
Comparative and Phylogenetic Analyses of the Complete Chloroplast Genomes of Four Ottelia Species

 ,
,
Abstract
1. Introduction
2. Results and Discussion
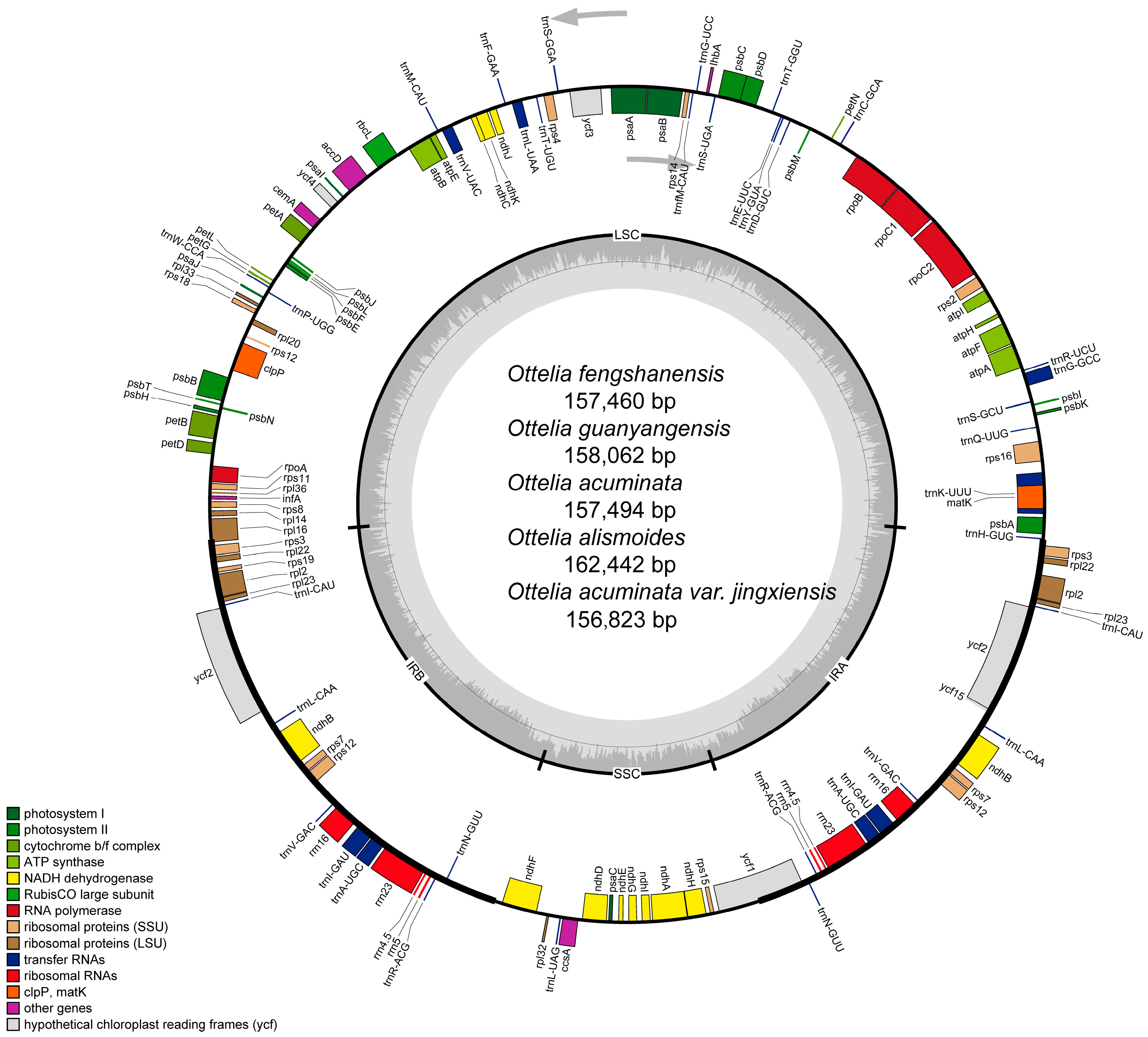
2.1. Chloroplast Genome Features of Ottelia
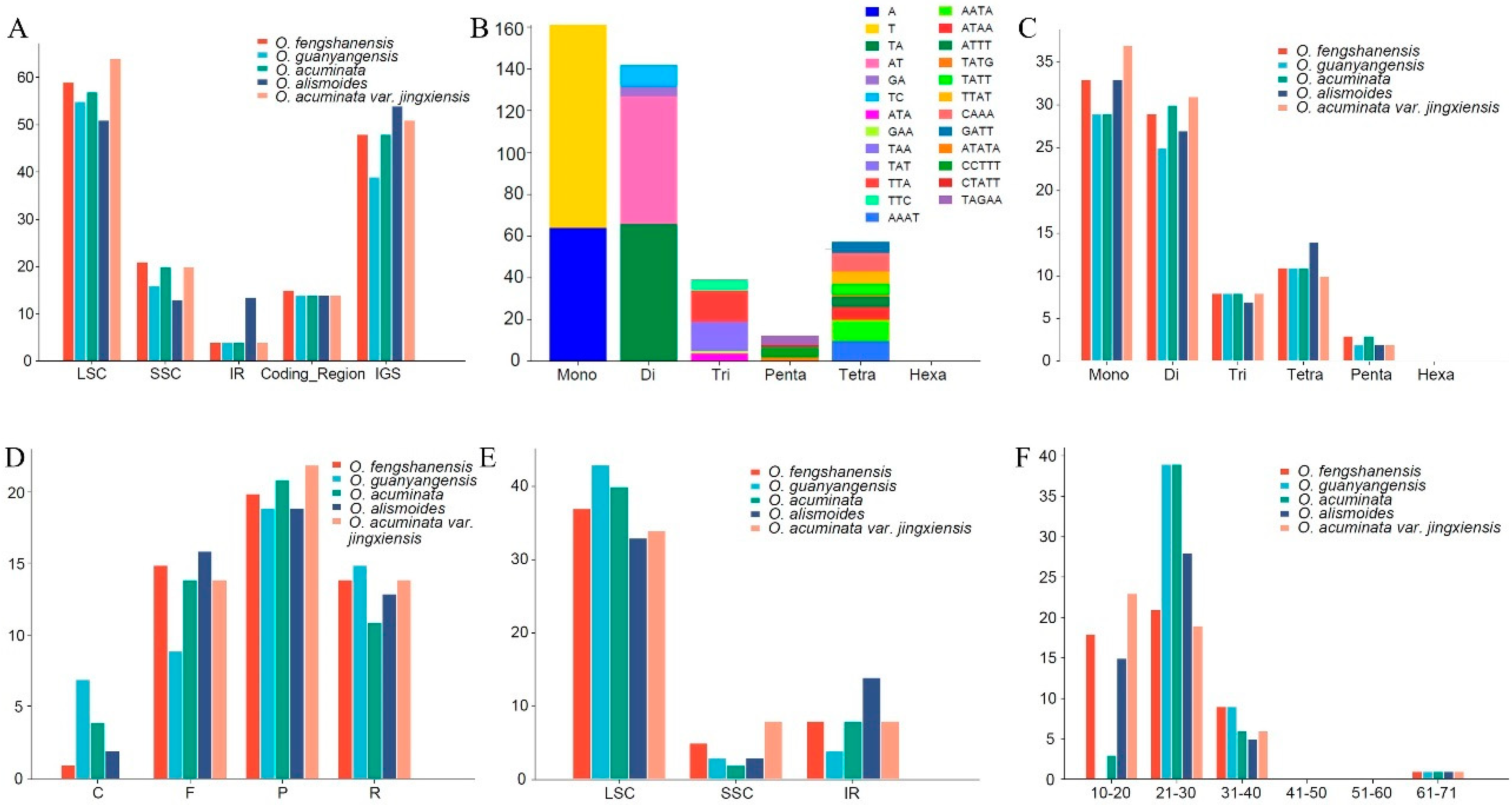
2.2. SSRs and Repeats
2.3. Codon Usage Analysis
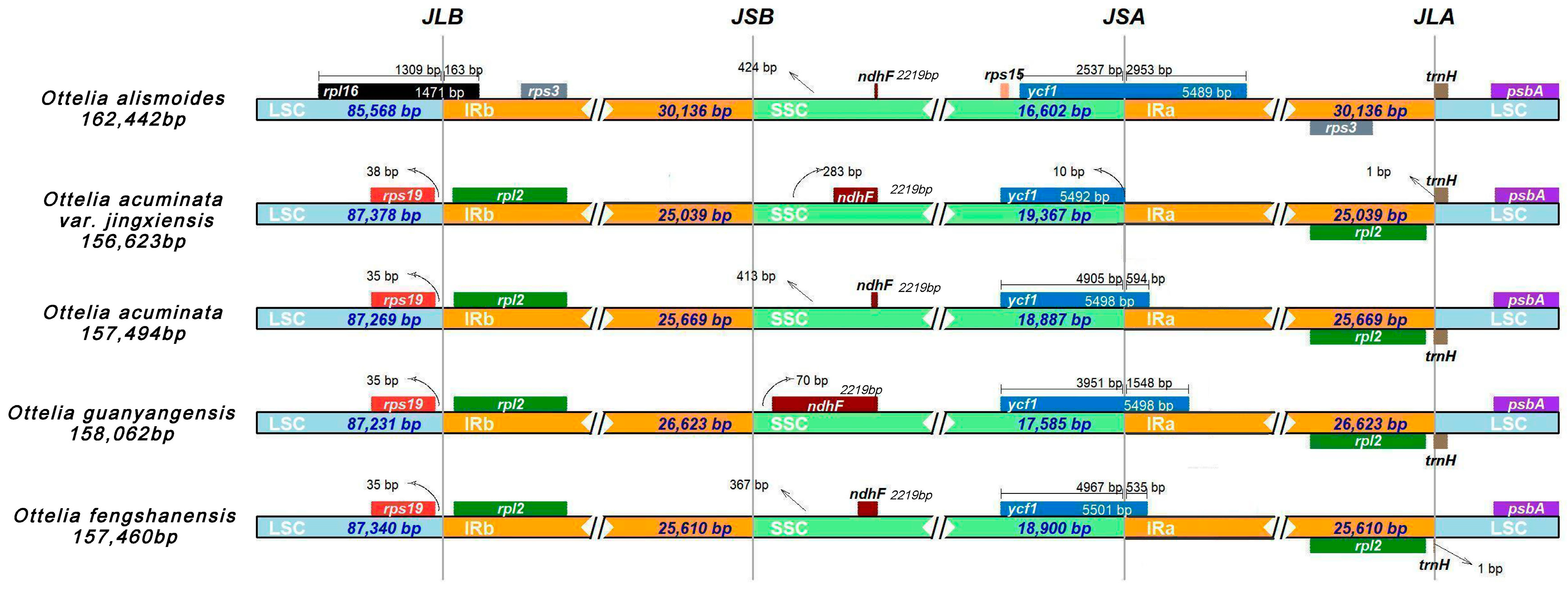
2.4. IR Expansion and Contraction
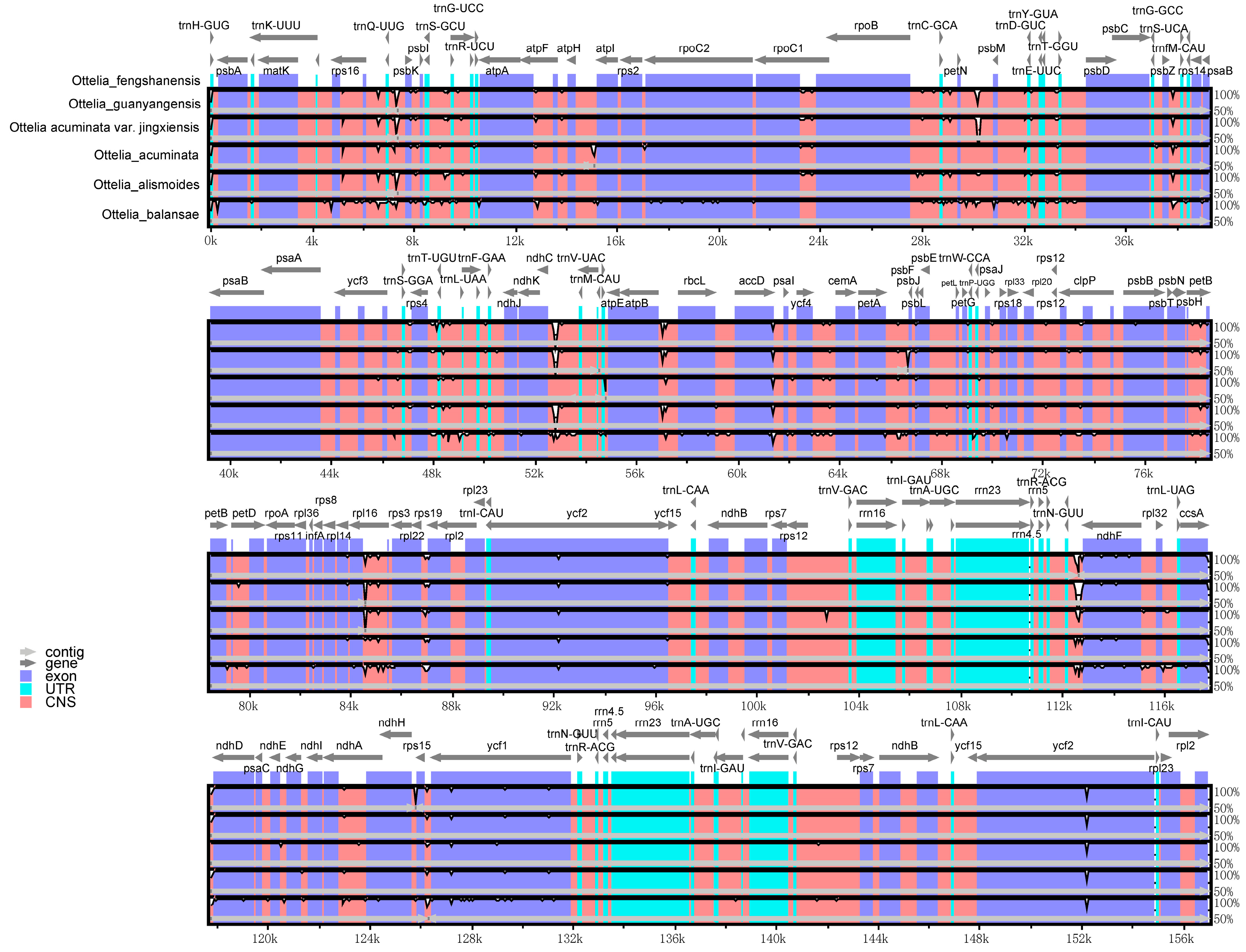
2.5. Variations within the cp Genome Synteny Analysis
2.6. Phylogenetic Analysis
3. Materials and Methods
3.1. Material Collection and DNA Extraction
3.2. Sequencing and Assembly of Chloroplast Genome
3.3. Chloroplast Genome Structural Analysis
3.4. Variations in the Chloroplast Genomes
3.5. Phylogenomic Reconstruction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cook, C.D.; Symoens, J.-J.; Urmi-König, K. A Revision of the Genus Ottelia (Hydrocharitaceae); Elsevier Science: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Ito, Y.; Tanaka, N.; Barfod, A.S.; Bogner, J.; Li, J.; Yano, O.; Gale, S.W. Molecular phylogenetic species delimitation in the aquatic genus Ottelia (Hydrocharitaceae) reveals cryptic diversity within a widespread species. J. Plant Res. 2019, 132, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Classification, distribution and phylogeny of the genus Ottelia. J. Syst. Evol. 1981, 19, 29. [Google Scholar]
- Li, Z.-Z.; Wu, S.; Zou, C.-Y.; Liu, Y.; Hu, G.-W.; Lehtonen, S.; Wang, Q.-F.; Chen, J.-M. Ottelia fengshanensis, a new bisexual species of Ottelia (Hydrocharitaceae) from southwestern China. PhytoKeys 2019, 135, 1. [Google Scholar] [CrossRef]
- Chen, J.-M.; Du, Z.-Y.; Long, Z.-C.; Gichira, A.W.; Wang, Q.-F. Molecular divergence among varieties of Ottelia acuminata (Hydrocharitaceae) in the Yunnan-Guizhou Plateau. Aquat. Bot. 2017, 140, 62–68. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, H.; Dao, Z.; Long, C. Ethnobotanical study on Ottelia acuminata, an aquatic edible plant occurring in Yunnan. J. Inn. Mong. Norm. Univ. 2010, 39, 163–168. [Google Scholar]
- Liang, S.; Li, G. Ottelia acuminata in Jingxi County. Wetl. Sci. Manag. 2007, 3, 15. [Google Scholar]
- Yang, J.; Li, J.; Li, J.; Li, M.; Zhang, Y. Summarize on the research of the hydro-bios and aquatic environment in Fuxian lake and Xingyun lake. Yunnan Geogr. Environ. Res. 2012, 24, 98–102. [Google Scholar]
- Qin, H.; Yang, Y.; Dong, S.; He, Q.; Jia, Y.; Zhao, L.; Yu, S.; Liu, H.; Liu, B.; Yan, Y. Threatened species list of China’s higher plants. Biodivers. Sci. 2017, 25, 696. [Google Scholar] [CrossRef]
- Tu, C.; Lin, H.; Wang, Q.; Wu, L.; Yang, Y.; Zou, L.; Wu, Q. The complete chloroplast genome of Ottelia acuminate var. crispa, an endangered aquatic herb with extremely narrow distribution. Mitochondrial DNA Part B 2021, 6, 1071–1072. [Google Scholar] [CrossRef]
- Guo, J.-L.; Yu, Y.-H.; Zhang, J.-W.; Li, Z.-M.; Zhang, Y.-H.; Volis, S. Conservation strategy for aquatic plants: Endangered Ottelia acuminata (Hydrocharitaceae) as a case study. Biodivers. Conserv. 2019, 28, 1533–1548. [Google Scholar] [CrossRef]
- He, J. Systematic Botanical and Biosystematic Studies on Ottelia in China; Wuhan University Press: Wuhan, China, 1991; Volume 1. [Google Scholar]
- Phillips, G.; Willby, N.; Moss, B. Submerged macrophyte decline in shallow lakes: What have we learnt in the last forty years? Aquat. Bot. 2016, 135, 37–45. [Google Scholar] [CrossRef]
- Zhang, Y.; Jeppesen, E.; Liu, X.; Qin, B.; Shi, K.; Zhou, Y.; Thomaz, S.M.; Deng, J. Global loss of aquatic vegetation in lakes. Earth-Sci. Rev. 2017, 173, 259–265. [Google Scholar] [CrossRef]
- Dandy, J.E. Notes on Hydrocharitaceae. 1. The delimitation and subdivision of Ottelia. J. Bot. 1934, 72, 132–137. [Google Scholar]
- Wang, Q.; Guo, Y.; Haynes, R.; Hellquist, C. Hydrocharitaceae. Flora China 2010, 23, 91–102. [Google Scholar]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Xing, S. Progress in chloroplast genome analysis. Prog. Biochem. Biophys. 2008, 35, 21–28. [Google Scholar]
- McCormack, J.E.; Hird, S.M.; Zellmer, A.J.; Carstens, B.C.; Brumfield, R.T. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol. Phylogenet. Evol. 2013, 66, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-L.; Li, Z.-M.; Yu, Y.-H.; Huang, Y.; Zhang, Y.-H. The complete chloroplast genome of Ottelia acuminata var. songmingensis, an endangered aquatic macrophyte in China. Mitochondrial DNA Part B 2019, 4, 1624–1625. [Google Scholar] [CrossRef]
- Wang, H.-X.; Guo, J.-L.; Li, Z.-M.; Yu, Y.-H.; Zhang, Y.-H. Characterization of the complete chloroplast genome of an endangered aquatic macrophyte, Ottelia cordata (Hydrocharitaceae). Mitochondrial DNA Part B 2019, 4, 1839–1840. [Google Scholar] [CrossRef]
- Li, Z.; Liao, K.; Zou, C.; Liu, Y.; Hu, G.; Wang, Q.; Chen, J. Ottelia guanyangensis (Hydrocharitaceae), a new species from southwestern China. Phytotaxa 2018, 361, 294–300. [Google Scholar] [CrossRef]
- Li, Z.-Z.; Lehtonen, S.; Martins, K.; Gichira, A.W.; Wu, S.; Li, W.; Hu, G.-W.; Liu, Y.; Zou, C.-Y.; Wang, Q.-F. Phylogenomics of the aquatic plant genus Ottelia (Hydrocharitaceae): Implications for historical biogeography. Mol. Phylogenet. Evol. 2020, 152, 106939. [Google Scholar] [CrossRef]
- Tang, J.; Zou, R.; Chen, T.; Pan, L.; Zhu, S.; Ding, T.; Chai, S.; Wei, X. Comparative Analysis of the Complete Chloroplast Genomes of Six Endangered Cycas Species: Genomic Features, Comparative Analysis, and Phylogenetic Implications. Forests 2023, 14, 2069. [Google Scholar] [CrossRef]
- Tang, J.; Zou, R.; Wei, X.; Li, D. Complete Chloroplast Genome Sequences of Five Ormosia Species: Molecular Structure, Comparative Analysis, and Phylogenetic Analysis. Horticulturae 2023, 9, 796. [Google Scholar] [CrossRef]
- Zhang, Q.; Shen, Z.; Li, F.; Li, G.; Shen, J. Complete chloroplast genome sequence of an endangered Ottelia cordata and its phylogenetic analysis. Mitochondrial DNA Part B 2020, 5, 2209–2210. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, L.; Qi, J.; Zhang, L. Complete chloroplast genome sequence of Hibiscus cannabinus and comparative analysis of the Malvaceae family. Front. Genet. 2020, 11, 227. [Google Scholar] [CrossRef]
- Shahzadi, I.; Mehmood, F.; Ali, Z.; Malik, M.S.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Comparative analyses of chloroplast genomes among three Firmiana species: Identification of mutational hotspots and phylogenetic relationship with other species of Malvaceae. Plant Gene 2019, 19, 100199. [Google Scholar]
- Wang, J.; Qian, J.; Jiang, Y.; Chen, X.; Zheng, B.; Chen, S.; Yang, F.; Xu, Z.; Duan, B. Comparative analysis of chloroplast genome and new insights into phylogenetic relationships of Polygonatum and tribe Polygonateae. Front. Plant Sci. 2022, 13, 882189. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Wang, L.; Lei, J.; Duan, B.; Ma, W.; Xiao, S.; Qi, H.; Wang, Z.; Liu, Y.; Shen, X. A comparative analysis of the chloroplast genomes of four Salvia medicinal plants. Engineering 2019, 5, 907–915. [Google Scholar] [CrossRef]
- Yaradua, S.S.; Alzahrani, D.A.; Albokhary, E.J.; Abba, A.; Bello, A. Complete chloroplast genome sequence of Justicia flava: Genome comparative analysis and phylogenetic relationships among Acanthaceae. BioMed Res. Int. 2019, 2019, 4370258. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dong, Y.; Liu, Y.; Yu, X.; Yang, M.; Huang, Y. Comparative analyses of Euonymus chloroplast genomes: Genetic structure, screening for loci with suitable polymorphism, positive selection genes, and phylogenetic relationships within Celastrineae. Front. Plant Sci. 2021, 11, 593984. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, T.; Zhou, Q.; Sheng, Q.; Zhu, Z. Complete Chloroplast Genomes and Phylogenetic Relationships of Bougainvillea spectabilis and Bougainvillea glabra (Nyctaginaceae). Int. J. Mol. Sci. 2023, 24, 13044. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Xue, Q.; Wang, H.; Xie, X.; Zhu, S.; Liu, W.; Ding, X. Mutational biases and GC-biased gene conversion affect GC content in the plastomes of Dendrobium genus. Int. J. Mol. Sci. 2017, 18, 2307. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Basak, S.; Ghosh, T.C. Nature of selective constraints on synonymous codon usage of rice differs in GC-poor and GC-rich genes. Gene 2007, 400, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.e.; Li, C. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, Z.; Shi, W.; Han, W.; Feng, Q.; Shi, C.; Engel, M.S.; Wang, S. Comparative analysis of complete chloroplast genomes of nine species of Litsea (Lauraceae): Hypervariable regions, positive selection, and phylogenetic relationships. Genes 2022, 13, 1550. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Han, L.; Chen, C.; Wang, Z. The complete chloroplast genome sequence of Epipremnum aureum and its comparative analysis among eight Araceae species. PLoS ONE 2018, 13, e0192956. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, F.; Shahzadi, I.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): Comparative analyses and identification of mutational hotspots. Genomics 2020, 112, 581–591. [Google Scholar]
- Cui, Y.; Nie, L.; Sun, W.; Xu, Z.; Wang, Y.; Yu, J.; Song, J.; Yao, H. Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family Zingiberaceae based on the complete chloroplast genome. Plants 2019, 8, 283. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, N.; Wu, S.; Jiang, C.; Xie, L.; Yang, F.; Yu, Z. A Comparative Analysis of the Chloroplast Genomes of Three Lonicera Medicinal Plants. Genes 2023, 14, 548. [Google Scholar] [CrossRef]
- Chen, J.; Hao, Z.; Xu, H.; Yang, L.; Liu, G.; Sheng, Y.; Zheng, C.; Zheng, W.; Cheng, T.; Shi, J. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front. Plant Sci. 2015, 6, 148371. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-J.; Cheng, C.-L.; Chang, C.-C.; Wu, C.-L.; Su, T.-M.; Chaw, S.-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef]
- Kim, H.T.; Kim, J.S.; Moore, M.J.; Neubig, K.M.; Williams, N.H.; Whitten, W.M.; Kim, J.-H. Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across orchids and associated instability of the inverted repeat/small single-copy region boundaries. PLoS ONE 2015, 10, e0142215. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Nguyen, V.B.; Dong, J.; Wang, Y.; Park, J.Y.; Lee, S.-C.; Yang, T.-J. Evolution of the Araliaceae family inferred from complete chloroplast genomes and 45S nrDNAs of 10 Panax-related species. Sci. Rep. 2017, 7, 4917. [Google Scholar] [CrossRef] [PubMed]
- Gielly, L.; Taberlet, P. The use of chloroplast DNA to resolve plant phylogenies: Noncoding versus rbcL sequences. Mol. Biol. Evol. 1994, 11, 769–777. [Google Scholar] [PubMed]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.-Y.; Gao, L.-Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Genome | LSC | IR | SSC | Gene Number | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Length (bp) | G+C Content (%) | Length (bp) | G+C Content (%) | Length (bp) | G+C Content (%) | Length (bp) | G+C Content (%) | Total | PCGs | tRNA | rRNA | |
| Ottelia guanyangensis | 158,062 | 36.68 | 87,231 | 34.42 | 26,623 | 42.53 | 17,585 | 30.12 | 130 | 85 | 37 | 8 |
| Ottelia fengshanensis | 157,460 | 36.65 | 87,340 | 34.40 | 25,564 | 43.09 | 18,992 | 29.70 | 130 | 85 | 37 | 8 |
| Ottelia acuminata var. jingxiensis | 156,823 | 36.63 | 87,378 | 34.34 | 25,039 | 43.23 | 19,367 | 29.95 | 130 | 85 | 37 | 8 |
| Ottelia acuminata | 157,494 | 36.64 | 87,269 | 34.39 | 25,669 | 43.01 | 18,887 | 29.73 | 130 | 85 | 37 | 8 |
| Ottelia alismoides | 162,442 | 36.41 | 85,568 | 34.45 | 30,136 | 41.10 | 16,602 | 29.54 | 133 | 88 | 37 | 8 |
| Type | Ottelia fengshanensis | Ottelia guanyangensis | Ottelia acuminata | Ottelia alismoides | Ottelia acuminata var. jingxiensis |
|---|---|---|---|---|---|
| TTT | 1.322 | 1.317 | 1.321 | 1.244 | 1.251 |
| TTC | 0.678 | 0.683 | 0.679 | 0.756 | 0.749 |
| TTA | 2.169 | 2.169 | 2.171 | 1.969 | 1.958 |
| TTG | 1.152 | 1.154 | 1.153 | 1.242 | 1.244 |
| TCT | 1.7 | 1.7 | 1.706 | 1.649 | 1.66 |
| TCC | 1.088 | 1.083 | 1.075 | 1.103 | 1.092 |
| TCA | 1.111 | 1.124 | 1.121 | 1.223 | 1.231 |
| TCG | 0.438 | 0.424 | 0.434 | 0.493 | 0.49 |
| TAT | 1.641 | 1.641 | 1.641 | 1.643 | 1.64 |
| TAC | 0.359 | 0.359 | 0.359 | 0.357 | 0.36 |
| TGT | 1.5 | 1.498 | 1.5 | 1.51 | 1.502 |
| TGC | 0.5 | 0.502 | 0.5 | 0.49 | 0.498 |
| TGG | 1 | 1 | 1 | 1 | 1 |
| CTT | 1.261 | 1.26 | 1.259 | 1.254 | 1.275 |
| CTC | 0.292 | 0.292 | 0.292 | 0.365 | 0.343 |
| CTA | 0.796 | 0.794 | 0.793 | 0.82 | 0.829 |
| CTG | 0.33 | 0.331 | 0.331 | 0.349 | 0.352 |
| CCT | 1.666 | 1.658 | 1.664 | 1.517 | 1.542 |
| CCC | 0.768 | 0.772 | 0.767 | 0.786 | 0.769 |
| CCA | 1.128 | 1.132 | 1.126 | 1.189 | 1.177 |
| CCG | 0.439 | 0.438 | 0.443 | 0.508 | 0.513 |
| CAT | 1.579 | 1.579 | 1.579 | 1.596 | 1.581 |
| CAC | 0.421 | 0.421 | 0.421 | 0.404 | 0.419 |
| CAA | 1.543 | 1.543 | 1.543 | 1.505 | 1.514 |
| CAG | 0.457 | 0.457 | 0.457 | 0.495 | 0.486 |
| CGT | 1.56 | 1.56 | 1.56 | 1.319 | 1.335 |
| CGC | 0.403 | 0.403 | 0.403 | 0.401 | 0.4 |
| CGA | 1.388 | 1.388 | 1.388 | 1.387 | 1.38 |
| CGG | 0.294 | 0.294 | 0.294 | 0.389 | 0.396 |
| ATT | 1.54 | 1.541 | 1.538 | 1.489 | 1.484 |
| ATC | 0.504 | 0.501 | 0.503 | 0.53 | 0.54 |
| ATA | 0.956 | 0.958 | 0.959 | 0.981 | 0.976 |
| ATG | 1 | 1 | 1 | 1 | 1 |
| ACT | 1.688 | 1.681 | 1.695 | 1.591 | 1.59 |
| ACC | 0.716 | 0.721 | 0.711 | 0.711 | 0.716 |
| ACA | 1.148 | 1.149 | 1.138 | 1.19 | 1.189 |
| ACG | 0.448 | 0.449 | 0.456 | 0.508 | 0.505 |
| AAT | 1.502 | 1.502 | 1.501 | 1.534 | 1.525 |
| AAC | 0.498 | 0.498 | 0.499 | 0.466 | 0.475 |
| AAA | 1.57 | 1.57 | 1.57 | 1.504 | 1.5 |
| AAG | 0.43 | 0.43 | 0.43 | 0.496 | 0.5 |
| AGT | 1.313 | 1.318 | 1.314 | 1.177 | 1.175 |
| AGC | 0.35 | 0.35 | 0.35 | 0.355 | 0.352 |
| AGA | 1.854 | 1.854 | 1.854 | 1.912 | 1.882 |
| AGG | 0.501 | 0.501 | 0.501 | 0.593 | 0.608 |
| GTT | 1.483 | 1.483 | 1.483 | 1.463 | 1.453 |
| GTC | 0.408 | 0.401 | 0.401 | 0.463 | 0.439 |
| GTA | 1.577 | 1.584 | 1.584 | 1.472 | 1.53 |
| GTG | 0.532 | 0.532 | 0.532 | 0.602 | 0.578 |
| GCT | 1.836 | 1.838 | 1.838 | 1.849 | 1.844 |
| GCC | 0.591 | 0.588 | 0.588 | 0.593 | 0.579 |
| GCA | 1.101 | 1.099 | 1.102 | 1.067 | 1.095 |
| GCG | 0.471 | 0.475 | 0.472 | 0.49 | 0.482 |
| GAT | 1.574 | 1.574 | 1.574 | 1.619 | 1.625 |
| GAC | 0.426 | 0.426 | 0.426 | 0.381 | 0.375 |
| GAA | 1.517 | 1.517 | 1.517 | 1.476 | 1.478 |
| GAG | 0.483 | 0.483 | 0.483 | 0.524 | 0.522 |
| GGT | 1.389 | 1.386 | 1.389 | 1.321 | 1.317 |
| GGC | 0.36 | 0.354 | 0.357 | 0.353 | 0.347 |
| GGA | 1.517 | 1.517 | 1.514 | 1.584 | 1.598 |
| GGG | 0.734 | 0.743 | 0.74 | 0.741 | 0.737 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Zou, R.; Huang, K.; Gao, L.; Tang, F.; Ding, T.; Jiang, Y.; Wei, X. Comparative and Phylogenetic Analyses of the Complete Chloroplast Genomes of Four Ottelia Species. Horticulturae 2024, 10, 603. https://doi.org/10.3390/horticulturae10060603
Tang J, Zou R, Huang K, Gao L, Tang F, Ding T, Jiang Y, Wei X. Comparative and Phylogenetic Analyses of the Complete Chloroplast Genomes of Four Ottelia Species. Horticulturae. 2024; 10(6):603. https://doi.org/10.3390/horticulturae10060603
Chicago/Turabian StyleTang, Jianmin, Rong Zou, Ke Huang, Limei Gao, Fengluan Tang, Tao Ding, Yunsheng Jiang, and Xiao Wei. 2024. "Comparative and Phylogenetic Analyses of the Complete Chloroplast Genomes of Four Ottelia Species" Horticulturae 10, no. 6: 603. https://doi.org/10.3390/horticulturae10060603
APA StyleTang, J., Zou, R., Huang, K., Gao, L., Tang, F., Ding, T., Jiang, Y., & Wei, X. (2024). Comparative and Phylogenetic Analyses of the Complete Chloroplast Genomes of Four Ottelia Species. Horticulturae, 10(6), 603. https://doi.org/10.3390/horticulturae10060603