
Joint Analysis of Small RNA and mRNA Sequencing Unveils miRNA-Mediated Regulatory Network in Response to Methyl Jasmonate in Apocynum venetum L.
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Stress Treatments
2.2. Physiological and Biochemical Analysis
2.3. Chromosomal Positioning of miRNA Precursor
2.4. Transcriptome Sequencing and Data Analysis
2.5. Utilization of Small RNA Sequencing Techniques Coupled with Comprehensive Data Analysis
2.6. qRT-PCR Analysis of miRNAs and Their Targeting mRNAs
2.7. Promoter Analysis of miRNAs in A. venetum
2.8. Double-Luciferase Reporter Gene Test
3. Results
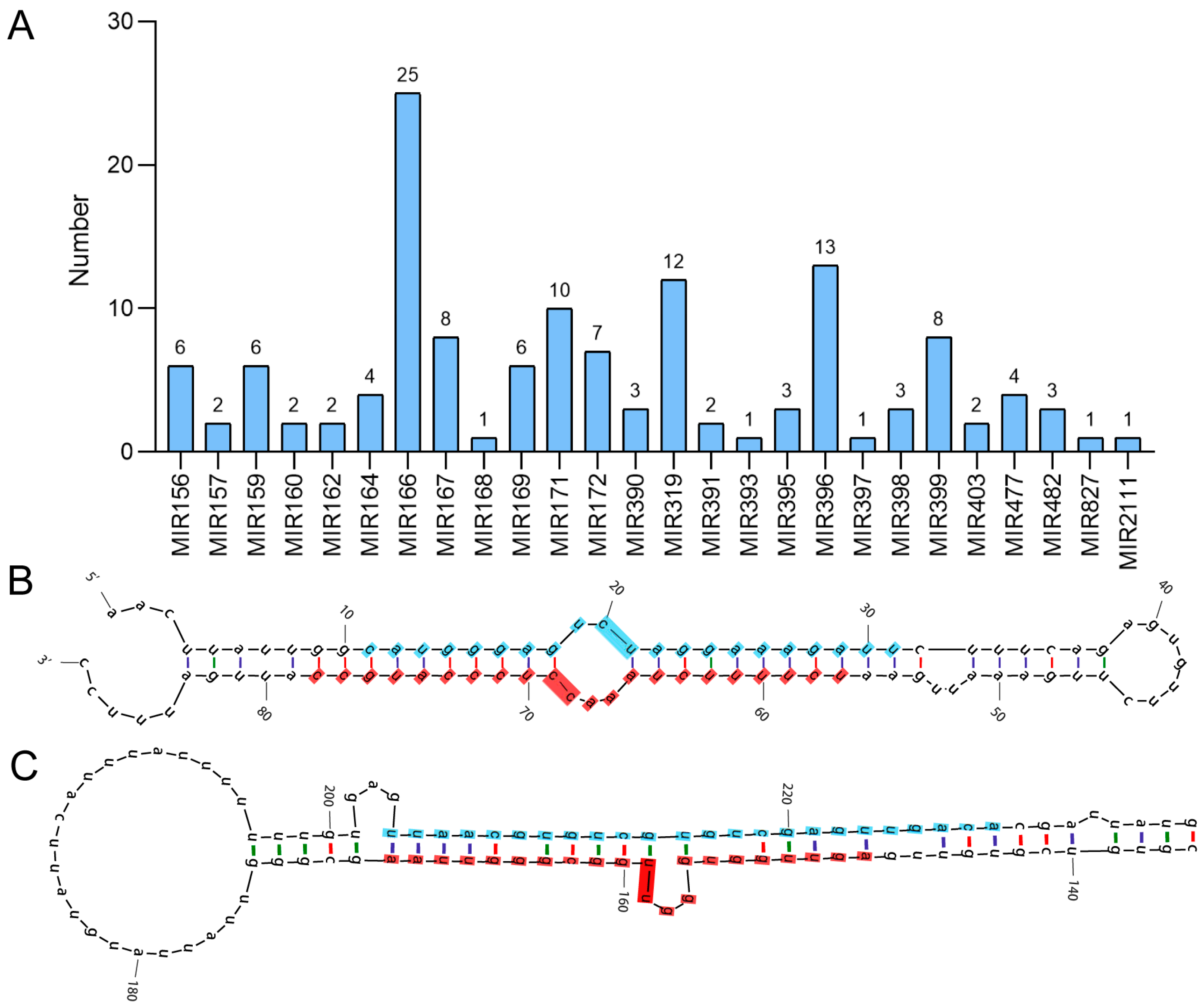
3.1. Identification and Characterization of Conserved and Species-Specific miRNAs in A. venetum
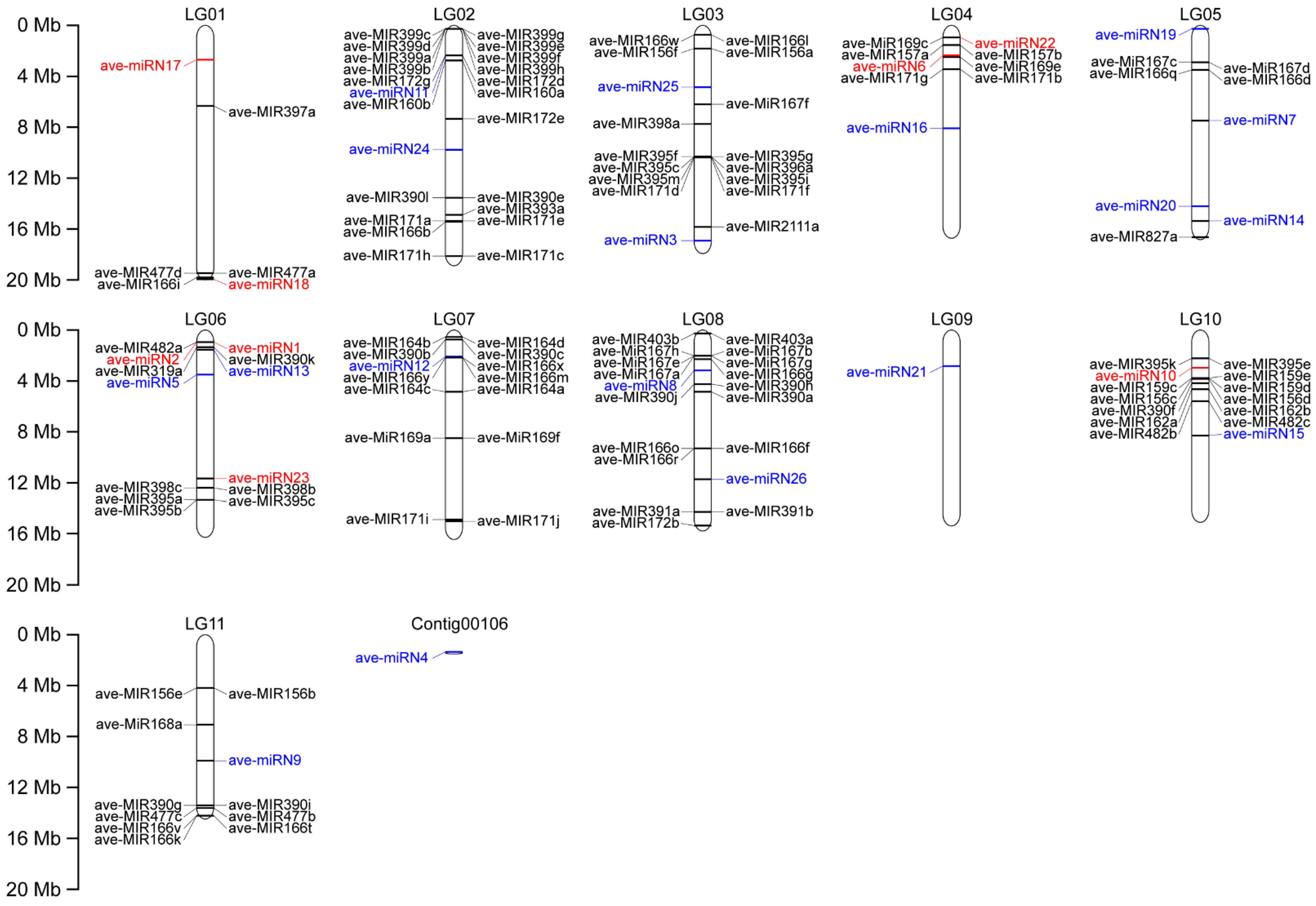
3.2. Chromosomal Localization of Identified miRNAs in A. venetum
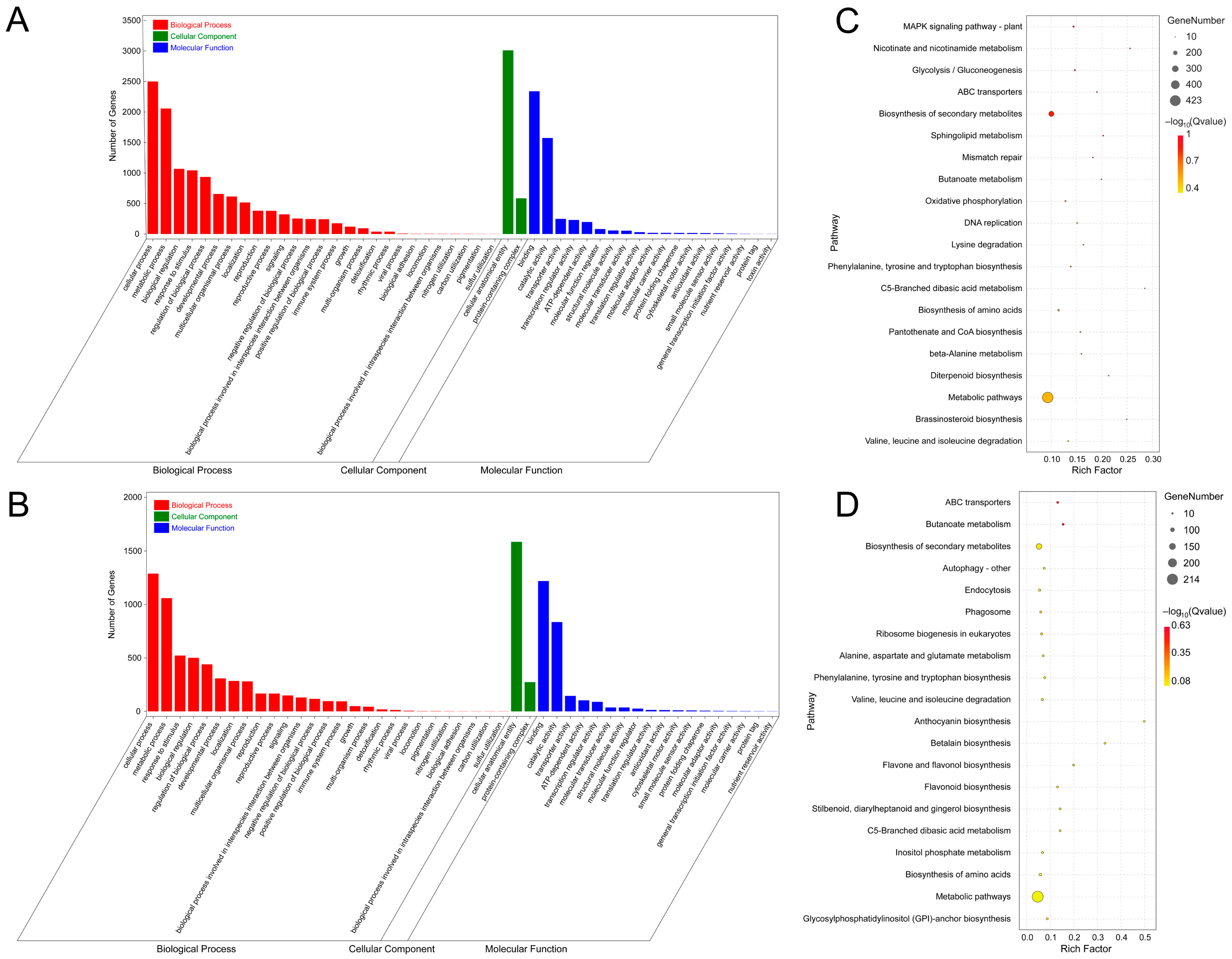
3.3. Prediction and Functional Analysis of Target Genes of miRNA in A. venetum
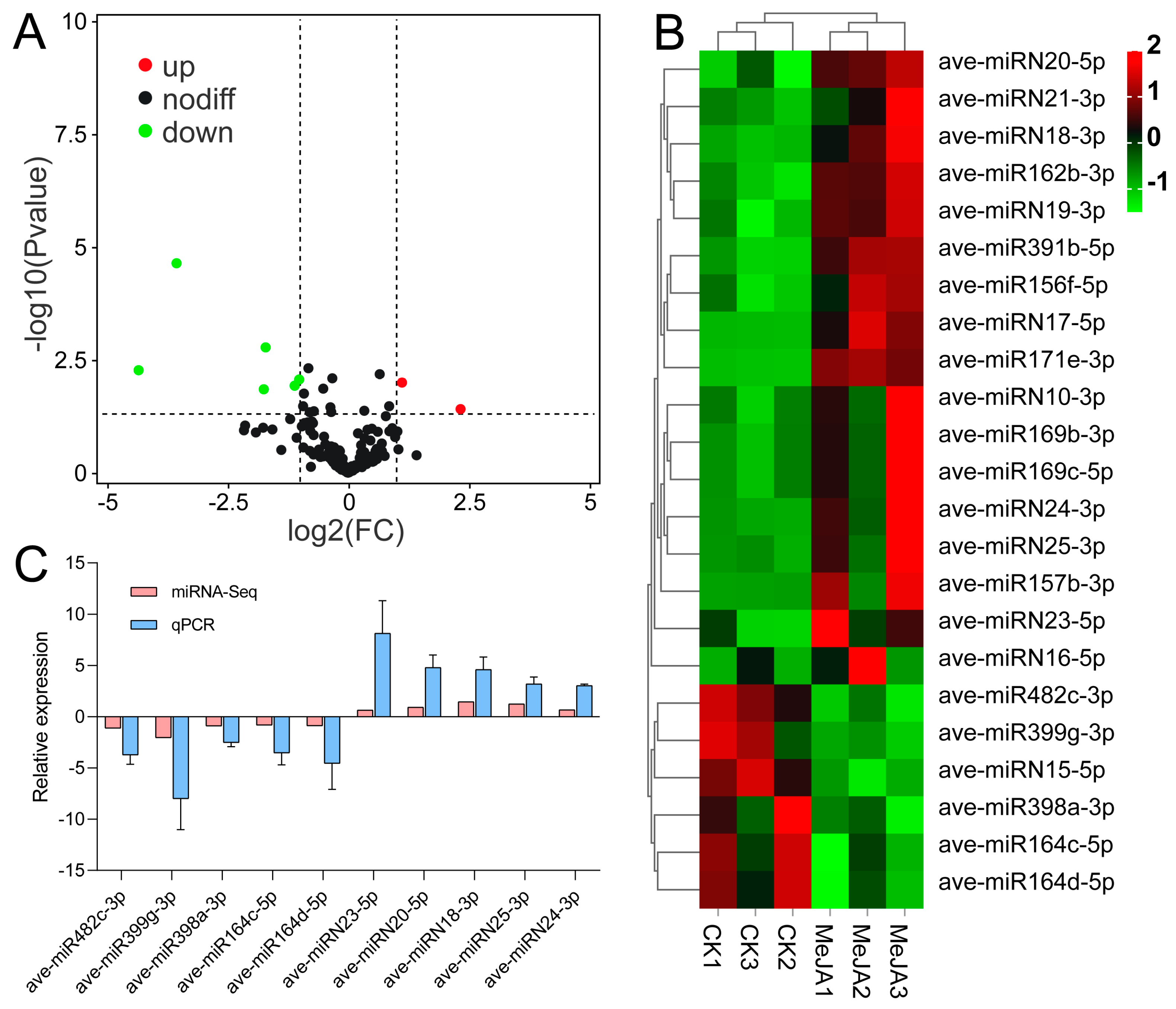
3.4. Identification of miRNAs Responding to MeJA Application in A. venetum
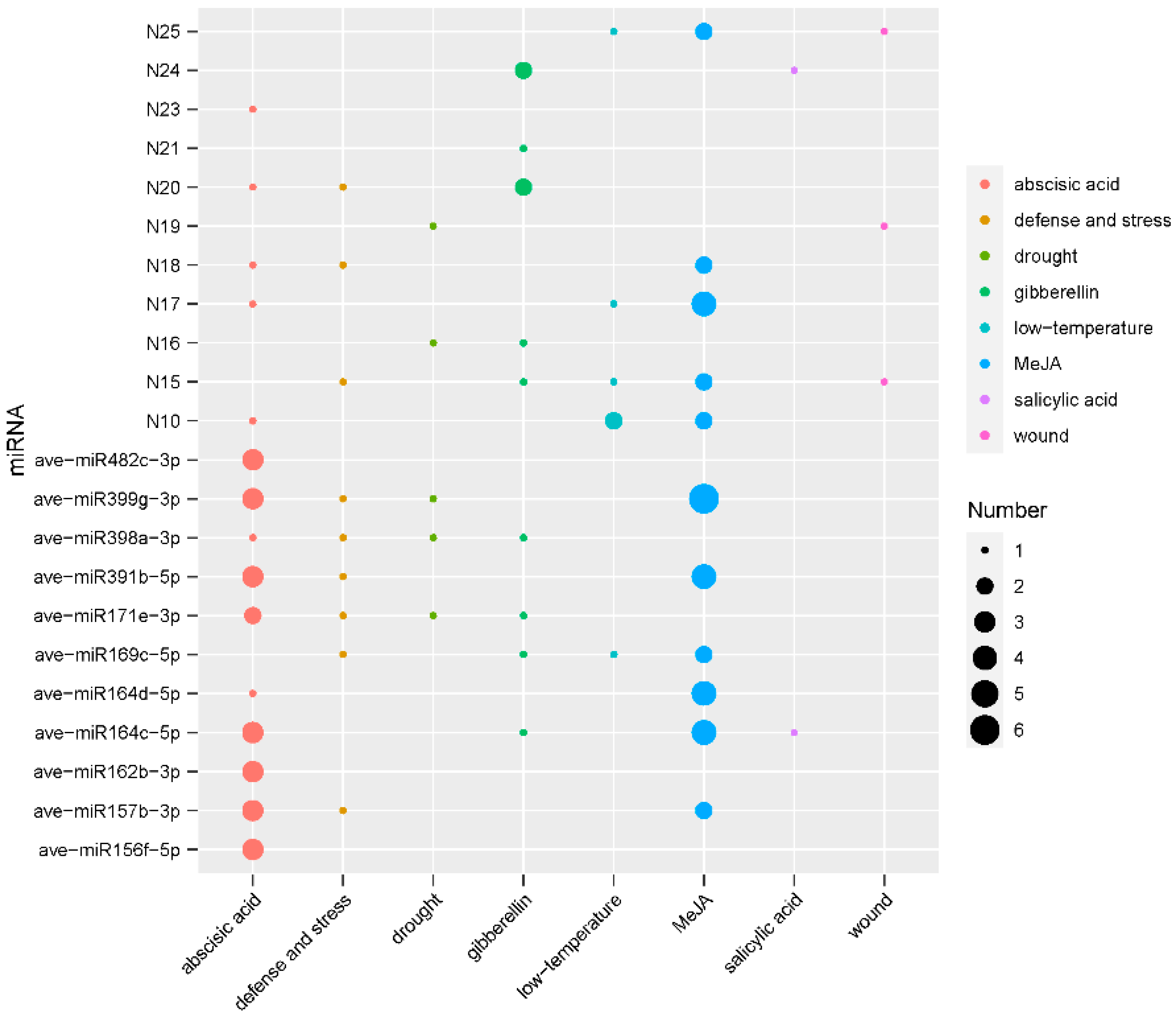
3.5. Promoter Analysis of MeJA-Responsive miRNAs
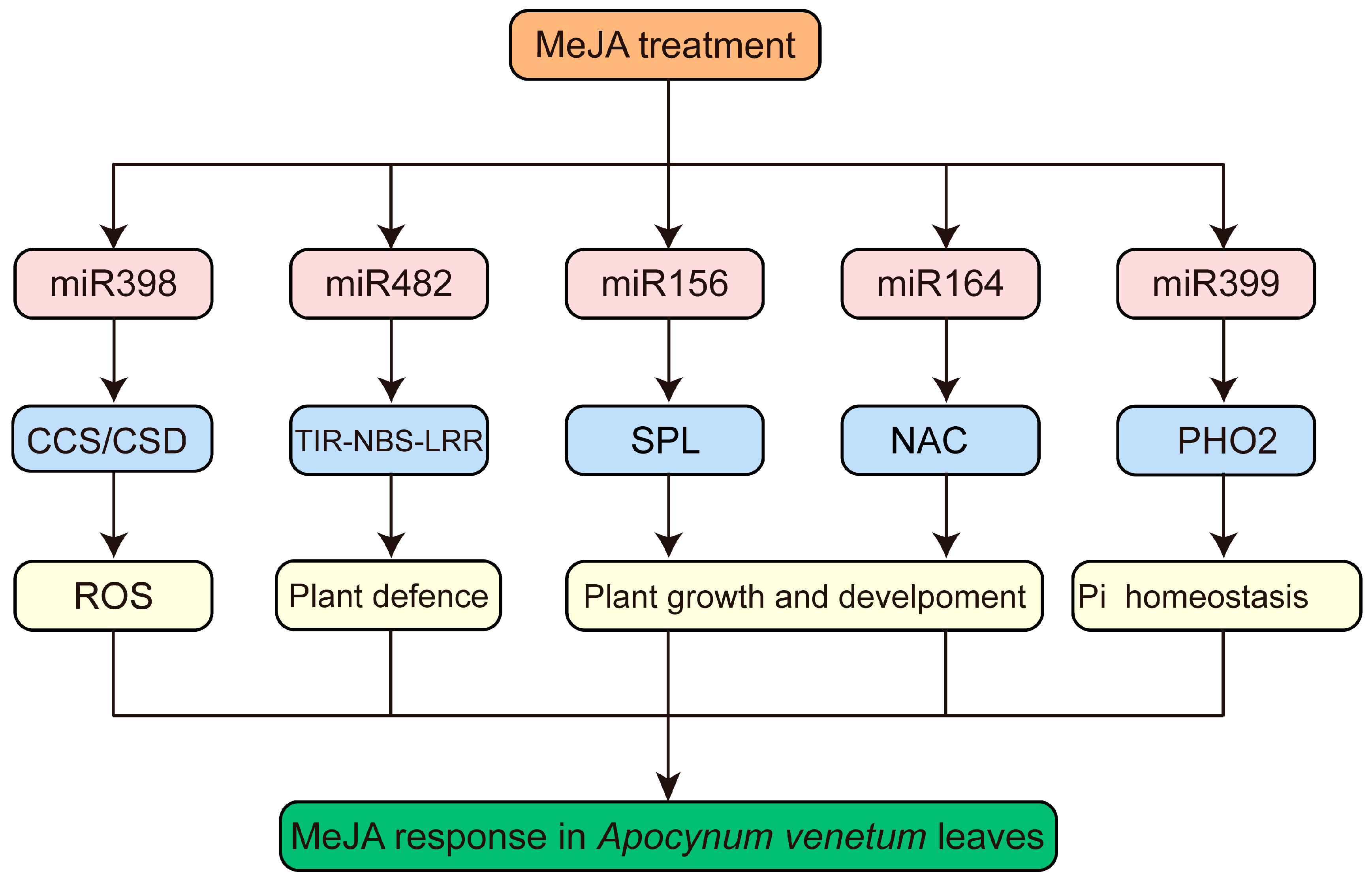
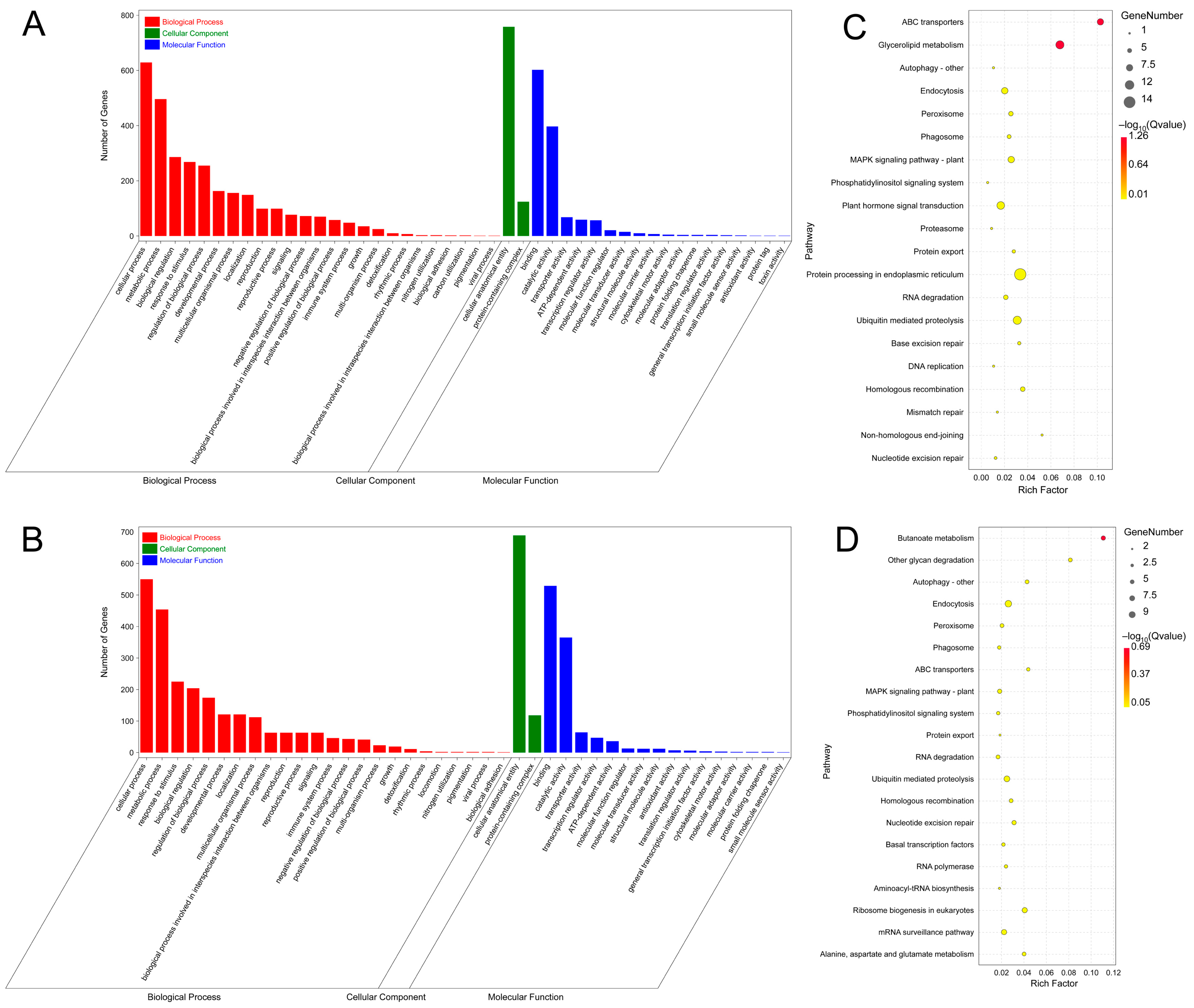
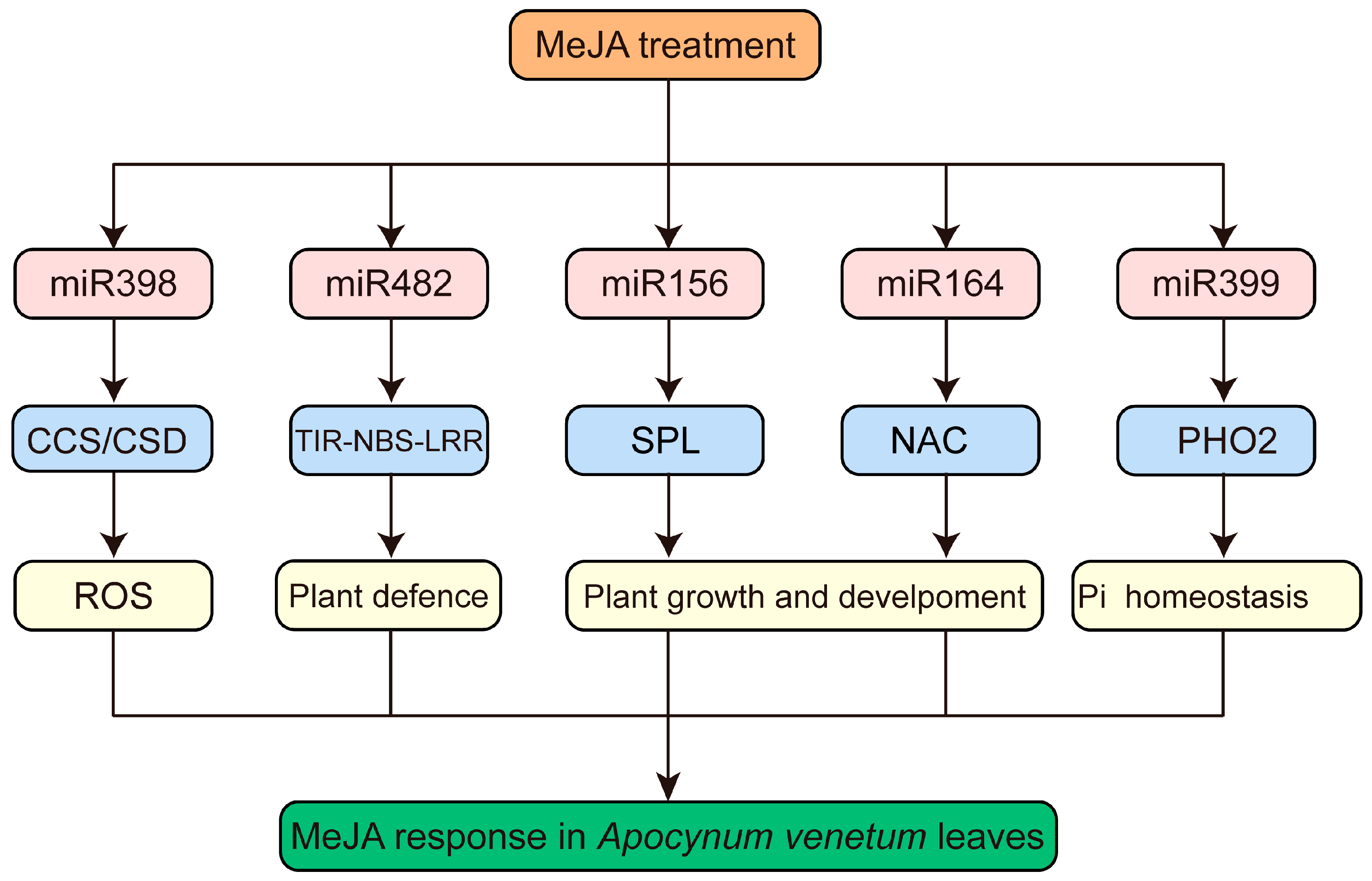
3.6. Functional Analysis of Targets of MeJA-Responsive miRNAs in A. venetum
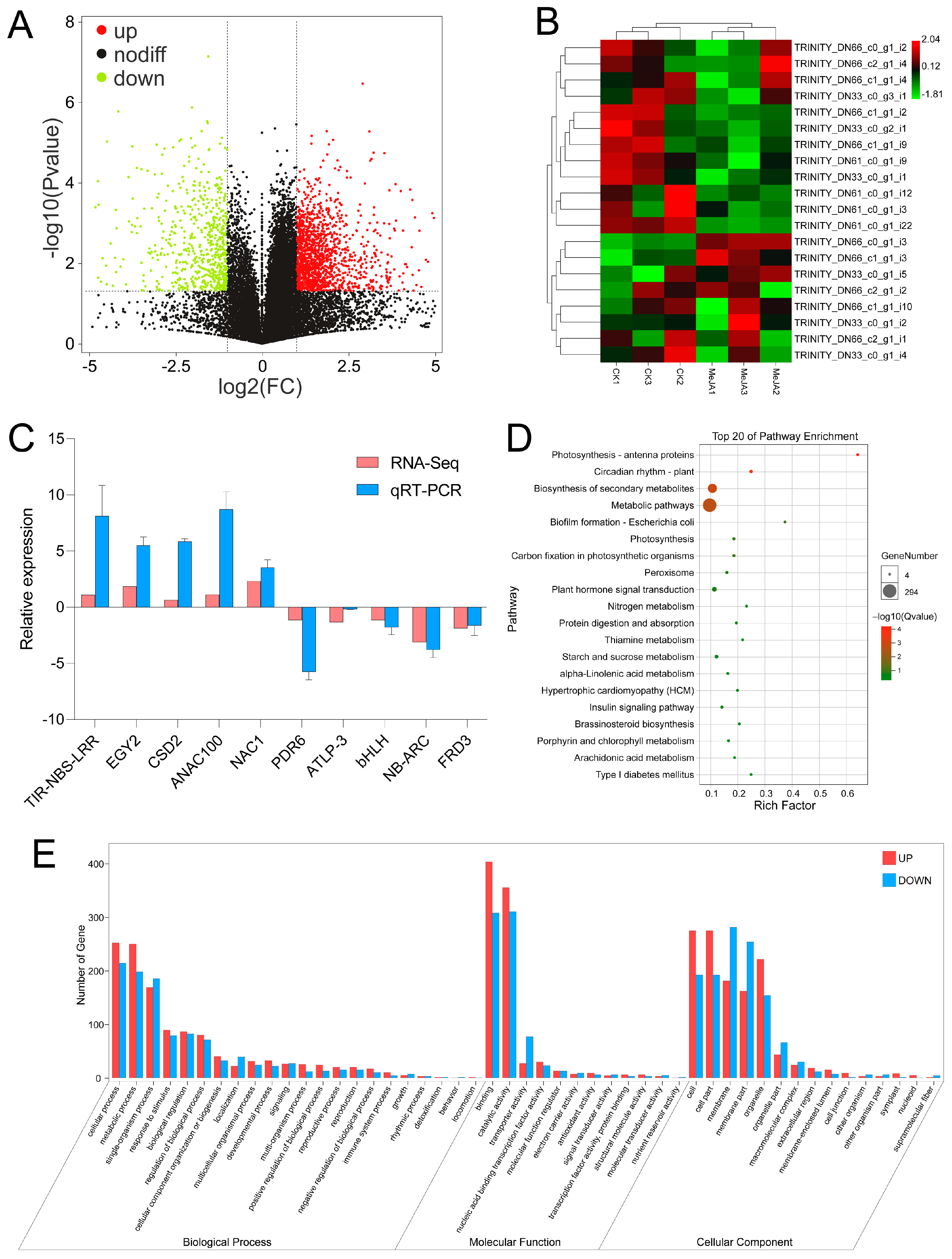
3.7. Transcriptome Analysis of the Response to MeJA in A. venetum
3.8. Joint Analysis of miRNA and mRNA Sequencing in A. venetum under MeJA Application
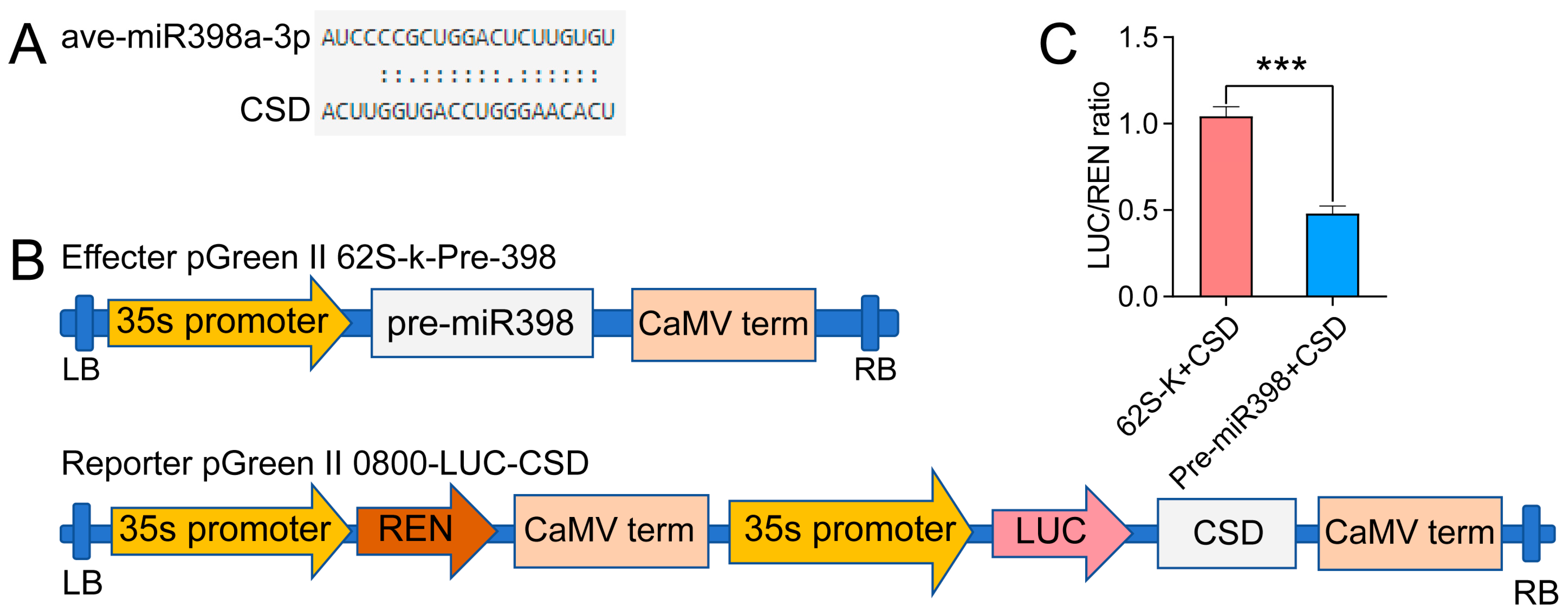
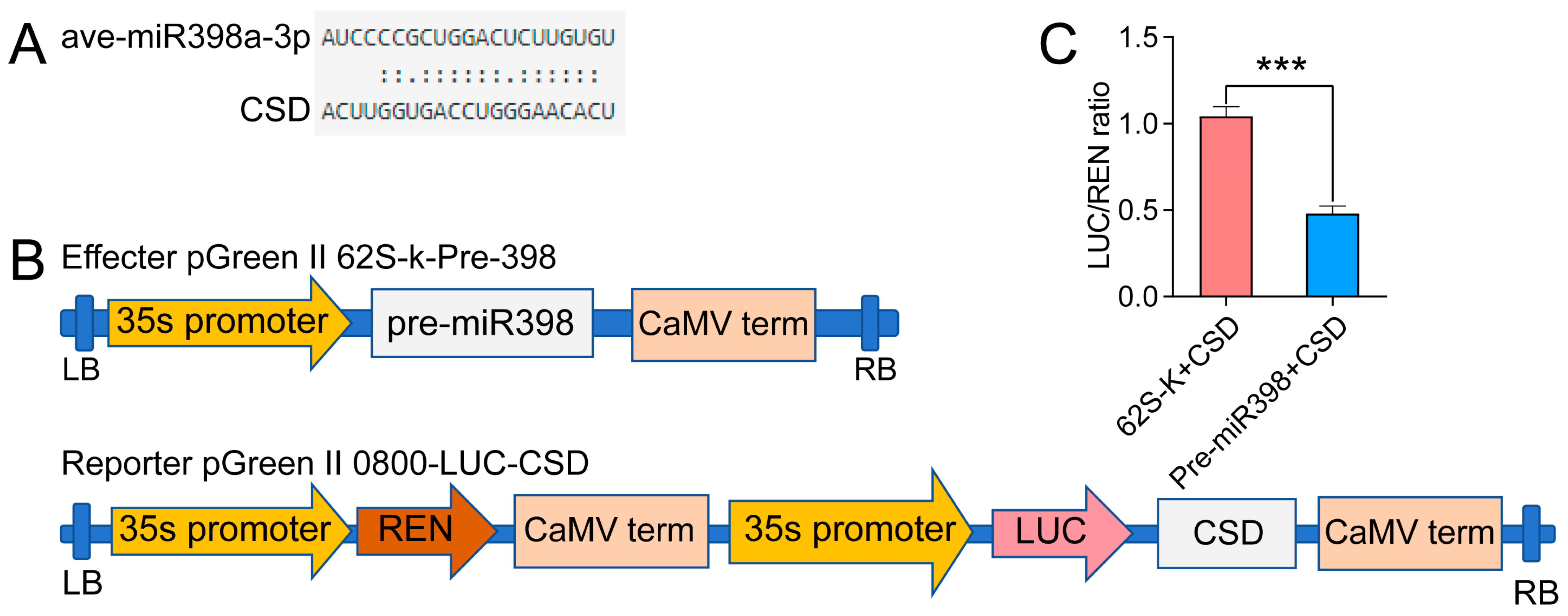
3.9. Dual-Luciferase Reporter Analysis Proving the Targeting Relation between miR398 and CSD2
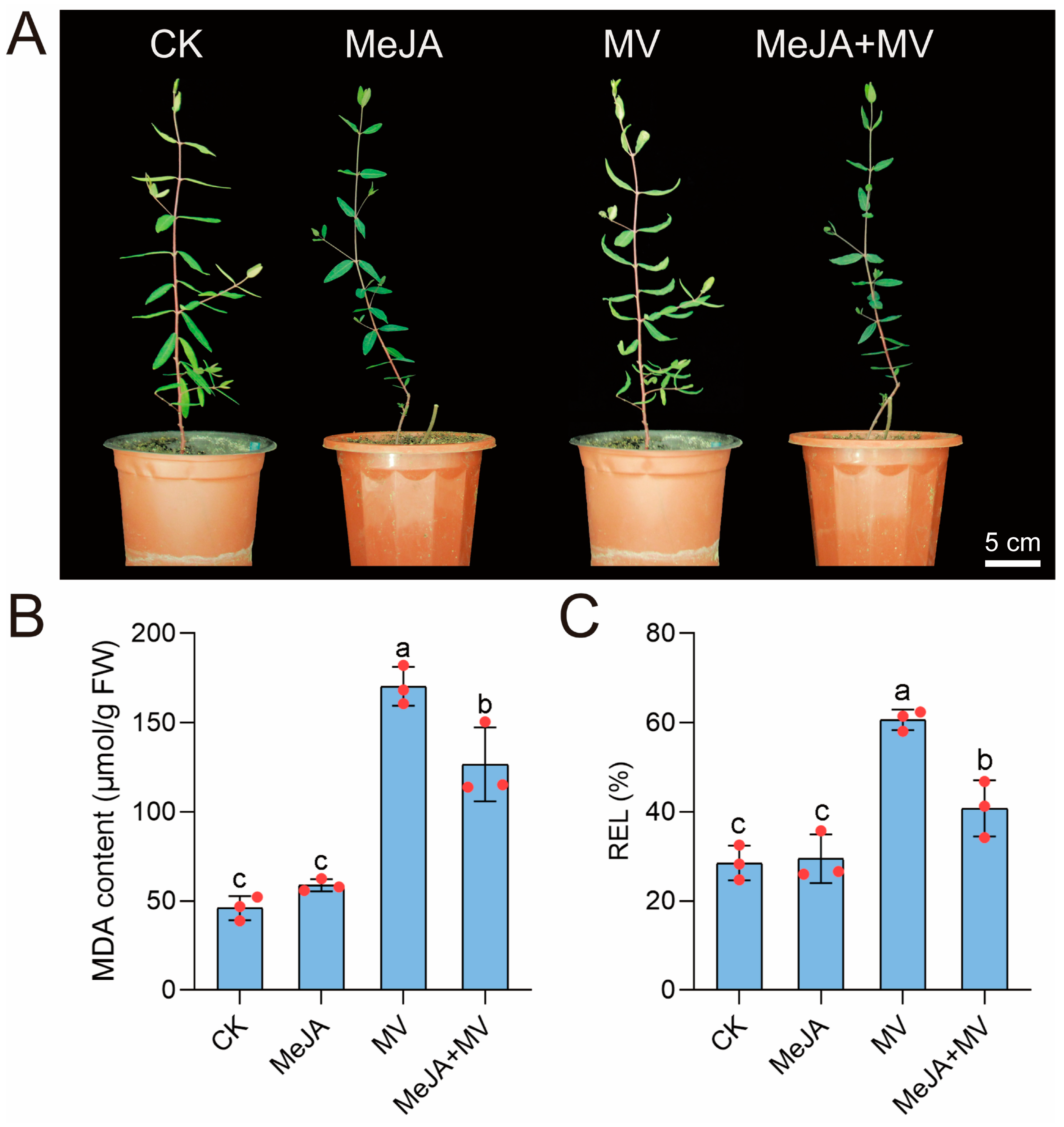
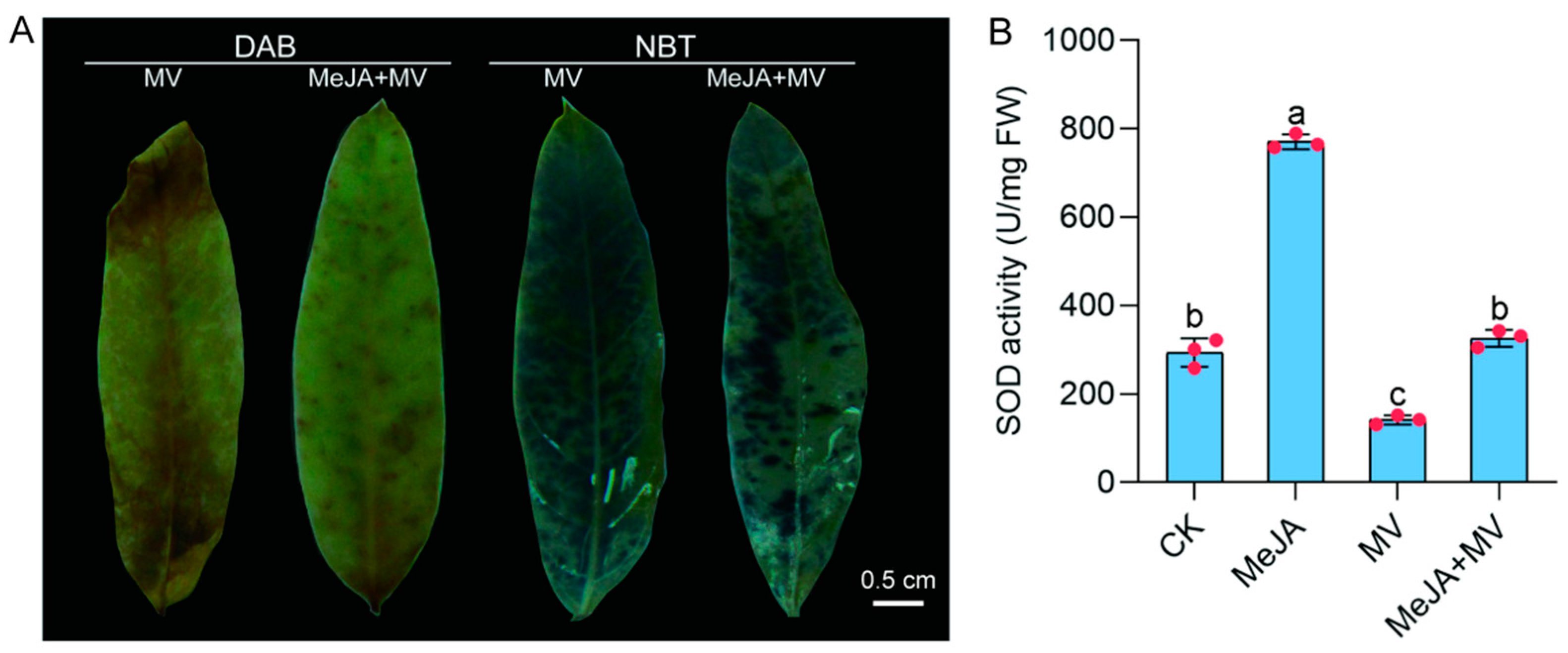
3.10. MeJA Treatment Improved Tolerance of A. venetum to Oxidative Stress
3.11. Tolerance to Oxidative Stress Induced by MeJA Is Related to the Elevation of SOD Enzyme Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xie, W.; Zhang, X.; Wang, T.; Hu, J. Botany, traditional uses, phytochemistry and pharmacology of Apocynum venetum L. (Luobuma): A review. J. Ethnopharmacol. 2012, 141, 1–8. [Google Scholar] [CrossRef]
- Lu, C.; Zhang, W.; Peng, X.; Gu, G.; Chen, M.; Tang, Z. Development of randomly amplified polymorphic DNA-Sequence characterized amplified region marker for identification of Apocynum venetum LINN. from A. pictum SCHRENK. Biol. Pharm. Bull. 2010, 33, 522–526. [Google Scholar] [CrossRef]
- Thevs, N.; Zerbe, S.; Kyosev, Y.; Rozi, A.; Tang, B.; Abdusalih, N.; Novitskij, Z. Apocynum venetum L. and Apocynum pictum Schrenk (Apocynaceae) as multi-functional and multi-service plant species in central asia: A review on biology, ecology, andutilization. J. Appl. Bot. Food Qual. 2012, 85, 159–167. Available online: https://hdl.handle.net/10863/178 (accessed on 2 December 2023).
- Yu, X.; Zhang, W.; Zhang, Y.; Zhang, X.; Lang, D.; Zhang, X. The roles of methyl jasmonate to stress in plants. Funct. Plant Biol. 2018, 46, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Wasternack, C.; Feussner, I. The oxylipin pathways: Biochemistry and function. Annu. Rev. Plant Biol. 2018, 69, 363–386. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chen, Y.; Zhao, L.; Chen, Y.; Zheng, L.; Zheng, K.; Zhang, J. Tripartite interactions between jasmonic/salicylic acid pathways, western flower thrips, and thrips-transmitted tomato zonate spot virus infection in Capsicuum annuum. Arthropod-Plant Interact. 2019, 13, 289–297. [Google Scholar] [CrossRef]
- Sun, D.; Lu, X.; Hu, Y.; Li, W.; Hong, K.; Mo, Y.; Xie, J. Methyl jasmonate induced defense responses increase resistance to Fusarium oxysporum f. sp. cubense race 4 in banana. Sci. Hortic. 2013, 164, 484–491. [Google Scholar] [CrossRef]
- Karimi, N.; Ghasempour, H.R. Salicylic acid and jasmonic acid restrains nickel toxicity by ameliorating antioxidant defense system in shoots of metallicolous and non-metallicolous Alyssum inflatum Náyr. Populations. Plant Physiol. Biochem. 2019, 135, 450–459. [Google Scholar] [CrossRef]
- Qiu, Y.; An, K.; Sun, J.; Chen, X.; Gong, X.; Ma, L.; Wang, Y. Investigating the effect of methyl jasmonate and melatonin on resistance of Malus crabapple ‘Hong Jiu’to ozone stress. Environ. Sci. Pollut. Res. 2019, 26, 27761–27768. [Google Scholar] [CrossRef]
- Kasschau, K.D.; Xie, Z.; Allen, E.; Llave, C.; Chapman, E.J.; Krizan, K.A.; Carrington, J.C. P1/HC-Pro. a viral suppressor of RNA silencing, interferes with Arabidopsis development and miRNA function. Dev. Cell 2002, 4, 205–217. [Google Scholar] [CrossRef]
- Llave, C.; Xie, Z.; Kasschau, K.D.; Carrington, J.C. Cleavage of scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 2002, 297, 2053–2056. [Google Scholar] [CrossRef]
- Song, X.; Li, Y.; Cao, X.; Qi, Y. MicroRNAs and their regulatory roles in plant-environment interactions. Annu. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, X.; Zhou, Y.; Tan, J.; Zhou, Y.; Gao, F. Small RNA sequencing revealed that miR4415, a legume-specific miRNA, was involved in the cold acclimation of Ammopiptanthus nanus by targeting an L-ascorbate oxidase gene and regulating the redox state of apoplast. Front. Genet. 2022, 13, 870446. [Google Scholar] [CrossRef]
- Stief, A.; Altmann, S.; Hoffmann, K.; Pant, B.D.; Scheible, W.R.; Baurle, I. Arabidopsis mir156 regulates tolerance to recurring environmental stress through spl transcription factors. Plant Cell 2014, 26, 1792. [Google Scholar] [CrossRef]
- Williams, L. Regulation of Arabidopsis shoot apical meristem and lateral organ formation by microRNA miR166g and its AtHD-ZIP target genes. Development 2005, 132, 3657–3668. [Google Scholar] [CrossRef]
- Val-Torregrosa, B.; Bundó, M.; San Segundo, B. Crosstalk between nutrient signaling pathways and immune responses in rice. Agriculture 2021, 11, 747. [Google Scholar] [CrossRef]
- Liao, L.; Xie, B.; Guan, P.; Jiang, N.; Cui, J. New insight into the molecular mechanism of miR482/2118 during plant resistance to pathogens. Front. Plant. Sci. 2022, 13, 1026762. [Google Scholar] [CrossRef]
- Sumbur, B.; Gao, F.; Liu, Q.; Feng, D.; Bing, J.; Dorjee, T.; Zhou, Y. The Characterization of R2R3-MYB Genes in Ammopiptanthus nanus uncovers that the miR858-AnaMYB87 Module mediates the accumulation of anthocyanin under osmotic stress. Biomolecules 2023, 13, 1721. [Google Scholar] [CrossRef]
- Sajad, S.; Dai, Q.; Yang, J.; Song, J. Identification of miRNAs involved in male fertility and pollen development in Brassica oleracea var. capitata L. by High-Throughput Sequencing. Horticulturae 2023, 9, 515. [Google Scholar] [CrossRef]
- Zheng, L.; Wu, W.; Gao, Y.; Wu, Y.; Xu, Y.; Zhang, G.; Zhou, Y. Integrated transcriptome, small RNA and degradome analysis provide insights into the transcriptional regulatory networks underlying cold acclimation in jojoba. Sci. Hortic. 2022, 299, 111050. [Google Scholar] [CrossRef]
- Lai, R.; Guan, Q.; Shen, C.; Feng, X.; Zhang, Y.; Chen, Y.; Wu, R. Integrated sRNA-seq and RNA-seq analysis reveals the regulatory roles of miRNAs in the low-temperature responses of Canarium album. Horticulturae 2022, 8, 667. [Google Scholar] [CrossRef]
- Cao, D.; Li, J.; Ma, L.; Liu, Y.; Huang, J.; Jin, X. Genome-Wide identification of selenium-responsive microRNAs in tea Plant (Camellia sinensis LO Kuntze). Horticulturae 2023, 9, 1278. [Google Scholar] [CrossRef]
- Lu, J.; Mao, X.; Xu, Y.; Liu, S.; Wang, L. MicroRNA identification and integrated network analyses for age-dependent flavonoid biosynthesis in Ginkgo biloba. Forests 2023, 14, 1706. [Google Scholar] [CrossRef]
- Yang, G.; Rhodes, D.; Joly, R.J. Effects of high temperature on membrane stability and chlorophyll fluorescence in glycinebetaine-deficient and glycinebetaine-containing maize lines. Funct. Plant Biol. 1996, 23, 437–443. [Google Scholar] [CrossRef]
- Yasar, F.; Ellialtioglu, S.; Yildiz, K. Effect of salt stress on antioxidant defense systems, lipid peroxidation, and chlorophyll content in green bean. Russ. J. Plant Physiol. 2008, 55, 782–786. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Meng, H.; Wen, H.; Fan, Y.; Zhao, J. Maize ABP9 enhances tolerance to multiple stresses in transgenic Arabidopsis by modulating ABA signaling and cellular levels of reactive oxygen species. Plant Mol. Biol. 2022, 75, 365–378. [Google Scholar] [CrossRef]
- Chao, J.; Kong, Y.; Wang, Q.; Sun, Y.; Gong, D.; Lv, J.; Liu, G. MapGene2Chrom, a tool to draw gene physical map based on Perl and SVG languages. Yi Chuan Hered. 2015, 37, 91–97. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Integrated nr database in protein annotation system and its localization. Nat. Commun. 2010, 6, 1–8. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Yeh, L.S.L. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Yamanishi, Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Sherlock, G. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- An, J.; Lai, J.; Sajjanhar, A.; Lehman, M.L.; Nelson, C.C. miRPlant: An integrated tool for identification of plant miRNA from RNA sequencing data. BMC Bioinform. 2014, 15, 275. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Li, W.; Zhao, F.A.; Fang, W.; Xie, D.; Hou, J.; Yang, X.; Lv, S. Identification of early salt stress responsive proteins in seedling roots of upland cotton (Gossypium hirsutum L.) employing iTRAQ-based proteomic technique. Front. Plant Sci. 2015, 6, 732. [Google Scholar] [CrossRef]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Higo, H. PLACE: A database of plant cis-acting regulatory DNA elements. Nucleic Acids Res. 1998, 26, 358–359. [Google Scholar] [CrossRef]
- Clément, T.; Salone, V.; Rederstorff, M. Dual luciferase gene reporter assays to study miRNA function. Methods Mol Biol. 2015, 1296, 187–198. [Google Scholar]
- Yoo, S.D.; Cho, Y.H.; Sheen, J. Arabidopsis mesophyll protoplasts: A versatile cell system for transient gene expression analysis. Nat. Protoc. 2007, 2, 1565–1572. [Google Scholar] [CrossRef]
- Chavez, M.R.A.; Rosas-Cárdenas, D.F.F.; De Paoli, E.; Accerbi, M.; Rymarquis, L.A.; Mahalingam, G.; De Folter, S. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat. Commun. 2014, 5, 3722. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, J.; Li, F.; Gu, J.; He, T.; Zhang, X.; Li, Y. MicroRNA identification based on sequence and structure alignment. Bioinformatics 2005, 21, 3610–3614. [Google Scholar] [CrossRef] [PubMed]
- Premathilake, A.T.; Ni, J.; Shen, J.; Bai, S.; Teng, Y. Transcriptome analysis provides new insights into the transcriptional regulation of methyl jasmonate-induced flavonoid biosynthesis in pear calli. BMC Plant Biol. 2020, 20, 388. [Google Scholar] [CrossRef]
- Yant, L.; Mathieu, J.; Dinh, T.T.; Ott, F.; Lanz, C.; Wollmann, H.; Schmid, M. Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell 2010, 22, 2156–2170. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Cai, W.J.; Wang, S.; Shan, C.M.; Wang, L.J.; Chen, X.Y. Temporal control of trichome distribution by microRNA156-targeted SPL genes in Arabidopsis thaliana. Plant Cell 2010, 22, 2322–2335. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Salinas, M.; Höhmann, S.; Berndtgen, R.; Huijser, P. miR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell 2010, 22, 3935–3950. [Google Scholar] [CrossRef]
- Wang, J.W.; Park, M.Y.; Wang, L.J.; Koo, Y.; Chen, X.Y.; Weigel, D.; Poethig, R.S. miRNA control of vegetative phase change in trees. PLoS Genet. 2011, 7, e1002012. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ruan, Q.; Zhu, X.; Wang, B.; Wei, B.; Wei, X. Identification of Alfalfa SPL gene family and expression analysis under biotic and abiotic stresses. Sci. Rep. 2023, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.I.; Chiang, S.F.; Lin, W.Y.; Chen, J.W.; Tseng, C.Y.; Chiou, W.T.J. Regulatory network of microrna399 and pho2 by systemic signaling. Plant Physiol. 2008, 147, 732–746. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Yang, Y.; Li, J.; Wang, M.; Jiang, J.; Wu, J.; Wang, Y. Functional Analysis of Bna-miR399c-PHO2 Regulatory Module Involved in Phosphorus Stress in Brassica napus. Life 2023, 13, 310. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Ahn, H.J.; Chiou, T.J.; Ahn, J.H. The role of the miR399-PHO2 module in the regulation of flowering time in response to different ambient temperatures in Arabidopsis thaliana. Mol. Cells 2011, 32, 83–88. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.; Li, X.; Wang, C.; Ding, G.; Xu, F.; Shi, L. Jasmonic acid participating in the systemic regulation of phosphate starvation response in Brassica napus. Plant Soil 2023. [Google Scholar] [CrossRef]
- Guo, H.S.; Xie, Q.; Fei, J.F.; Chua, N.H. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell 2005, 17, 1376–1386. [Google Scholar] [CrossRef]
- Fang, Y.; Xie, K.; Xiong, L. Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. J. Exp. Bot. 2014, 65, 2119–2135. [Google Scholar] [CrossRef]
- Wang, W.Q.; Wang, J.; Wu, Y.Y.; Li, D.W.; Allan, A.C.; Yin, X.R. Genome-wide analysis of coding and non-coding RNA reveals a conserved miR164-NAC regulatory pathway for fruit ripening. N. Phytol. 2020, 225, 1618–1634. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Liu, X.F.; Fu, B.L.; Zhang, Q.Y.; Tong, Y.; Wang, J.; Yin, X.R. Methyl jasmonate enhances ethylene synthesis in kiwifruit by inducing NAC genes that activate ACS1. J. Agric. Food Chem. 2020, 68, 3267–3276. [Google Scholar] [CrossRef]
- Ellis, J.B.; Everhart, B.M. The North American species of Gloeosporium. J. Mycol. 1885, 1, 109–119. [Google Scholar] [CrossRef]
- Martin, G. The Phyllostictas of North America. J. Mycol. 1886, 2, 13–20. [Google Scholar] [CrossRef]
- Horst, R.K. Field Manual of Diseases on Garden and Greenhouse Flowers; Springer: Dordrecht, The Netherlands, 2013. [Google Scholar]
- González, V.M.; Müller, S.; Baulcombe, D. Evolution of NBS-LRR gene copies among dicot plants and its regulation by members of the miR482/2118 superfamily of miRNAs. Mol. Plant 2015, 8, 329–331. [Google Scholar] [CrossRef]
- Zhai, J.; Jeong, D.H.; De Paoli, E.; Park, S.; Rosen, B.D.; Li, Y.; Meyers, B.C. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Gene Dev. 2011, 25, 2540–2553. [Google Scholar] [CrossRef]
- Shivaprasad, P.V.; Chen, H.M.; Patel, K.; Bond, D.M.; Santos, B.A.; Baulcombe, D.C. A microRNA superfamily regulates nucleotide binding site–leucine-rich repeats and other mRNAs. Plant Cell 2012, 24, 859–874. [Google Scholar] [CrossRef]
- Zhu, Q.H.; Fan, L.; Liu, Y.; Xu, H.; Llewellyn, D.; Wilson, I. miR482 regulation of NBS-LRR defense genes during fungal pathogen infection in cotton. PLoS ONE 2013, 8, e84390. [Google Scholar] [CrossRef]
- You, J.; Chan, Z. ROS regulation during abiotic stress responses in crop plants. Front. Plant Sci. 2015, 6, 1092. [Google Scholar] [CrossRef]
- Dreyer, A.; Dietz, K.J. Reactive oxygen species and the redox-regulatory network in cold stress acclimation. Antioxidants 2018, 7, 169. [Google Scholar] [CrossRef]
- Lu, Y.; Feng, Z.; Bian, L.; Xie, H.; Liang, J. miR398 regulation in rice of the responses to abiotic and biotic stresses depends on CSD1 and CSD2 expression. Funct. Plant Biol. 2010, 38, 44–53. [Google Scholar] [CrossRef]
- Leng, X.; Wang, P.; Zhu, X.; Li, X.; Zheng, T.; Shangguan, L.; Fang, J. Ectopic expression of CSD1 and CSD2 targeting genes of miR398 in grapevine is associated with oxidative stress tolerance. Funct. Integr. Genomics 2017, 17, 697–710. [Google Scholar] [CrossRef]
- Yan, G.; Hua, Y.; Jin, H.; Huang, Q.; Zhou, G.; Xu, Y.; Zhu, Z. Sly-miR398 participates in cadmium stress acclimation by regulating antioxidant system and cadmium transport in tomato (Solanum lycopersicum). Int. J. Mol. Sci. 2023, 24, 1953. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, J.; Han, X.; Liu, Q.; Dorjee, T.; Zhou, Y.; Sun, H.; Gao, F. Joint Analysis of Small RNA and mRNA Sequencing Unveils miRNA-Mediated Regulatory Network in Response to Methyl Jasmonate in Apocynum venetum L. Horticulturae 2024, 10, 173. https://doi.org/10.3390/horticulturae10020173
Tan J, Han X, Liu Q, Dorjee T, Zhou Y, Sun H, Gao F. Joint Analysis of Small RNA and mRNA Sequencing Unveils miRNA-Mediated Regulatory Network in Response to Methyl Jasmonate in Apocynum venetum L. Horticulturae. 2024; 10(2):173. https://doi.org/10.3390/horticulturae10020173
Chicago/Turabian StyleTan, Jinhua, Xiaowei Han, Qi Liu, Tashi Dorjee, Yijun Zhou, Huigai Sun, and Fei Gao. 2024. "Joint Analysis of Small RNA and mRNA Sequencing Unveils miRNA-Mediated Regulatory Network in Response to Methyl Jasmonate in Apocynum venetum L." Horticulturae 10, no. 2: 173. https://doi.org/10.3390/horticulturae10020173
APA StyleTan, J., Han, X., Liu, Q., Dorjee, T., Zhou, Y., Sun, H., & Gao, F. (2024). Joint Analysis of Small RNA and mRNA Sequencing Unveils miRNA-Mediated Regulatory Network in Response to Methyl Jasmonate in Apocynum venetum L. Horticulturae, 10(2), 173. https://doi.org/10.3390/horticulturae10020173