral Meristem by Auxin Signaling. Plant Physiol 2019, 180, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Xie, D.J.; Tang, Z.S.; Shi, D.Q.; Yang, W.C. PINOID regulates floral organ development by modulating auxin transport and interacts with MADS16 in rice. Plant Biotechnol. J. 2020, 18, 1778–1795. [Google Scholar] [CrossRef]
- Wang, H.; Wu, T.; Liu, J.; Cong, L.; Zhu, Y.; Zhai, R.; Yang, C.; Wang, Z.; Ma, F.; Xu, L. PbGA20ox2 Regulates Fruit Set and Induces Parthenocarpy by Enhancing GA4 Content. Front. Plant Sci. 2020, 11, 113. [Google Scholar] [CrossRef]
- Lee, J.; Lee, I. Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 2010, 61, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.A.; Iacuone, S.; Li, S.F.; Parish, R.W. The MYB80 Transcription Factor Is Required for Pollen Development and the Regulation of Tapetal Programmed Cell Death in Arabidopsis thaliana. Plant Cell 2011, 23, 2209–2224. [Google Scholar] [CrossRef]
- Millar, A.A.; Gubler, F. The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are MicroRNA-regulated genes that redundantly facilitate anther development. Plant Cell 2005, 17, 705–721. [Google Scholar] [CrossRef]
- Gocal, G.F.W.; Sheldon, C.C.; Gubler, F.; Moritz, T.; Bagnall, D.J.; MacMillan, C.P.; Li, S.F.; Parish, R.W.; Dennis, E.S.; Weigel, D.; et al. GAMYB-like genes, flowering, and gibberellin signaling in Arabidopsis. Plant Physiol 2001, 127, 1682–1693. [Google Scholar] [CrossRef]
- Kumimoto, R.W.; Zhang, Y.; Siefers, N.; Holt, B.F. NF-YC3, NF-YC4 and NF-YC9 are required for CONSTANS-mediated, photoperiod-dependent flowering in Arabidopsis thaliana. Plant J. 2010, 63, 379–391. [Google Scholar] [CrossRef]
- Hackenberg, D.; Wu, Y.F.; Voigt, A.; Adams, R.; Schramm, P.; Grimm, B. Studies on Differential Nuclear Translocation Mechanism and Assembly of the Three Subunits of the Arabidopsis thaliana Transcription Factor NF-Y. Mol. Plant 2012, 5, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Murmu, J.; Bush, M.J.; DeLong, C.; Li, S.; Xu, M.; Khan, M.; Malcolmson, C.; Fobert, P.R.; Zachgo, S.; Hepworth, S.R. Arabidopsis basic leucine-zipper transcription factors TGA9 and TGA10 interact with floral glutaredoxins ROXY1 and ROXY2 and are redundantly required for anther development. Plant Physiol 2010, 154, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, Z.; Guo, H.; Ye, N.; Lyu, T.; Yang, W.; Wang, J.; Wang, J.T.; Wu, B.; Li, J.; et al. Comparative genomics analysis reveals gene family expansion and changes of expression patterns associated with natural adaptations of flowering time and secondary metabolism in yellow Camellia. Funct. Integr. Genom. 2018, 18, 659–671. [Google Scholar] [CrossRef]
- Wei, B.; Wang, L.; Bosland, P.W.; Zhang, G.; Zhang, R. Comparative transcriptional analysis of Capsicum flower buds between a sterile flower pool and a restorer flower pool provides insight into the regulation of fertility restoration. BMC Genom. 2019, 20, 837. [Google Scholar] [CrossRef]
- Alagna, F.; Cirilli, M.; Galla, G.; Carbone, F.; Daddiego, L.; Facella, P.; Lopez, L.; Colao, C.; Mariotti, R.; Cultrera, N.; et al. Transcript Analysis and Regulative Events during Flower Development in Olive (Olea europaea L.). PLoS ONE 2016, 11, e0152943. [Google Scholar] [CrossRef] [PubMed]
- Andrés, F.; Kinoshita, A.; Kalluri, N.; Fernández, V.; Falavigna, V.S.; Cruz, T.; Jang, S.; Chiba, Y.; Seo, M.; Mettler-Altmann, T.; et al. The sugar transporter SWEET10 acts downstream of FLOWERING LOCUS T during floral transition of Arabidopsis thaliana. BMC Plant Biol. 2020, 20, 1–14. [Google Scholar] [CrossRef]
- Jing, T.; Zhang, N.; Gao, T.; Wu, Y.; Zhao, M.; Jin, J.; Du, W.; Schwab, W.; Song, C. UGT85A53 promotes flowering via mediating abscisic acid glucosylation and FLC transcription in Camellia sinensis. J. Exp. Bot. 2020, 71, 7018–7029. [Google Scholar] [CrossRef]
- Ding, L.N.; Li, M.; Wang, W.J.; Cao, J.; Tan, X.L. Advances in plant GDSL lipases: From sequences to functional mechanisms. Acta Physiol. Plant. 2019, 41, 151. [Google Scholar] [CrossRef]
- An, X.; Dong, Z.; Tian, Y.; Xie, K.; Wu, S.; Zhu, T.; Zhang, D.; Zhou, Y.; Niu, C.; Ma, B.; et al. ZmMs30 Encoding a Novel GDSL Lipase Is Essential for Male Fertility and Valuable for Hybrid Breeding in Maize. Mol. Plant 2019, 12, 343–359. [Google Scholar] [CrossRef]
- Zhao, J.; Long, T.; Wang, Y.; Tong, X.; Tang, J.; Li, J.; Wang, H.; Tang, L.; Li, Z.; Shu, Y.; et al. RMS2 Encoding a GDSL Lipase Mediates Lipid Homeostasis in Anthers to Determine Rice Male Fertility. Plant Physiol. 2020, 182, 2047–2064. [Google Scholar] [CrossRef]
- Mayfield, J.A.; Fiebig, A.; Johnstone, S.E.; Preuss, D. Gene families from the Arabidopsis thaliana pollen coat proteome. Science 2001, 292, 2482–2485. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.X.; Bergonci, T.; Zhao, Y.L.; Zou, Y.; Du, S.; Liu, M.C.; Luo, X.; Ruan, H.; García-Valencia, L.E.; Zhong, S.; et al. Arabidopsis pollen tube integrity and sperm release are regulated by RALF-mediated signaling. Science 2017, 358, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Delong, A.; Calderonurrea, A.; Dellaporta, S.L. Sex determination gene tasselseed2 of maize encodes a short-chain alcohol-dehydrogenase required for stage-specific floral organ abortion. Cell 1993, 74, 757–768. [Google Scholar] [CrossRef]
- Muller, N.A.; Kersten, B.; Leite Montalvao, A.P.; Mähler, N.; Bernhardsson, C.; Bräutigam, K.; Carracedo Lorenzo, Z.; Hoenicka, H.; Kumar, V.; Pakull, B.; et al. A single gene underlies the dynamic evolution of poplar sex determination. Nat. Plants 2020, 6, 630–637. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).