
Metabolic Engineering of Escherichia coli Nissle 1917 for the Production of Heparosan Using Mixed Carbon Sources


Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Plasmids
2.2. Deletion of E. coli Chromosomal Genes
2.3. Plasmid Construction
2.4. Culture Conditions for Shake-Flask Experiments
2.5. Analytical Methods
3. Results and Discussion
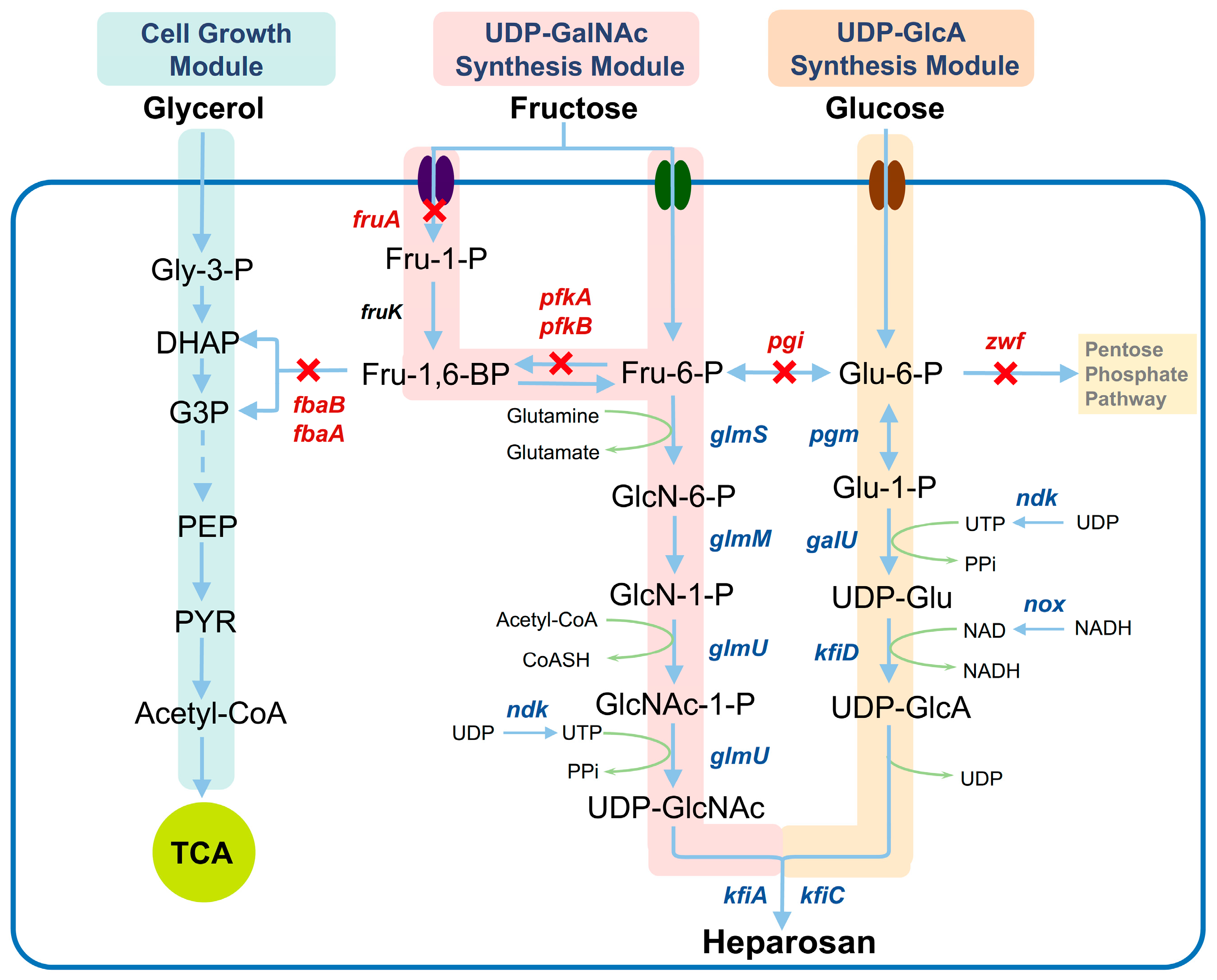
3.1. Design of Co-Utilization Pathways for Glucose, Fructose, and Glycerol
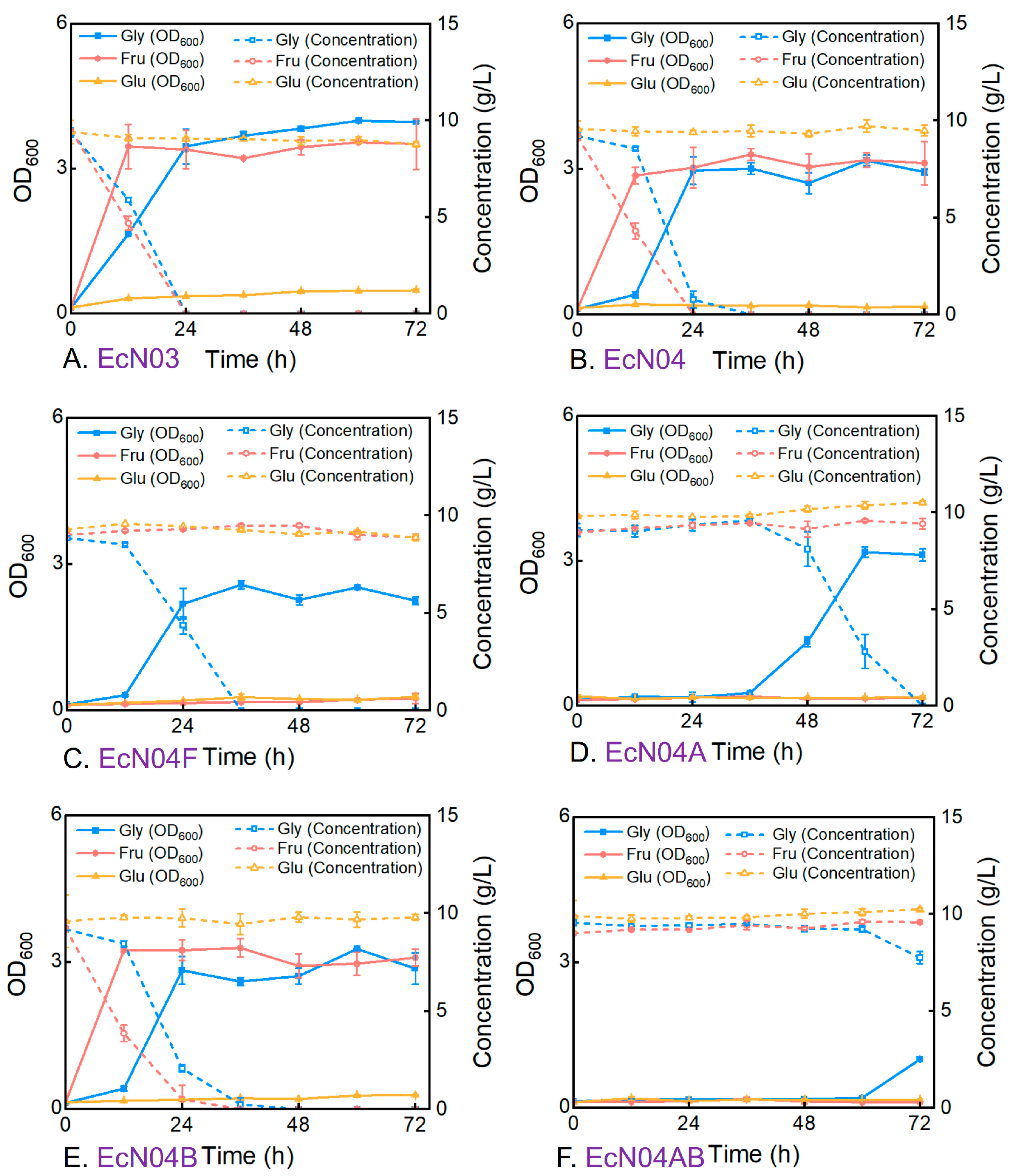
3.2. Metabolic Characteristics of Engineered Mutants During Cultivation on a Single Carbon Source
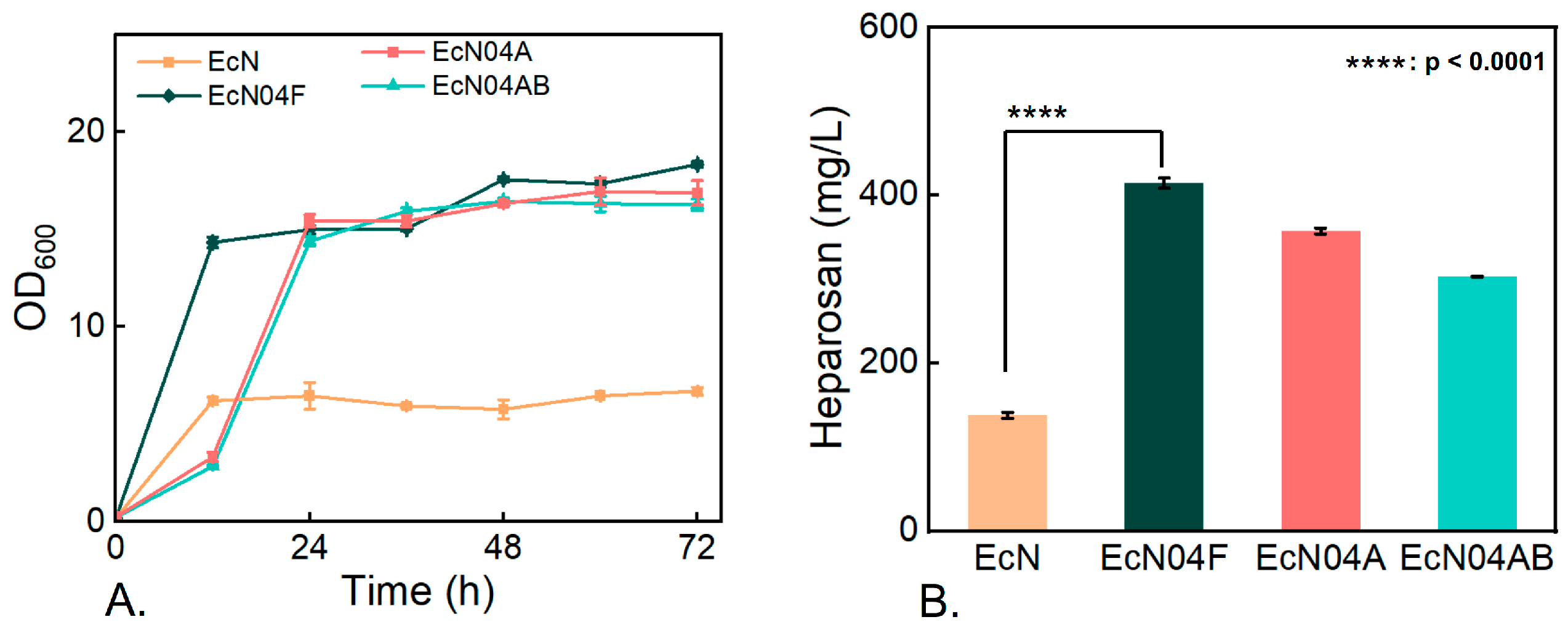
3.3. Metabolic Characteristics of Engineered Mutants During Cultivation on Mixed Carbon Sources
3.4. Effect of Carbon-Flux Allocation on Heparosan Production
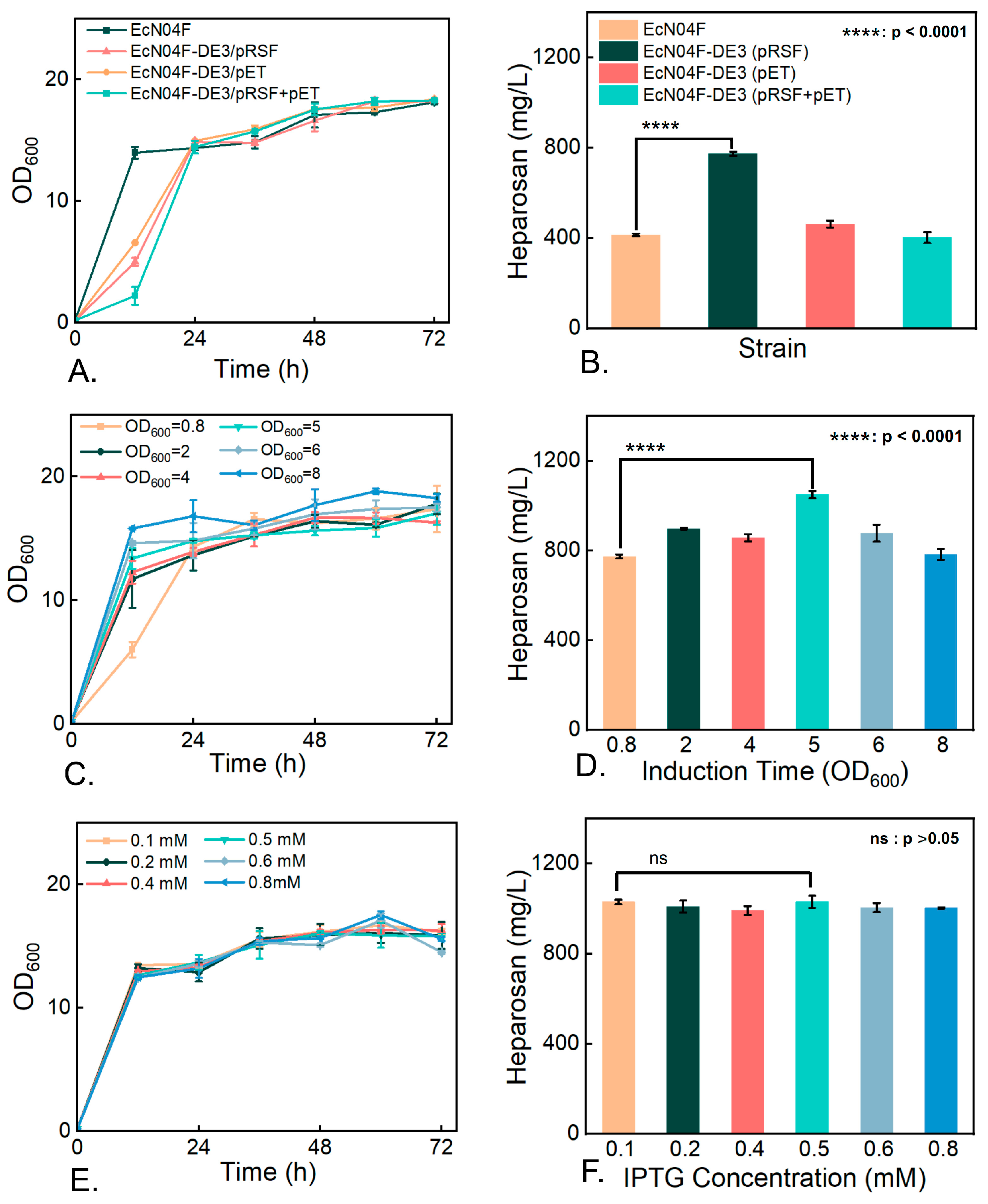
3.5. Impact of Key Enzyme Overexpression on Heparosan Biosynthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sultana, R.; Kamihira, M. Bioengineered heparin: Advances in production technology. Biotechnol. Adv. 2024, 77, 108456. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.L.; Adachi, R. Protease–proteoglycan complexes of mouse and human mast cells and importance of their β-tryptase–heparin complexes in inflammation and innate immunity. Immunol. Rev. 2007, 217, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Wander, R.; Kaminski, A.M.; Wang, Z.; Stancanelli, E.; Xu, Y.; Pagadala, V.; Li, J.; Krahn, J.M.; Pham, T.Q.; Liu, J.; et al. Structural and substrate specificity analysis of 3-O-sulfotransferase isoform 5 to synthesize heparan sulfate. ACS Catal. 2021, 11, 14956–14966. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ishii-Watabe, A.; Hashii, N.; Nakagawa, Y.; Takahashi, T.; Ebisawa, A.; Nishi, S.; Fujita, N.; Bando, A.; Sekimoto, Y.; et al. The establishment and validation of efficient assays for anti-IIa and anti-Xa activities of heparin sodium and heparin calcium. Biologicals 2013, 41, 415–423. [Google Scholar] [CrossRef]
- Hogwood, J.; Mulloy, B.; Lever, R.; Gray, E.; Page, C.P. Pharmacology of heparin and related drugs: An update. Pharmacol. Rev. 2023, 75, 328–379. [Google Scholar] [CrossRef]
- Wang, P.; Chi, L.; Zhang, Z.; Zhao, H.; Zhang, F.; Linhardt, R.J. Heparin: An old drug for new clinical applications. Carbohydr. Polym. 2022, 295, 119818. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Zhou, Z.; Wang, P.; Xi, X.; Hu, S.; Xu, R.; Du, G.; Li, J.; Chen, J.; et al. Synthesis of bioengineered heparin by recombinant yeast Pichia pastoris. Green Chem. 2022, 24, 3180–3192. [Google Scholar] [CrossRef]
- Vann, W.F.; Schmidt, M.A.; Jann, B.; Jann, K. The structure of the capsular polysaccharide (K5 antigen) of urinary-tract-infective Escherichia coli 010:K5:H4. A polymer similar to desulfo-heparin. Eur. J. Biochem. 1981, 116, 359–364. [Google Scholar] [CrossRef]
- Datta, P.; Fu, L.; Brodfuerer, P.; Dordick, J.S.; Linhardt, R.J. High density fermentation of probiotic E. coli Nissle 1917 towards heparosan production, characterization, and modification. Appl. Microbiol. Biotechnol. 2021, 105, 1051–1062. [Google Scholar] [CrossRef]
- DeAngelis, P.L.; White, C.L. Identification and molecular cloning of a heparosan synthase from Pasteurella multocida type D. J. Biol. Chem. 2002, 277, 7209–7213. [Google Scholar] [CrossRef]
- Sheng, L.L.; Cai, Y.M.; Li, Y.; Huang, S.L.; Sheng, J.Z. Advancements in heparosan production through metabolic engineering and improved fermentation. Carbohydr. Polym. 2024, 331, 121881. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.Q.; Li, Y.; Wang, Y.J.; Cao, Y.L.; Xin, S.Y.; Li, X.Y.; Xi, R.M.; Wang, F.S.; Sheng, J.Z. Biosynthetic production of anticoagulant heparin polysaccharides through metabolic and sulfotransferases engineering strategies. Nat. Commun. 2024, 15, 3755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, L.; Teng, L.; Chen, J.; Liu, J.; Li, J.; Du, G.; Chen, J. Metabolic engineering of Escherichia coli BL21 for biosynthesis of heparosan, a bioengineered heparin precursor. Metab. Eng. 2012, 14, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Barreteau, H.; Richard, E.; Drouillard, S.; Samain, E.; Priem, B. Production of intracellular heparosan and derived oligosaccharides by lyase expression in metabolically engineered E. coli K-12. Carbohydr. Res. 2012, 360, 19–24. [Google Scholar] [CrossRef]
- Williams, A.; Gedeon, K.S.; Vaidyanathan, D.; Yu, Y.; Collins, C.H.; Dordick, J.S.; Linhardt, R.J.; Koffas, M.A.G. Metabolic engineering of Bacillus megaterium for heparosan biosynthesis using Pasteurella multocida heparosan synthase, PmHS2. Microb. Cell Fact. 2019, 18, 132. [Google Scholar] [CrossRef]
- Jin, P.; Zhang, L.; Yuan, P.; Kang, Z.; Du, G.; Chen, J. Efficient biosynthesis of polysaccharides chondroitin and heparosan by metabolically engineered Bacillus subtilis. Carbohydr. Polym. 2016, 140, 424–432. [Google Scholar] [CrossRef]
- Hu, L.; Wang, Y.; Hu, Y.; Yin, J.; Wang, L.; Du, G.; Chen, J.; Kang, Z. Biosynthesis of non-sulfated high-molecular-weight glycosaminoglycans and specific-sized oligosaccharides. Carbohydr. Polym. 2022, 295, 119829. [Google Scholar] [CrossRef]
- Grozdanov, L.; Raasch, C.; Schulze, J.; Sonnenborn, U.; Gottschalk, G.; Hacker, J.; Dobrindt, U. Analysis of the genome structure of the nonpathogenic probiotic Escherichia coli strain Nissle 1917. J. Bacteriol. 2004, 186, 5432–5441. [Google Scholar] [CrossRef]
- Sonnenborn, U.; Schulze, J. The non-pathogenic Escherichia coli strain Nissle 1917—features of a versatile probiotic. Microb. Ecol. Health Dis. 2009, 21, 122–158. [Google Scholar]
- Zhang, Q.; Yao, R.; Chen, X.; Liu, L.; Xu, S.; Chen, J.; Wu, J. Enhancing fructosylated chondroitin production in Escherichia coli K4 by balancing the UDP-precursors. Metab. Eng. 2018, 47, 314–322. [Google Scholar] [CrossRef]
- Yan, H.; Bao, F.; Zhao, L.; Yu, Y.; Tang, J.; Zhou, X. Cyclic AMP (cAMP) receptor protein-cAMP complex regulates heparosan production in Escherichia coli strain Nissle 1917. Appl. Environ. Microbiol. 2015, 81, 7687–7696. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Zhao, L.; Hu, L.; Xi, X.; Zhang, Y.; Wang, Y.; Chen, J.; Chen, J.; Kang, Z. Engineering the probiotic bacterium Escherichia coli Nissle 1917 as an efficient cell factory for heparosan biosynthesis. Enzyme. Microb. Technol. 2022, 158, 110038. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, H.L. Routes for fructose utilization by Escherichia coli. J. Mol. Microbiol. Biotechnol. 2001, 3, 355–359. [Google Scholar]
- Prior, T.I.; Kornberg, H.L. Nucleotide sequence of fruA, the gene specifying enzyme IIfru of the phosphoenolpyruvate-dependent sugar phosphotransferase system in Escherichia coli K12. J. Gen. Microbiol. 1988, 134, 2757–2768. [Google Scholar] [CrossRef]
- Shimizu, K.; Matsuoka, Y. Redox rebalance against genetic perturbations and modulation of central carbon metabolism by the oxidative stress regulation. Biotechnol. Adv. 2019, 37, 107441. [Google Scholar] [CrossRef]
- Abernathy, M.H.; Zhang, Y.; Hollinshead, W.D.; Wang, G.; Baidoo, E.E.K.; Liu, T.; Tang, Y.J. Comparative studies of glycolytic pathways and channeling under in vitro and in vivo modes. AIChE J. 2019, 65, 483–490. [Google Scholar] [CrossRef]
- Martínez-Gómez, K.; Flores, N.; Castañeda, H.M.; Martínez-Batallar, G.; Hernández-Chávez, G.; Ramírez, O.T.; Gosset, G.; Encarnación, S.; Bolivar, F. New insights into Escherichia coli metabolism: Carbon scavenging, acetate metabolism and carbon recycling responses during growth on glycerol. Microb. Cell Fact. 2012, 11, 46. [Google Scholar] [CrossRef]
- Macomber, L.; Elsey, S.P.; Hausinger, R.P. Fructose-1,6-bisphosphate aldolase (class II) is the primary site of nickel toxicity in Escherichia coli. Mol. Microbiol. 2011, 82, 1291–1300. [Google Scholar] [CrossRef]
- Yu, H.; Rao, X.; Zhang, K. Nucleoside diphosphate kinase (Ndk): A pleiotropic effector manipulating bacterial virulence and adaptive responses. Microbiol. Res. 2017, 205, 125–134. [Google Scholar] [CrossRef]
- Gao, H.; Li, J.; Sivakumar, D.; Kim, T.-S.; Patel, S.K.S.; Kalia, V.C.; Kim, I.-W.; Zhang, Y.-W.; Lee, J.-K. NADH oxidase from Lactobacillus reuteri: A versatile enzyme for oxidized cofactor regeneration. Int. J. Biol. Macromol. 2019, 123, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Ding, W.; An, Z.; Zhang, H.; Wei, X.; Xu, J.; Liu, H.; Fang, H. Increased distribution of carbon metabolic flux during de novo cytidine biosynthesis via attenuation of the acetic acid metabolism pathway in Escherichia coli. Microb. Cell Fact. 2025, 24, 36. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Liu, Y.Q.; Xu, Y.S.; Li, Z.J.; Wang, Y.Z.; Zhang, Z.X.; Sun, X.M. Regulating the T7 RNA polymerase expression in E. coli BL21 (DE3) to provide more host options for recombinant protein production. Microb. Cell Fact. 2021, 20, 189. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gong, B.; Wang, H.; Yang, G.; Zhou, X. Chromosome evolution of Escherichia coli Nissle 1917 for high-level production of heparosan. Biotechnol. Bioeng. 2023, 120, 1081–1096. [Google Scholar] [CrossRef]
- Wang, Z.; Dordick, J.S.; Linhardt, R.J. Escherichia coli K5 heparosan fermentation and improvement by genetic engineering. Bioeng. Bugs 2011, 2, 63–67. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains/Plasmids | Description | References |
|---|---|---|
| Strains | ||
| E. coli DH5α | Wild-type strain for gene cloning | Takara Bio |
| E. coli Nissle 1917 | Wild-type strain for heparosan production | DSMZ * |
| Bacillus subtilis 168 | Wild-type strain for cloning galU gene | DSMZ |
| E. coli EcN03 | Nissle 1917 Δzwf ΔpfkB ΔpfkA | This study |
| E. coli EcN04 | Nissle 1917 Δzwf ΔpfkB ΔpfkA Δpgi | This study |
| E. coli EcN04F | Nissle 1917 Δzwf ΔpfkB ΔpfkA Δpgi ΔfruA | This study |
| E. coli EcN04A | Nissle 1917 Δzwf ΔpfkB ΔpfkA Δpgi ΔfbaA | This study |
| E. coli EcN04B | Nissle 1917 Δzwf ΔpfkB ΔpfkA Δpgi ΔfbaB | This study |
| E. coli EcN04AB | Nissle 1917 Δzwf ΔpfkB ΔpfkA Δpgi ΔfbaA ΔfbaB | This study |
| E. coli EcN04F-DE3 | Nissle 1917 Δzwf ΔpfkB ΔpfkA Δpgi ΔfruA ΔpoxB::DE3 | This study |
| Plasmids | ||
| pKD13 | Plasmid harboring KanR and FLP recognition target | Yale CGSC ** |
| pKD46 | λ-Red recombinase expression helper plasmid | Yale CGSC |
| pCP20 | FLP recombinase helper plasmid | Yale CGSC |
| pEC-XK99E | Expression vector, trc promoter, KanR | Sangon Biotech |
| pETDuet | Expression vector, T7 promoter, AmpR | Sangon Biotech |
| pRSFDuet | Expression vector, T7 promoter, KanR | Sangon Biotech |
| pEC-kfiD | pEC-XK99E derived, harboring kfiD from EcN | This study |
| pEC-glmM | pEC-XK99E derived, harboring glmM from EcN | This study |
| pEC-glmS | pEC-XK99E derived, harboring glmS from EcN | This study |
| pEC-pgm | pEC-XK99E derived, harboring pgm from EcN | This study |
| pEC-galU | pEC-XK99E derived, harboring galU from B. subtilis 168 | This study |
| pEC-kfiA | pEC-XK99E derived, harboring kfiA from EcN | This study |
| pEC-kfiC | pEC-XK99E derived, harboring kfiC from EcN | This study |
| pEC-ndk | pEC-XK99E derived, harboring ndk from EcN | This study |
| pEC-nox1 | pEC-XK99E derived, harboring nox1 from L. rhamnosus GG | This study |
| pEC-nox2 | pEC-XK99E derived, harboring nox2 from L. rhamnosus GG | This study |
| pEC-nox3 | pEC-XK99E derived, harboring nox3 from L. rhamnosus GG | This study |
| pEC-nox4 | pEC-XK99E derived, harboring nox4 from L. rhamnosus GG | This study |
| pET-kfiD-kfiA | pETDuet derived, harboring kfiA and kfiC | This study |
| pRSF-glmM-pgm-galU | pRSFDuet derived, harboring glmM, pgm, and galU | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, F.; Wu, R.; Li, Z.-J. Metabolic Engineering of Escherichia coli Nissle 1917 for the Production of Heparosan Using Mixed Carbon Sources. Fermentation 2025, 11, 289. https://doi.org/10.3390/fermentation11050289
Shao F, Wu R, Li Z-J. Metabolic Engineering of Escherichia coli Nissle 1917 for the Production of Heparosan Using Mixed Carbon Sources. Fermentation. 2025; 11(5):289. https://doi.org/10.3390/fermentation11050289
Chicago/Turabian StyleShao, Fangqi, Ruiji Wu, and Zheng-Jun Li. 2025. "Metabolic Engineering of Escherichia coli Nissle 1917 for the Production of Heparosan Using Mixed Carbon Sources" Fermentation 11, no. 5: 289. https://doi.org/10.3390/fermentation11050289
APA StyleShao, F., Wu, R., & Li, Z.-J. (2025). Metabolic Engineering of Escherichia coli Nissle 1917 for the Production of Heparosan Using Mixed Carbon Sources. Fermentation, 11(5), 289. https://doi.org/10.3390/fermentation11050289