Abstract
Fungal endophytes exhibit symbiotic relationships with their host plants and convert inorganic selenium to organoselenium and Se0. In order to elucidate how Epichloë sinensis from Festuca sinensis adapts to different concentrations of sodium selenate, the dynamic changes of mycelial enzyme activities and metabolic changes at the transcriptional level were documented over a period of 36 h. The activity of enzymes (superoxide dismutase, glutathione reductase, glutathione S-transferase, cysteine synthetase, and methionine synthesis) in mycelia increased in the presence of increased Se concentrations during the cultivation period. The strain with selenium enrichment showed differential changes in gene expression compared to the strain without selenium enrichment, with more changes observed at higher Se concentrations over time. Notably, genes related to ribosomes or ribosome biogenesis in eukaryotes showed significant expression differences among certain groups, with up-regulation of genes involved in oxidoreductase activity, superoxide dismutase, and siderophore biosynthetic processes, and down-regulation of genes involved in steroid biosynthesis. These findings contribute to a better understanding of the transcriptional response of Epichloë sinensis to selenium.
1. Introduction
Selenium (Se) is acknowledged as a vital micronutrient for various organisms ranging from bacteria to mammals, with a narrow safe intake range (40–400 μg/d) in humans [1]. It plays a critical role in selenoproteins like glutathione peroxidase and thioredoxin reductase, exhibiting antioxidant properties [2]. However, at elevated concentrations, it can generate free radicals, leading to cell apoptosis, primarily through DNA double-strand breaks without vigor [3]. Selenium exists in multiple oxidation states (−2, 0, +4, and +6), and certain microorganisms metabolize it through processes such as oxidation, reduction, methylation, demethylation, and selenoprotein synthesis [4,5]. The conversion of selenite to elemental selenium can be facilitated by enzymes like glutathione reductase (GR), nitrite reductase, and superoxide dismutase (SOD) in specific microorganisms [6,7,8,9]. Both bacteria and fungi are now known to be capable of producing methylated selenium species through methyltransferase, which utilizes S-adenosyl methionine as the methyl donor [10,11]. Notably, some microorganisms incorporate Se into proteins by replacing sulfur-containing compounds with organic Se forms, leading to the formation of selenomethionine, selenocysteine, and selenoproteins catalyzed by key enzymes like cysteine synthetase (CS), methionine synthetase (MTR), selenocysteine methyltransferase (SMT), and S-adenosylmethionine synthetase [12,13,14,15,16]. Generally, the microbial detoxification of Se may involve the reduction of oxidized forms to Se0, the production of volatile Se in significant amounts, or the conversion of selenium to organoselenium compounds [17,18].
Endophytes, consisting of bacteria, actinomycetes, and fungi, inhabit the intracellular spaces of plant tissues. They are generally considered benign or advantageous to host plants, and they can act as microbial agents in agriculture [19,20,21], food security [22], and environmental restoration [23]. Recently, endophytic microbes have shown proficiency in Se accumulation and have been utilized in the production of red elemental selenium (Se0) and selenoproteins in vitro [24,25,26], and also to impact plant Se metabolism [27]. For example, the endophyte Alternaria tenuissima predominantly accumulates organic C-Se-C and Se0 when provided with selenate or selenite, respectively, and it influences its’ host’s growth, Se speciation, and tissue distribution [28].
Festuca sinensis is a perennial bunchgrass widely distributed in the cool and semi-arid regions of Qinghai Province. It exhibits adaptation to stress, thereby playing a crucial role in the restoration of degraded meadows [29]. An increasing body of research indicates that this species frequently forms symbiotic relationships with Epichloë endophytes, which enhance its ability to resist or tolerate various stressors such as pathogens [30], cold [31], drought [32], waterlogging [33], heavy metals [34], and reverse degradation [35]. Studies have identified Epichloës sinensis as the fungal endophyte species infecting F. sinensis, with these strains demonstrating antioxidant capacity, element accumulation, hormone and alkaloid production, and tolerance to metal(loid)s in vitro [36,37]. Recent investigations involving liquid and solid cultures of Epichloës sinensis exposed to varying concentrations of sodium selenite have shown changes in the abundance of certain metabolites [38,39]. Transcriptome reveals all of the mRNAs, gene constituents, and functions in a given organism. RNA-Seq has recently become a popular tool for quantitative transcriptome profiling because it provides a more precise measurement of transcriptional levels than other methods [12]. However, only a limited amount of research has delved into the mechanisms underlying selenium tolerance in Epichloës sinensis using high-throughput RNA-Seq technology. This study treated Epichloës sinensis with different concentrations of Na2SeO3 to explore key enzyme activities involved in Na2SeO3 metabolism and to conduct mycelial transcriptional profiling.
2. Materials and Methods
2.1. Preparation of Mycelia
The mycelia were cultivated in a petri dish with a diameter of 9 cm, containing PD agar medium composed of potato infusion (200 g), dextrose (20 g), agar (20 g), and distilled water (1000 mL), and maintained at 25 °C for a duration of 30 days. Subsequently, three mycelial agar plugs (4 mm in diameter) were transferred to a flask containing 100 mL of liquid PD medium (potato infusion 200 g, dextrose 20 g, and distilled water 1000 mL) for further cultivation over a period of 15 days at 25 °C. The liquid seed was then inoculated at a 10% (v/v) ratio into 100 mL of fermentation medium containing 3% (m/v) sucrose, 0.25% (m/v) yeast extract, and 0.1% (m/v) peptone, with final concentrations of Na2SeO3 at 0, 0.1, or 0.2 mM, respectively [38]. The mycelia were cultured at 25 °C for 2 h, 12 h, and 36 h of fermentation, respectively. Following cultivation, the mycelia were harvested by centrifugation at 4000× g for 10 min at 4 °C, washed twice with 10 mL of distilled water, immediately frozen in liquid nitrogen, and stored at −80 °C prior to RNA isolation and biochemical analyses.
2.2. Analysis of Enzymatic Activities
In the pathways of selenium metabolism, six key enzymes (superoxide dismutase, glutathione reductase, glutathione S-transferase, cysteine synthetase, methionine synthetase, and selenocysteine methyltransferase) were selected for activity analysis using colorimetric detection or ELISA kits. These assay kits for microbiology were supplied by Shanghai Jining Industry Co., Ltd. (Shanghai, China, www.shjning.com, accessed on 5 December 2023). The specific kits used were: the superoxide dismutase (SOD) kit, with product number S8511N793379L and item number JN24918; the glutathione S-transferase (GST) kit, with product number S9622N248822L and item number JN24287; the glutathione reductase (GR) kit, with product number S0733N804478C and item number JN24288; the methionine synthetase (MTR) ELISA kit, with item number JN724602; the cysteine synthetase (CS) ELISA kit, with item number JN724606; and the selenocysteine methyltransferase (SMT) ELISA kit, with item number JN724608.
2.3. RNA Extraction
Using liquid nitrogen in a mortar, 200 mg of mycelia from each sample were pulverized into powder. Total RNA extraction was performed utilizing the TRIzol® RNA kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. The quantity of RNA was determined through agarose gel electrophoresis, and the RNA concentration was measured using a spectrophotometer (NanoPhotometer; Implen, Munich, Germany). Equivalent amounts of RNA obtained from individual replicates of each isolate were combined and employed for RNA-Seq analysis.
2.4. RNA Sequencing and Data Analysis
A total of 1.0 µg of RNA per sample was utilized for the construction of cDNA libraries. The construction of the libraries, quality assessment, and subsequent sequencing procedures were conducted by Azenta, Inc. (South Plainfield, NJ, USA, Suzhou, China) employing an Illumina HiSeq/Illumina Novaseq/MGI2000 platform following established protocols. The obtained clean reads were aligned and mapped to the reference genome of Epichloë sinensis (provided by Prof. Pei Tian, Lanzhou University, Lanzhou, China) using Hisat2 software (version 2.2.1). For the analysis of gene expression, HTSEQ software (version 0.6.1) was employed to determine the read count for each gene, and the fragments per kilobase per million (FPKM) for each gene was calculated based on the gene length and the mapped read counts [40]. Principal component analysis of the samples was carried out using R software (version 3.3.1). The molecular functions of the identified genes were annotated through Gene Ontology (GO) analysis using GOSeq software (version 1.34.1) [41]. Custom scripts were developed in-house to enrich the significant differentially expressed genes (DEGs) in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (http://en.wikipedia.org/wiki/KEGG, accessed on 1 December) [42]. The gene expression comparisons in the mycelia were performed under different conditions. Genes exhibiting an adjusted p-value below 0.05 and a fold-change exceeding 2 were considered differentially expressed in the mycelia of the two samples, and the DEGs were identified utilizing the DESeq2 bioconductor package (version 1.26.0). One-way analysis of variance was performed on enzymatic activities, with p < 0.05 considered to be significant.
3. Results
3.1. Changes in Enzyme Activities from Mycelia of Epichloë sinensis in Response to Selenium Conditions
Table 1 presents the activities of SOD, GR, GST, CS, MTR, and SMT in mycelia with and without Se. The activities of SOD, GR, GST, CS, and MTR increased with higher Se concentrations during the cultivation period. The GST, CS, MTR, and SMT activities in mycelia treated with 0.1 and 0.2 mM Se were significantly higher (p < 0.05) than those in the control groups at the same culture time. An Se concentration of 0.2 mM significantly increased SOD and GR activities compared to the control mycelia, while there was no significant difference between the control and 0.1 mM Se groups at 12 h or 36 h.

Table 1.
The effect of Se concentration on the enzyme activities of mycelia.
3.2. Transcriptome Sequencing of Epichloë sinensis
3.2.1. Quality Assessment and Reference Train Election
The mycelia’s total reads were 6.65 × 107 under standard conditions at 2 h (MZH10), 5.16 × 107 under 0.1 mM Se at 2 h (MZH11), 5.56 × 107 under 0.2 mM Se at 2 h (MZH12), 5.52 × 107 under standard conditions at 12 h (MZH20), 7.00 × 107 under 0.1 mM Se at 12 h (MZH21), 6.97 × 107 under 0.2 mM Se at 12 h (MZH22), 4.38 × 107 under standard conditions at 36 h (MZH30), 5.64 × 107 under 0.1 mM Se at 36 h (MZH31), 4.67 × 107 under 0.2 mM Se at 36 h (MZH32) of the fermentation process (Table S1). RNA sequencing generated clean reads ranging from 4.34 × 107 to 6.97 × 107 per sample and bases ranging from 6.39 × 109 to 1.03 × 1010 bp. The G + C contents of these samples ranged from 52.19% to 53.37%. Overall, the RNA-Seq data exhibited high quality for subsequent transcriptome analysis, with a clean read rate exceeding 98.85%.
Approximately 50 million reads per sample were acquired and aligned to the Epichloë sinensis reference genome, achieving total mapping rates ranging from 79.49% to 90.03% (Table S2). Uniquely mapped reads accounted for 47.82% to 65.52%, while multiple mapped reads comprised 20.37% to 42.83%. For the analysis, only the uniquely mapped reads were taken into consideration. The distribution of reads across regions and chromosomes of the reference genome is presented in Supplementary Figure S1 and Supplementary Figure S2, respectively. The majority of reads were found to be distributed among four chromosomes, namely tig00000001, tig00000002, tig00000016, tig00000030, as well as exon regions (33.90–46.03%), and intergenic regions (45.05–52.30%).
3.2.2. Analysis of Gene Expression and Differentially Expressed Genes (DEGs)
The FPKM method was utilized to determine the expression level of each unigene. A total of 12,774–12,825 genes was identified in the mycelial samples (Table S3). The highest number of genes was observed in the MZH12, MZH22, and MZH31 samples. Specifically, at 2 h and 12 h, 8483 genes (66.45%) and 8409 genes (65.87%) in the control samples (2 h, 12 h) exhibited FPKM values ≥ 15. However, selenium (Se) led to a decrease in the expression levels of some genes.
The variances in gene expression between selenium-treated samples from the mycelia of Epichloë sinensis at various time points were elucidated using PCA (Figure 1). The first and second principal components explained 44% and 16% of the overall sample variance, respectively, suggesting a distinct separation among the different groups.
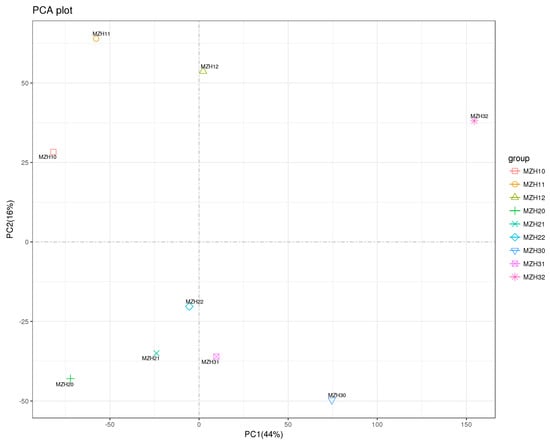
Figure 1.
Principle component analysis (PCA) score plot of first and second PCs from nine mycelial samples. MZH10, MZH11, and MZH12 respectively represent the mycelia treated with 0, 0.1, 0.2 mM Na2SeO3 for 2 h. MZH20, MZH21, and MZH22 respectively represent the mycelia treated with 0, 0.1, 0.2 mM Na2SeO3 for 12 h. MZH30, MZH31, and MZH32 respectively represent the mycelia treated with 0, 0.1, 0.2 mM Na2SeO3 for 36 h.
The differential gene expression patterns of endophytes among different groups are presented in Figure 2. The comparison between MZH10 and MZH11 groups revealed the lowest number of DEGs, with four up-regulated and four down-regulated genes. Conversely, the MZH22 versus MZH32 group exhibited the highest number of DEGs, with 508 up-regulated and 547 down-regulated genes. In the comparison between the 0.2 mM selenium-treated and control groups at 2 h, 319 DEGs were identified, with 287 up-regulated and 32 down-regulated genes. At 12 h, 19 significantly different genes were found between the 0.1 mM selenium-treated and control groups, comprising 12 up-regulated and seven down-regulated genes. Additionally, 377 differential genes were observed between the 0.2 mM selenium-treated and control groups, including 354 up-regulated and 23 down-regulated genes. Furthermore, 533 and 564 DEGs were up-regulated under the 0.1 and 0.2 mM Se conditions, respectively, while 208 and 221 DEGs were down-regulated compared to the control at 36 h in the presence of 0.1 and 0.2 mM Se, respectively. These results indicate that 0.2 mM Na2SeO3 had a more pronounced effect on the gene transcription level of endophytes, particularly at 36 h.
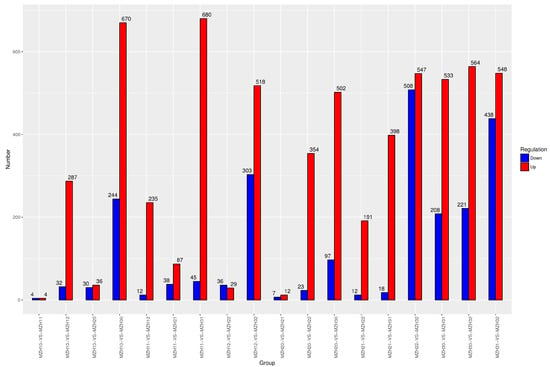
Figure 2.
Number of DEGs in comparison groups. X-axis represents the groups. Y-axis displays the number of DEGs. MZH10-VS-MZH11 represents the comparison of DEGs in the mycelia supplied with and without 0.1 mM Na2SeO3 at 2 h; MZH20-VS-MZH21 represents the comparison of DEGs in the mycelia supplied with and without 0.1 mM Na2SeO3 at 12 h; MZH30-VS-MZH31 represents the comparison of DEGs in the mycelia supplied with and without 0.1 mM Na2SeO3 at 36 h; MZH10-VS-MZH20 represented the comparison of DEGs in the mycelia in the absence of Se at 2 h and 12 h; MZH11-VS-MZH21 represents the comparison of DEGs in the mycelia supplied with 0.1 mM Na2SeO3 at 2 h and 12 h; MZH12-VS-MZH22 represents the comparison of DEGs in the mycelia supplied with 0.2 mM Na2SeO3 at 2 h and 12 h. And so on.
The time-course analysis revealed the dynamics of up-regulated and down-regulated genes, as presented in Figure 2. In comparison to the 2-h fermentation period, mycelia exhibited differential expression of 66 transcripts at 12 h under standard conditions, which escalated to 914 transcripts at 36 h. Similarly, under the 0.1 mM Se treatment, the number of differentially expressed transcripts was 125 at 12 h and increased to 725 at 36 h. Furthermore, a total of 65 DEGs were identified between the MZH12 and MZH22 groups, with 29 up-regulated and 36 down-regulated genes. Notably, a total of 821 DEGs were observed between the MZH12 and MZH32 groups, comprising 518 up-regulated and 303 down-regulated DEGs. These findings suggest a rapid and time-dependent response of mycelia to varying Se concentrations.
3.3. GO Annotation of Differentially Expressed Genes (DEGs)
Based on the results of the GO functional enrichment analysis, Table 2 presents the enriched GO terms associated with DEGs. At 2 h, 9 DEGs in the MZH10-VS-MZH11 comparison were significantly enriched in 9 GO terms, all of which were up-regulated. In contrast, a total of 58 DEGs in the MZH10-VS-MZH12 comparison were significantly enriched in 33 GO terms, comprising 28 up-regulated and 30 down-regulated DEGs. Moving to the 12-h time point, 40 terms were identified in the MZH20-VS-MZH21 comparison, with 34 up-regulated and 18 down-regulated DEGs. Similarly, 24 terms were observed in the MZH20-VS-MZH22 comparison, consisting of one down-regulated and 38 up-regulated DEGs. At 36 h, 38 up-regulated and 20 down-regulated DEGs were significantly enriched in 30 terms in the MZH30-VS-MZH31 group. In comparison, the MZH30-VS-MZH32 group exhibited 34 up-regulated DEGs and 18 down-regulated DEGs significantly enriched in 22 terms.
Table 2.
Number of DEGs based on GO and KEGG analysis.
An impact of the duration of culture on the quantity of DEGs was observed, as shown in Table 2. A comparison with the 2-h control mycelia revealed that in the MZH20 and MZH30 groups, 31 terms (23 up-regulated and 12 down-regulated DEGs) and 59 terms (100 up-regulated and 52 down-regulated DEGs) were identified, respectively. Upon the application of 0.1 mM Na2SeO3, 43 DEGs (22 up-regulated and 21 down-regulated) in the MZH11-VS-MZH21 group were significantly associated with 38 GO terms. Similarly, in the MZH11-VS-MZH31 group, 47 DEGs (45 up-regulated and 2 down-regulated) were significantly linked to 24 terms. Conversely, in the MZH12-VS-MZH22 group, 2 up-regulated and 3 down-regulated DEGs were notably associated with five terms, while in the MZH12-VS-MZH32 group, 29 up-regulated and 31 down-regulated DEGs were significantly linked to 27 terms.
A total of 561 DEGs was annotated in the GO database. Among the various GO results, ribosome, oxidoreductase activity, superoxide dismutase activity, ATPase activity, oxidative phosphorylation, monooxygenase activity, iron ion binding, sterol biosynthetic process, and signal transduction were highly represented (Figure 3). However, only a few genes associated with scytalone dehydratase activity, ferricrocin biosynthetic processes, protein-lysine N-methyltransferase activity, and toxin activity were identified among the Se groups. Furthermore, chaperone-mediated protein folding requiring cofactor, unfolded protein binding, chaperone binding, ATPase activity, RNA helicase activity, and oxidoreductase activity were prominently represented among the time groups. Nevertheless, only a limited number of genes related to L-methionine-(R)-S-oxide reductase activity, methionine-R-sulfoxide reductase activity, isopenicillin-N epimerase activity, cutinase activity, signaling adaptor activity, scytalone dehydratase activity, superoxide dismutase activity, and the MAPK cascade involved in osmosensory signaling pathway were identified among the time groups.
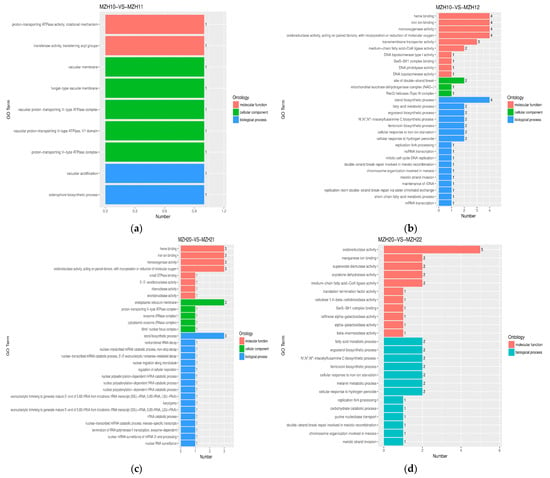
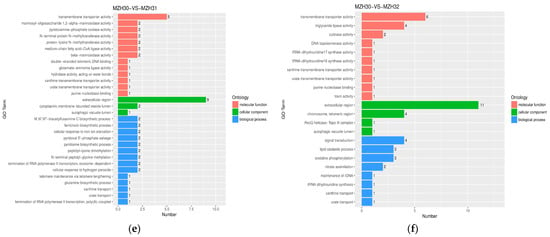
Figure 3.
GO functional classification of DEGs among the different Se-treated groups. X-axis represents the number of DEGs involved in the distinct GO terms. Y-axis displays the descriptions of GO terms. (a) MZH10-VS-MZH11 group; (b) MZH10-VS-MZH12 group; (c) MZH20-VS-MZH21 group; (d) MZH20-VS-MZH22 group; (e) MZH30-VS-MZH31 group; (f) MZH30-VS-MZH32 group.
The GO terms associated with the DEGs exhibited distinct variations in mycelia in response to different selenite concentrations. The enrichment analysis of GO terms revealed that the DEGs were implicated in various biological processes and molecular functions. For instance, in the biological process category, the DEGs were found to be involved in the siderophore biosynthetic process and vacuolar acidification in the MZH10-VS-MZH11 comparison, the sterol biosynthetic process in the MZH10-VS-MZH12 and MZH20-VS-MZH21 groups, and signal transduction in the MZH30-VS-MZH32 comparison. In terms of molecular function, the DEGs were associated with functions such as iron ion binding, monooxygenase activity, heme binding, and oxidoreductase activity in various comparisons. Notably, oxidoreductase activity exhibited significant variations in the MZH20-VS-MZH22 comparison. Additionally, transmembrane transporter activity was observed in the MZH30-VS-MZH31 and MZH30-VS-MZH32 comparisons. Regarding the cellular component categories, significant terms included the endoplasmic reticulum membrane in the MZH20-VS-MZH21 comparison and the extracellular region in the MZH30-VS-MZH31 and MZH30-VS-MZH32 comparisons. These findings highlight the diverse roles of DEGs in response to selenite concentrations in mycelia.
Distinct patterns of GO terms associated with the DEGs varied across different time intervals when mycelia were exposed to either common or Se conditions (Figure 4). In terms of biological processes, certain DEGs were linked to chaperone-mediated protein folding requiring cofactor (8 genes, 2.07–2.73 fold up-regulated) and protein refolding (5 genes, 2.07–2.80 fold up-regulated) in the MZH10-VS-MZH30 comparison; vesicle-mediated transport, protein import into mitochondrial matrix (4 genes, 2.01–4.03 fold up-regulated; 1 gene, 0.43 fold down-regulated), and oxidative phosphorylation (4 genes, 2.31–3.67 fold up-regulated) in the MZH12-VS-MZH32 comparison. The majority of DEGs associated with molecular functions were more prevalent, including ATPase activity (9 genes, 2.8–3.20 fold up-regulated; 9 genes, 0.36–0.48 fold down-regulated), unfolded protein binding (10 genes, 2.07–3.20 fold up-regulated), chaperone binding (6 genes, 2.08–2.80 fold up-regulated), and RNA helicase activity (6 genes, 0.44–0.48 fold down-regulated) in the MZH10-VS-MZH30 comparison; oxidoreductase activity (6 genes, 2.01–7.51 fold up-regulated; 1 gene, 0.49 fold down-regulated) in the MZH11-VS-MZH31 comparison; and sequence-specific DNA binding RNA polymerase II transcription factor activity (5 genes, 0.41–0.50 fold down-regulated) in the MZH12-VS-MZH32 comparison. Within the cellular component categories, a higher number of DEGs was implicated in the extracellular region (2 genes, 2.02–2.44 fold up-regulated; 2 genes, 0.30–0.49 fold down-regulated) in the MZH11-VS-MZH21 comparison.
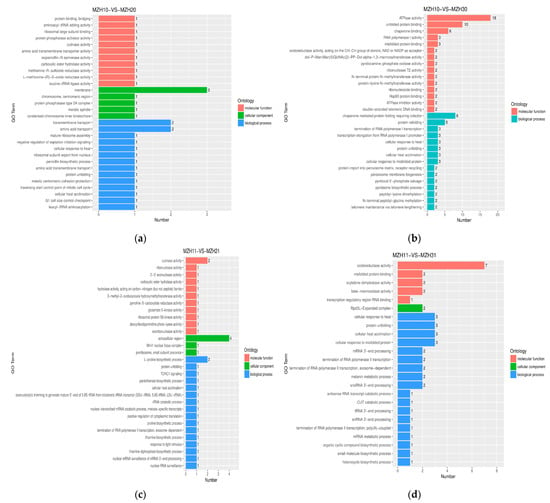
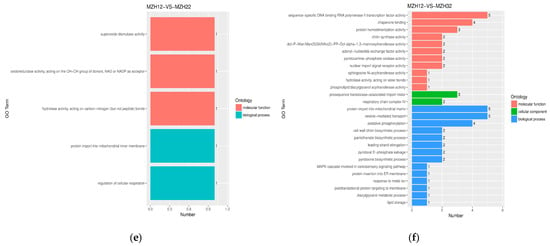
Figure 4.
GO functional classification of DEGs among the different culture time groups. X-axis represents the number of DEGs involved in the distinct GO terms. Y-axis displays the descriptions of GO terms. (a) MZH10-VS-MZH20 group; (b) MZH10-VS-MZH30 group; (c) MZH11-VS-MZH21 group; (d) MZH11-VS-MZH31 group; (e) MZH12-VS-MZH22 group; (f) MZH12-VS-MZH32 group.
3.4. KEGG Annotation of Differentially Expressed Genes (DEGs)
A KEGG pathway enrichment analysis of DEGs was conducted to elucidate the transcriptional alterations in metabolic pathways associated with the endophyte’s response to varying selenium levels at different time points during cultivation. A total of 505 DEGs were aligned with the reference pathways in the KEGG database to delineate the biological pathways distinguishing the different experimental groups. Among these, two pathways exhibited differences between MZH10 and MZH11, with four down-regulated DEGs identified (Table 2, Figure 5). MZH10 and MZH12 displayed six pathways, consisting of six up-regulated and eight down-regulated DEGs. Four pathways were shared between MZH20 and MZH21, encompassing nine up-regulated and two down-regulated DEGs. Additionally, MZH20 and MZH22 shared 11 pathways, involving 13 up-regulated DEGs. The comparison between MZH30 and MZH31 revealed five pathways, with five up-regulated and four down-regulated DEGs. Similarly, MZH30 and MZH32 shared four pathways, which included two up-regulated and seven down-regulated DEGs.
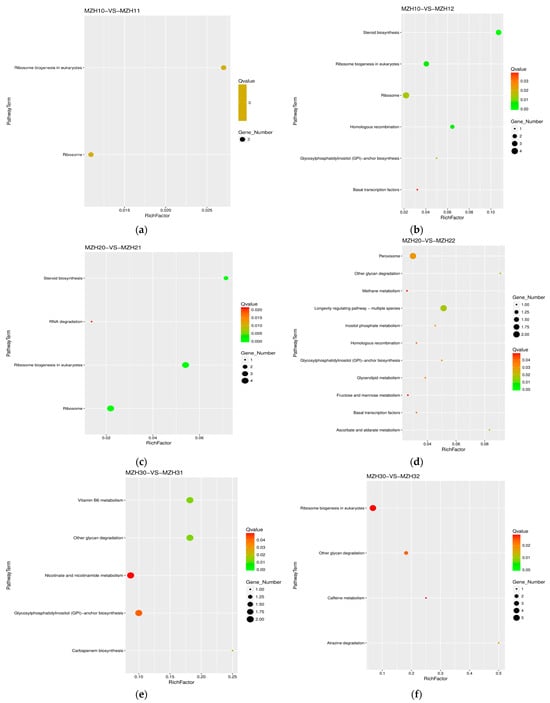
Figure 5.
Scatter plot of KEGG pathway annotation for DEGs among the different Se-treated groups. X-axis represents the number of DEGs involved in the distinct pathway terms. Y-axis displays the descriptions of pathway terms. (a) MZH10-VS-MZH11 group; (b) MZH10-VS-MZH12 group; (c) MZH20-VS-MZH21 group; (d) MZH20-VS-MZH22 group; (e) MZH30-VS-MZH31 group; (f) MZH30-VS-MZH32 group.
The comparison between MZH10 and MZH20 revealed 11 pathways (Table 2, Figure 6), with six up-regulated and nine down-regulated DEGs. In contrast, the MZH10 versus MZH30 comparison showed two pathways, with four up-regulated and 11 down-regulated DEGs. The comparison between MZH11 and MZH21 demonstrated 17 pathways, with 21 up-regulated and 13 down-regulated DEGs. Similarly, MZH11 versus MZH31 exhibited 5 pathways, with eight up-regulated DEGs. The comparison between MZH12 and MZH22 displayed 10 pathways, with four up-regulated and eight down-regulated DEGs. However, the comparison between MZH12 and MZH32 did not show the number of DEGs.
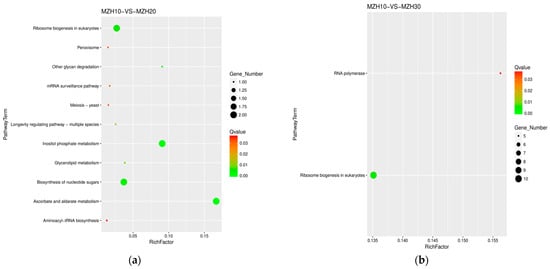
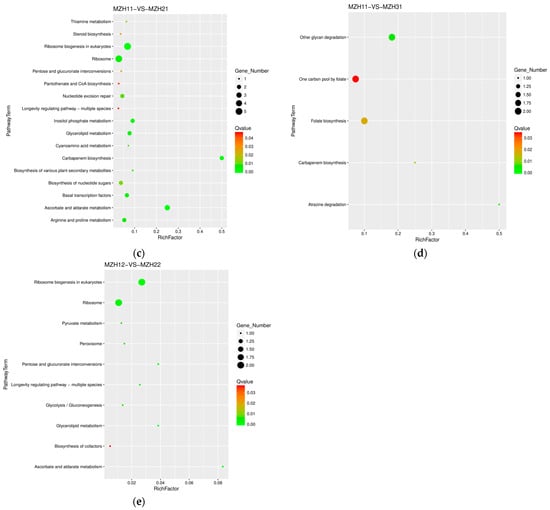
Figure 6.
Scatter plot of KEGG pathway annotation for DEGs among the different culture time groups. X-axis represents the number of DEGs involved in the distinct pathway terms. Y-axis displays the descriptions of pathway terms. (a) MZH10-VS-MZH20 group; (b) MZH10-VS-MZH30 group; (c) MZH11-VS-MZH21 group; (d) MZH11-VS-MZH31 group; (e) MZH12-VS-MZH22 group.
The majority of DEGs exhibited significant enrichment in various biological processes, such as ribosome and steroid biosynthesis, ribosome biogenesis in eukaryotes in the MZH10-VS-MZH12 comparison, ribosome and ribosome biogenesis in eukaryotes in the MZH20-VS-MZH21 and MZH11-VS-MZH21 comparisons, ribosome biogenesis in eukaryotes in the MZH30-VS-MZH32 comparison, and ribosome biogenesis in eukaryotes and RNA polymerase in the MZH10-VS-MZH30 comparison. Overall, a considerable number of DEGs involved in the response of mycelia to selenium and culture duration lacked functional annotations in the GO and KEGG databases.
3.5. Differentially Expressed Genes Involved in Ribosome and Peroxisome
At 2 h, tig00002100G000020 and tig00002101G005230, annotated as LSUrRNA and involved in ribosome (ko03010) or ribosome biogenesis in eukaryotes (ko03008), were down-regulated by 0.44–0.46-fold in the endophyte under the 0.1 mM selenium supply (Table 3). On the other hand, tig00002100G000010 and tig00000015G000020 were up-regulated by 2.54–4.87-fold, while tig00002101G005240 and tig00002100G000020 were down-regulated by 0.28–0.30 in response to 0.2 mM Se, which is potentially involved in large subunit ribosomal RNA. By the 12-h mark, the ribosome and ribosome biogenesis pathways in eukaryotes showed higher expression in the MZH20-VS-MZH21 groups, including tig00002101G005240, tig00002100G000020, tig00002101G005250, and tig00002100G000010. Simultaneously, tig00000001G030720 and tig00000107G004080, involved in peroxisome (ko04146), were up-regulated by 2.64–3.04-fold in the MZH20-VS-MZH22 group, encoding superoxide dismutase, which is in the Fe-Mn family. By 36 h, five genes, such as tig00002101G005240, tig00002100G000020, tig00002100G000010, tig00002101G005230, and tig00000015G006270 (RPP1, RPP30), were down-regulated by 0.31–0.49-fold in response to 0.2 mM Se.
Table 3.
The selected DEGs involved in KEGG pathways among different groups.
Compared to the 2-h control mycelia, the expression levels of tig00002101G005240 and tig00000016G015310 (EIF6) were down-regulated at 12 h. At 36 h, tig00002101G004790 (IMP3) and tig00000105G002750 (CSNK2B) were up-regulated. Meanwhile, eight genes, including tig00002100G000020, tig00002101G005250, tig00002101G005240, tig00000030G011540 (UTP12), tig00000016G003870 (NOG1), tig00000002G011500 (BMS1), and tig00000002G011250 (NOG1), were down-regulated. Furthermore, under 0.1 mM Se, five genes (tig00002100G000020, tig00002101G005250, tig00002101G005230, tig00002101G005240, and tig00002100G000010) showed a significant increase in ribosome or ribosome biogenesis in eukaryotes. Notably, tig00002101G005250 was up-regulated and tig00002100G000010 was down-regulated under 0.2 mM Se at 12 h compared to the 2-h samples. In the peroxisome pathway (ko04146), tig00000094G002550, encoding alpha-methylacyl-CoA racemase, exhibited higher expression at 12 h than at 2 h in the absence of Se. Conversely, when 0.2 mM Na2SeO3 was administered, tig00000001G030720 at 2 h showed significantly higher expression levels than at 12 h.
3.6. Differentially Expressed Genes Involved in Carbohydrate Metabolism
At 12 h, tig00000122G002730 (MIOX, inositol oxygenase) was 2.21-fold up-expressed in ascorbate and aldarate metabolism (ko00053) or inositol phosphate metabolism upon 0.2 mM Se (Table 3). Moreover, tig00000099G005510 was 2.42-fold up-expressed in fructose and mannose metabolism (ko00051), which is annotated as DAK/TKFC and regulates triose/dihydroxyacetone kinase/FAD-AMP lyase (cyclizing). These DEGs were enhanced in response to selenium.
The genes tig00000105G003930 (MIOX, inositol oxygenase) and tig00000122G002730 were 0.27–0.44 and 0.44–0.50 fold down-expressed in ascorbate and aldarate metabolism (ko00053) or inositol phosphate metabolism in the MZH10-VS-MZH20 and MZH11-VS-MZH21 groups.
3.7. Differentially Expressed Genes Involved in Glycan Biosynthesis and Metabolism
PIGG or GPI7 (tig00000002G007450), which encodes ethanolamine phosphate transferase 2 subunit G, was 4.50, 7.78, and 322.50-fold up-expressed in glycosylphosphatidylinositol (GPI)-anchor biosynthesis (ko00563) in the MZH10-VS-MZH12, MZH20-VS-MZH22, and MZH30-VS-MZH31 groups, respectively. However, at 36 h, tig00000122G003550 (PIGH, GPI15), which is associated with phosphatidylinositol N-acetylglucosaminyltransferase subunit H, was decreased by 0.47-fold in response to 0.1 mM Se.
At 12 h, tig00000016G001160 (MANBA), which is involved in beta-mannosidase, was 2.73-fold up-expressed in other glycan degradation (ko00511) in 0.2 mM Se. At 36 h, tig00000002G008510 (MANBA) and tig00000016G001160 were 2.66 and 5.08-fold, respectively, up-expressed in 0.1 mM Se. Also at 36 h, tig00000016G001160 was 6.27-fold up-expressed in 0.2 mM Se, but tig00000001G036310 (AGA), which is associated with N4-(beta-N-acetylglucosaminyl)-L-asparaginase, was decreased by 0.49-fold in other glycan degradation (ko00511) in response to 0.2 mM Se.
As compared with the 2 h samples which had the same selenium concentration added, tig00000001G036310’s (AGA) expression was increased by 2.88-fold in other glycan degradation (ko00511) at 12 h in the absence of Se. When 0.1 mM Na2SeO3 was used, tig00000002G008510 and tig00000016G001160 increased 2.77-fold and 3.21-fold at 36 h.
3.8. Differentially Expressed Genes Involved in Lipid Metabolism
After 2 h of Se treatment, tig00000002G007490, tig00000002G018450, and tig00000030G008490 (annotated as CYP51) were 0.42–0.47-fold down-regulated in steroid biosynthesis (ko00100) in endophytes under 0.2 mM Se (Table 3). Similarly, after 12 h, tig00000030G008490 and tig00000002G007490 were 0.33–0.42-fold down-regulated in response to 0.1 mM Se. However, tig00000099G005510 (DAK/TKFC), which is related to glycerolipid metabolism (ko00561), was 2.42-fold up-regulated in response to 0.2 mM Se.
Regarding the variance observed per time period, the expression of tig00000030G008490 involved in steroid biosynthesis (ko00100) was lower at 12 h than at 2 h under 0.1 mM Se. In glycerolipid metabolism (ko00561), the expressions of tig00000077G000990 (glpK), tig00000094G003040 (AKR1A1), and tig00000122G003720 (AKR1A1) were down-regulated by 0.46–0.50-fold in MZH10-VS-MZH20, MZH11-VS-MZH21, and MZH12-VS-MZH22, respectively. However, tig00002100G000060 (LPIN) encoding phosphatidate phosphatase was 2.40-fold up-expressed compared with the 2 h sample at 12 h in response to 0.1 mM.
3.9. Differentially Expressed Genes Involved in the Metabolism of Cofactors and Vitamins
During the 36 h of cultivation, compared with the control group, the expressions of tig00000016G004440 and tig00000002G011990 (pdxH) involved in pyridoxamine 5′-phosphate oxidase were significantly decreased by 0.46 and 0.50-fold in vitamin B6 metabolism (ko00750) in 0.1 mM Se (Table 3). In addition, tig00002100G002750 (nadC) was down-expressed by 0.49-fold in 0.1 mM Se, which was associated with nicotinate-nucleotide pyrophosphorylase (carboxylating) in nicotinate and nicotinamide metabolism (ko00760). However, 0.1 mM Se increased the expression of tig00000030G016720 (NNT) involved in H+-translocating NAD(P) transhydrogenase by 2.26-fold.
3.10. Differentially Expressed Genes Involved in the Biosynthesis of Other Secondary Metabolites
After cultured for 36 h, tig00000001G029780 (proB) involved in glutamate 5-kinase was 2.20-fold up-regulated in carbapenem biosynthesis (ko00332) in endophytes in response to 0.1 mM Se (Table 3), and tig00000077G005930 (uaZ) associated with urate oxidase in caffeine metabolism (ko00232) was 2.05-fold up-expressed in 0.2 mM Se. These DEGs were enhanced in response to selenium.
When compared with the 2 h samples, tig00000001G038060 (proA) encoding glutamate-5-semialdehyde dehydrogenase in carbapenem biosynthesis (ko00332) was 2.18–2.75 fold up-regulated at 12 h and 36 h in media supplemented with 0.1 mM Se, but tig00000001G029780 (proB) was 0.45-fold down-regulated at 12 h.
4. Discussion
To investigate the biological impacts of selenium, we examined the enzymatic activity related to selenium metabolism and the transcriptomic response of Epichloë sinensis following selenite treatment. The selenium treatment resulted in changes in enzymatic activities and gene expression in pathways such as ribosome function, carbohydrate metabolism, steroid biosynthesis, glycan degradation, and superoxide dismutase activity, among others.
Selenium possesses antioxidant properties in living organisms. Microorganisms trigger a range of antioxidative enzymes to uphold cellular redox balance, such as catalase, SOD, GR, and ascorbate peroxidase. SOD, as a crucial enzyme, eliminates oxygen free radicals, significantly impacting a strain’s stress resistance [43,44]. GR plays a vital role in the ascorbate-glutathione cycle by scavenging active oxygen. The activities of SOD and GR increase with rising Se concentrations and culture duration, with no notable difference between the control group and the 0.1 mM Se group at 12 h or 36 h. The antioxidative enzymatic activities varied among different strains or over time. Previous studies observed increased SOD and GR activities with rising sodium selenite concentration in Saccharomyces cerevisiae [45,46], contrasting with decreased GR activity in Lachancea thermotolerans. Furthermore, no significant differences in SOD activity were noted between the control and 5 μg/mL Se groups for Starmerella bacillaris and S. cerevisiae [46]. The activity of GR increased with selenium concentration in Candida utilis and S. cerevisiae, with no significant differences at lower selenium concentrations [47]. By contrast, Penicillium expansum exhibited decreased SOD activity over time (1 d–3 d) with 15 mg/L sodium selenite [48]. In addition, Candida utilis, simultaneously encountering moderate acid stress and 15 mg/L Na2SeO3, produces more γ-glutamylcysteine synthetase activity at 9 h than it does at 18 h, although severe acid stress exerts a negative effect on the activity [12]. High doses of selenium compounds such as Na2SeO3 (diacetophenonyl selenide) were found to enhance antioxidant activity in Aspergillus niger mycelium in the 7th day of cultivation [49]. These findings indicate that the effect of Se supplementation on antioxidant enzyme activity was influenced by the species of microorganisms, the forms and concentrations of selenium, and environmental conditions. Selenium also modulates the expression of genes encoding antioxidant enzymes in various microorganisms. For instance, the exposure of Fructobacillus tropaeoli to 5 and 100 ppm of Se led to the overexpression of glutathione reductases by 1.8–3.1-fold [50]. In this study, Se was observed to enhance the expression levels of genes encoding superoxide dismutase (2.64–3.04-fold) and oxidoreductase activity (2.00–85.16-fold). In Escherichia coli, exposure to selenite salts led to the induction of several genes encoding antioxidant enzymes such as sodA, gor, and trxB [51]. Ralstonia metallidurans exhibited overexpression of Fe-containing superoxide dismutase in the presence of selenite [52]. Additionally, A. oryzae demonstrated the overexpression of oxidoreductase genes in response to bis(2-pyridyl-1-oxide) diselenide, which is associated with oxidative stress [53,54].
In this investigation, the inclusion of 0.1 and 0.2 mM of Se in the growth medium resulted in elevated activities of GST, CS, MTR, and SMT in the Se-enriched mycelia (refer to Table 1), as compared to the control mycelia. These findings led to the hypothesis that the microorganisms under study utilized an enzymatic pathway for selenium metabolism. GST plays a vital role in the metabolism, physiology, redox balance, and versatile detoxification processes in certain microorganisms [55,56]. For example, Kieliszek et al. [47] observed an increase in GST activity in the yeast strains S. cerevisiae and C. utilis with rising selenium concentrations, with no significant differences noted in S. cerevisiae between the 0 and 10 mg/L Se groups. Furthermore, they demonstrated a significant increase in GST activity in C. utilis in the presence of 30 mg/L of selenium [57]. The compound bis(2-pyridyl-1-oxide) diselenide was found to enhance the expression of oxidoreductases and GST in A. oryzae [53]. Limited information is available regarding the crucial enzymes MTR and SMT, which are involved in microbial selenium detoxification. Comparative analysis of RNA-Seq data indicated that key enzymes such as methionine synthase, cysteine synthase, and S-adenosylmethionine synthase, were up-regulated in Se-enriched C. utilis under moderate acid stress [12]. In a prior investigation, S. cerevisiae augmented Seleno-methylselenocysteine (SeMCys) through the overexpression of S-adenosylmethionine synthetase (SAM). Yin et al. [58] documented that Bacillus subtilis achieved higher SeMCys levels by upregulating SMT and SAM. E. coli, expressing the bacterial enzyme thiopurine methyltransferase, methylated selenate to form dimethylselenide (DMSe) and dimethyldiselenide (DMDSe) [10]. These findings collectively suggest that these enzymes are easily affected by some factors, and the synthesis of organic selenium compounds is linked to the genetic regulation of key enzymes involved in selenium metabolism, potentially serving as a tolerance mechanism in microbes.
Previous studies have reported that the ribosome pathways were significantly down-regulated in heat-treated Metarhizium anisopliae conidia [59]. In the analysis of DEGs in the macrofungus Auricularia cornea cultivated in a substrate supplied with 100 µg/g selenium during the long period of culture, Li et al. [60] identified numerous genes associated with the ribosome pathway involved in translation that exhibited high expression levels during the budding stage but decreased expression levels during the mature stage. In a study comparing Lactobacillus brevis JLD715 treated with or without 0.5 mM Na2SeO3 for 24 h, certain DEGs were found to be enriched in the ribosome pathway (ko03010) [61]. We observed up-regulated unigenes in the ribosome pathway (ko03010) or ribosome biogenesis in eukaryotes (ko03008) in response to a 0.1 mM selenium supply at 12 h. However, the ribosome pathway exhibited decreased expression levels under 0.1 mM Se at 2 h and under 0.2 mM Se at 36 h, indicating the importance of protein synthesis during these time intervals. Furthermore, certain genes related to the ribosome pathway, such as EIF6, LSUrRNA, IMP3, CSNK2B, UTP12, NOG1, and BMS1, displayed dynamic expression patterns that varied with both time and the level of selenium exposure.
The current investigation examined the impact of sodium selenite at concentrations of 0.1 or 0.2 mM on the down-regulation of certain genes associated with steroid biosynthesis and linked to lipid metabolism after 2 h and 12 h of cultivation. This finding contrasts with the results of Li et al., where steroid biosynthesis (ko00100) was up-regulated in A. cornea during the budding stage in response to long term selenium treatment [60]. Specifically, the addition of 0.2 mM Na2SeO3 at 2 or 12 h resulted in the up-regulation of the fatty acid metabolic process and ergosterol biosynthetic process (GO:0006696). Ergosterol is a crucial component of plasma membranes in certain fungi, with several ergosterol biosynthesis genes showing significant increases in A. fumigatus under hypoxic conditions [62]. Additionally, a positive relationship was observed between ergosterol biosynthesis and the capacity for oxidative stress protection in S. cerevisiae [63]. Notably, ergosterol biosynthesis enzymes necessitate iron as a cofactor, and the rise in iron uptake transcripts aligns with an elevated iron requirement under hypoxia [62]. At 36 h, supplementation with 0.1 mM Na2SeO3 increased medium-chain fatty acid-CoA ligase activity and inhibited hydrolase activity targeting ester bonds. Furthermore, selenium up-regulated the genes PIGG, GPI7, MANBA, and the melanin metabolic process, while downregulating the gene AGA, which encodes N4-(beta-N-acetylglucosaminyl)-L-asparaginase, which are closely associated with cell wall biosynthesis and organization. The presence of melanin in the fungal cell wall enhances species survival in challenging environments [64]. Additionally, the study noted that selenium supplementation led to an up-regulation of fructose and mannose metabolism, consistent with a previous study demonstrating increased expression of fructose and mannose metabolism in Bacillus amyloliquefaciens [65].
Siderophores play crucial roles in the survival and iron acquisition of many fungal species [66,67,68]. Recent evidence has also indicated that certain environmental cues can trigger the synthesis of siderophores [69,70,71]. At 2 h, genes associated with siderophore synthesis showed increased expression levels in response to selenium supplementation, therefore leading to the enhancement of the triacetylfusarinine C biosynthetic process and ferricrocin biosynthetic process in the MZH20 vs. MZH22 and MZH30 vs. MZH31 groups. However, at 2 and 12 h, some genes involved in iron ion binding were down-regulated in the presence of 0.2 or 0.1 mM Se. Triacetylfusarinine C and ferricrocin are common intracellular siderophores utilized as iron sources in various fungi, such as A. fumigatus [72,73], A. nidulans [74], and Metarhizium robertsii [75], and are known to be involved in fungal development and oxidative stress resistance [74,76]. In this context, the addition of 0.1 mM Se enhanced the ferricrocin biosynthetic process and the cellular response to iron ion deficiency in Epichloë sinensis at the final time point. The regulation of iron homeostasis through siderophores can detect the absence of ferricrocin [76], thereby impacting the biosynthesis process of ferricrocin and iron homeostasis. Through the analysis of these differentially expressed genes, it was revealed that Epichloë sinensis regulated some metabolic pathways for selenium condition during cultivation time.
5. Conclusions
In this study, we found that some of the enzyme activities of Epichloë sinensis increased with increasing Na2SeO3 concentrations over time. Transcriptional changes in ribosomes were a major cellular response to Se. A substantial number of transcription factors were up-regulated in response to Se, including the known siderophore synthesis, superoxide dismutase, oxidoreductase activity, and ergosterol biosynthesis. However, we also observed a concomitant decrease in steroid biosynthesis. Therefore, further studies are required to verify the gene function of genes related to Se metabolism.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/fermentation10090445/s1, Table S1: Read quality of RNA sequencing; Table S2: Clean reads map to the Epichloë sinensis reference genome; Table S3: Gene FPKM density under different conditions; Table S4: Overrepresented KEGG pathways among different groups; Figure S1: Reads distribution on chromosomes of the reference genome; Figure S2: The distributions of reads in region of the reference genome; Figure S3: Gene FPKM density under different conditions; Figure S4: A heatmap showing the log10 (FPKM) values of the differentially expressed genes in each sample.
Author Contributions
Conceptualization, L.Z.; methodology, F.Q.; software, H.X. and Q.L.; validation, Y.L. and Y.M.; formal analysis, Y.L.; data curation, Y.M.; writing—original draft preparation, L.Z. and H.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (grant number 32260345), the Science and Technology Program of Qinghai Province (grant number 2022-ZJ-740).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available within the article.
Acknowledgments
We would like to thank Suzhou Azenta Biotech Co., Ltd. for the technical support in high-throughput sequencing. We also thank Pei Tian for the whole gene of Epichloë sinensis.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Winkel, L.H.E.; Johnson, C.A.; Lenz, M.; Grundl, T.; Leupin, O.X.; Amini, M.; Charlet, L. Environmental selenium research: From microscopic processes to global under standing. Environ. Sci. Technol. 2012, 46, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Kieliszek, M. Selenium-fascinating microelement, properties and sources in food. Molecules 2019, 24, 1298. [Google Scholar] [CrossRef]
- Mániková, D.; Vlasáková, D.; Loduhová, J.; Letavayová, L.; Vigašová, D.; Krascsenitsová, E.; Vlcková, V.; Brozmanová, J.; Chovanec, M. Investigations on the role of base excision repair and non-homologous end-joining pathways in sodium selenite-induced toxicity and mutagenicity in Saccharomyces cerevisiae. Mutagenesis 2009, 25, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Kieliszek, M.; Błażejak, S.; Gientka, I.; Bzducha-Wróbel, A. Accumulation and metabolism of selenium by yeast cells. Appl. Microbiol. Biotechnol. 2015, 99, 5373–5382. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Rensing, C.; Zheng, S.X. Microbial reduction and resistance to selenium: Mechanisms, applications and prospects. J. Hazard. Mater. 2022, 421, 126684. [Google Scholar] [CrossRef]
- Kessi, J.; Hanselmann, K.M. Similarities between the abiotic reduction of selenite with glutathione and the dissimilatory reaction mediated by Rhodospirill rubrum and Escherichia coli. J. Biol. Chem. 2004, 279, 50662–50669. [Google Scholar] [CrossRef]
- Kessi, J. Enzymic systems proposed to be involved in the dissimilatory reduction of selenite in the purple non-sulfur bacteria Rhodospirillum rubrum and Rhodobacter capsulatus. Microbiology 2006, 152, 731–743. [Google Scholar] [CrossRef]
- Eswayah, A.S.; Smith, T.J.; Gardiner, P.H.E. Microbial transformations of selenium species of relevance to bioremediation. Appl. Environ. Microbiol. 2016, 82, 4848–4859. [Google Scholar] [CrossRef]
- Tugarova, A.V.; Kamnev, A.A. Proteins in microbial synthesis of selenium nanoparticles. Talanta 2017, 174, 539–547. [Google Scholar] [CrossRef]
- Ranjard, L.; Prigent-Combaret, C.; Nazaret, S.; Cournoyer, B. Methylation of inorganic and organic selenium by the bacterial thiopurine methyltransferase. J. Bacteriol. 2002, 184, 3146–3149. [Google Scholar] [CrossRef]
- Ranjard, L.; Prigent-Combaret, C.; Favre-Bonté, S.; Monnez, C.; Nazaret, S.; Cournoyer, B. Characterization of a novel selenium methyltransferase from freshwater bacteria showing strong similarities with the calicheamicin methyltransferase. BBA—Gene Struct. Expr. 2004, 1679, 80–85. [Google Scholar] [CrossRef]
- Zhang, G.C.; Wang, D.H.; Wangg, D.H.; Wei, G.Y. The mechanism of improved intracellular organic selenium and glutathione contents in selenium-enriched Candida utilis by acid stress. Appl. Microbiol. Biotechnol. 2017, 101, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Mapelli, V.; Hillestrøm, P.R.; Kápolna, E.; Larsen, E.H.; Olsson, L. Metabolic and bioprocess engineering for production of selenized yeast with increased content of seleno-methylselenocysteine. Metab. Eng. 2011, 13, 282–293. [Google Scholar] [CrossRef]
- Lacourciere, G.M.; Levine, R.L.; Stadtman, T.C. Direct detection of potential selenium delivery proteins by using an Escherichia coli strain unable to incorporate selenium from selenite into proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 9150–9153. [Google Scholar] [CrossRef]
- Malkowski, M.G.; Quartley, E.; Friedman, A.E.; Babulski, J.; Kon, Y.; Wolfley, J.; Said, M.; Luft, J.R.; Phizicky, E.M.; DeTitta, G.T.; et al. Blocking S-adenosylmethionine synthesis in yeast allows selenomethionine incorporation and multiwavelength anomalous dispersion phasing. Proc. Natl. Acad. Sci. USA 2007, 104, 6678–6683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gladyshev, V.N. Comparative genomics of trace elements: Emerging dynamic view of trace element utilization and function. Chem. Rev. 2009, 109, 4828–4861. [Google Scholar] [CrossRef]
- Rosenfeld, C.E.; Kenyon, J.A.; James, B.R.; Santelli, C.M. Selenium (IV, VI) reduction and tolerance by fungi in an oxic environment. Geobiology 2017, 15, 441–452. [Google Scholar] [CrossRef]
- Staicu, L.C.; Ackerson, C.J.; Cornelis, P.; Ye, L.; Berendsen, R.L.; Hunter, W.J.; Noblitt, S.D.; Henry, C.S.; Cappa, J.J.; Montenieri, R.L.; et al. Pseudomonas moraviensis subsp. stanleyae: A bacterial endophyte capable of efficient selenite reduction to elemental selenium under aerobic conditions. J. Appl. Microbiol. 2015, 119, 400–410. [Google Scholar]
- Johnson, L.J.; de Bonth, C.; Briggs, L.R.; Caradus, J.R.; Finch, S.C.; Fleetwood, D.J.; Fletcher, L.R.; Hume, D.E.; Johnson, R.D.; Popay, A.J.; et al. The exploitation of epichloae endophytes for agricultural benefit. Fungal Divers. 2013, 60, 171–188. [Google Scholar] [CrossRef]
- Zamani, N.; Sabzalian, M.R.; Afyuni, M. Elevated atmospheric CO2 combined with Epichloë endophyte may improve growth and Cd phytoremediation potential of tall fescue (Festuca arundinacea L.). Environ. Sci. Pollut. Res. 2024, 31, 8164–8185. [Google Scholar] [CrossRef]
- Adeleke1, B.S.; Babalola, O.O. Roles of plant endosphere microbes in agriculture-a review. J. Plant Growth Regul. 2022, 41, 1411–1428. [Google Scholar] [CrossRef]
- Dwibedi, V.; Rath, S.K.; Joshi, M.; Kaur, R.; Kaur, G.; Singh, D.; Kaur, G.; Kaur, S. Microbial endophytes: Application towards sustainable agriculture and food security. Appl. Microbiol. Biotechnol. 2022, 106, 5359–5384. [Google Scholar] [CrossRef]
- Creamer, C.A.; Leewis, M.C.; Kracmarova-Farren, M.; Papik, J.; Kacur, S.; Freeman, J.; Uhlik, O.; Foster, A.L. A combined compost, dolomite, and endophyte addition is more effective than single amendments for improving phytorestoration of metal contaminated mine tailings. Plant Soil. 2024, 497, 219–240. [Google Scholar] [CrossRef]
- Nassar, A.R.A.; Eid, A.M.; Atta, H.M.E.; EI Naghy, W.S.; Fouda, A. Exploring the antimicrobial, antioxidant, anticancer, biocompatibility, and larvicidal activities of selenium nanoparticles fabricated by endophytic fungal strain Penicillium verhagenii. Sci. Rep. 2023, 13, 9054. [Google Scholar] [CrossRef]
- Ni, X.C.; Tian, J.B.; Chen, C.M.; Huang, L.; Lei, J.; Yu, X.J.; Wang, X.G. Multiple exposures to high concentrations of selenate significantly improve selenate tolerability, red elemental selenium (Se0) and selenoprotein biosynthesis in Herbaspirillum camelliae WT00C. World J. Microbl Biotechnol. 2022, 38, 5. [Google Scholar] [CrossRef]
- Hussein, H.G.; El-Sayed, E.S.R.; Younis, N.A.; Hamdy, A.H.A.; Easa, S.M. Harnessing endophytic fungi for biosynthesis of selenium nanoparticles and exploring their bioactivities. AMB Expr. 2022, 12, 68. [Google Scholar] [CrossRef]
- Lindbloma, S.D.; Valdez-Barillas, J.R.; Fakra, S.C.; Marcus, M.A.; Wangelinec, A.L.; Pilon-Smits, E.A.H. Influence of microbial associations on selenium localization and speciation in roots of Astragalus and Stanleya hyperaccumulators. Environ. Exp. Bot. 2013, 88, 33–42. [Google Scholar] [CrossRef]
- Lindblom, S.D.; Wangeline, A.L.; Valdez Barillas, J.R.; Devibiss, B.; Fakra, S.C.; Pilon-Smits, E.A.H. Fungal endophyte Alternaria tenuissima can affect growth and selenium accumulation in its hyperaccumulator host Astragalus bisulcatus. Front. Plant Sci. 2018, 9, 1213. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Yin, Y.L.; Song, J.Q.; Li, S.X. Mixed sowing improves plant and soil bacterial community restoration in the degraded alpine meadow. Plant Soil. 2024, 49, 379–392. [Google Scholar] [CrossRef]
- Wang, Y.B.; Luo, Y.; Tian, P.; Peng, H.; Feng, J. Preliminary evaluation of the disease resistance of Festuca sinensis infected by Epichloë sinensis. J. Phytopathol. 2021, 169, 623–629. [Google Scholar] [CrossRef]
- Zhou, L.Y.; Li, C.J.; Zhang, X.X.; Johnson, R.; Bao, G.S.; Yao, X.; Chai, Q. Effects of cold shocked Epichloë infected Festuca sinensis on ergot alkaloid accumulation. Fungal Ecol. 2015, 14, 99–104. [Google Scholar] [CrossRef]
- Xu, W.B.; Li, M.M.; Lin, W.H.; Nan, Z.B.; Tian, P. Effects of Epichloë sinensis endophyte and host ecotype on physiology of Festuca sinensis under different soil moisture conditions. Plants 2021, 10, 1649. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Zhou, Y.P.; Lin, W.H.; Li, M.M.; Wang, M.N.; Wang, Z.G.; Kuang, Y.; Tian, P. Effect of an Epichloë endophyte on adaptability to water stress in Festuca sinensis. Fungal Ecol. 2017, 30, 39–47. [Google Scholar] [CrossRef]
- Wang, M.N.; Tian, P.; Gao, M.; Li, M.M. The promotion of Festuca sinensis under heavy metal treatment mediated by Epichloë endophyte. Agronomy 2021, 11, 2049. [Google Scholar] [CrossRef]
- Yao, X.; Christensen, M.J.; Bao, G.S.; Zhang, C.P.; Li, X.Z.; Li, C.J.; Nan, Z.B. A toxic endophyte-infected grass helps reverse degradation and loss of biodiversity of over-grazed grasslands in northwest China. Sci. Rep. 2015, 5, 18527. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Xu, W.B.; Li, C.J.; Song, H.; Wang, M.N.; Schardl, C.L.; Nan, Z.B. Phylogenetic relationship and taxonomy of a hybrid Epichloë species symbiotic with Festuca sinensis. Mycol. Prog. 2020, 19, 1069–1081. [Google Scholar] [CrossRef]
- Luo, Y.; Tian, P. Growth and characteristics of two different Epichloë sinensis strains under different cultures. Front. Microbiol. 2021, 12, 726935. [Google Scholar] [CrossRef]
- Zhou, L.Y.; Xie, H.C.; Ma, X.L.; Ju, J.S.; Luo, Q.Y.; Qiao, F. Effect of sodium selenite concentration and culture time on extracellular and intracellular metabolite profiles of Epichloë sp. isolated from Festuca sinensis in liquid culture. Agriculture 2022, 12, 1423. [Google Scholar] [CrossRef]
- Zhou, L.Y.; Jiao, L.; Ju, J.S.; Ma, X.L. Effect of sodium selenite on the metabolite profile of Epichloë sp. mycelia from Festuca sinensis in solid culture. Biol. Trace Elem. Res. 2022, 200, 4865–4879. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCUe, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Oncology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Mohamadhasani, F.; Rahimi, M. Growth response and mycoremediation of heavy metals by fungus Pleurotus sp. Sci. Rep. 2022, 12, 19947. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Q.; Xia, C.C.; Yang, F.; Xu, N.; Wu, Q.; Hu, Y.; Xia, L.S.; Wang, C.; Zhou, M.Z. Effect of selenium supplements on the antioxidant activity and nitrite degradation of lactic acid bacteria. World J. Microbiol. Biotechnol. 2019, 35, 61. [Google Scholar] [CrossRef]
- Kaur, T.; Bansal, M.P. Selenium enrichment and anti-oxidant status in baker’s yeast, Saccharomyces cerevisiae at different sodium selenite concentrations. Nutr. Hosp. 2006, 21, 704–708. [Google Scholar]
- Assunção, M.; Martins, L.L.; Mourato, M.P.; Baleiras-Couto, M.M. Effect of selenium on growth and antioxidant enzyme activities of wine related yeasts. World J. Microbiol. Biotechnol. 2015, 31, 1899–1906. [Google Scholar] [CrossRef]
- Kieliszek, M.; Błażejak, S.; Bzducha-Wróbel, A.; Kot, A.M. Effect of selenium on growth and antioxidative system of yeast cells. Mol. Biol. Rep. 2019, 46, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.L.; Yin, X.B.; Lin, Z.Q.; Bañuelos, G.S.; Yuan, L.X.; Liu, Y.; Li, M. Inhibitory effect of selenium against Penicillium expansum and its possible mechanisms of action. Curr. Microbiol. 2014, 69, 192–201. [Google Scholar] [CrossRef]
- Poluboyarinov, P.A.; Kuznetsova, A.V.; Moiseeva, I.Y.; Mikulyak, N.I.; Kaplun, A.P. Induction of antioxidant activity by selenium compounds in the Aspergillus niger mycelium. Russ. J. Bioorg. Chem. 2023, 49, 823–835. [Google Scholar] [CrossRef]
- Martínez, F.G.; Moreno-Martin, G.; Mozzi, F.; Madrid, Y.; Pescuma, M. Selenium stress response of the fruit origin strain Fructobacillus tropaeoli CRL 2034. Appl. Microbiol. Biotechnol. 2023, 107, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Bébien, M.; Lagniel, G.; Garin, J.; Touati, D.; Verméglio, A.; Labarre, J. Involvement of superoxide dismutases in the response of Escherichia coli to selenium oxides. J. Bacteriol. 2002, 184, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Roux, M.; Covés, J. The iron-containing superoxide dismutase of Ralstonia metallidurans CH34. FEMS Microbiol. Lett. 2002, 210, 129–133. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zalepkina, S.A.; Smirnova, V.F.; Borisovb, A.V.; Matsulevich, Z.V. Genomic profiling of the response of Aspergillus oryzae to the treatment with bis(2-pyridine-1-oxide) diselenide. Russ. J. Genet. 2019, 55, 301–308. [Google Scholar] [CrossRef]
- Juby, S.; Soumya, P.; Jayachandran, K.; Radhakrishnan, E.K. Morphological, metabolomic and genomic evidences on drought stress protective functioning of the endophyte Bacillus safensis Ni7. Curr. Microbiol. 2024, 81, 209. [Google Scholar] [CrossRef]
- Zhang, J.; Chi, Y.; Feng, L. The mechanism of degradation of alizarin red by a white-rot fungus Trametes gibbosa. BMC Biotechnol. 2021, 21, 64. [Google Scholar] [CrossRef]
- Senabio, J.A.; de Campos Pereira, F.; Pietro-Souza, W.; Sousa, T.F.; Silva, G.F.; Soares, M.A. Enhanced mercury phytoremediation by Pseudomonodictys pantanalensis sp. nov. A73 and Westerdykella aquatica P71. Braz. J. Microbiol. 2023, 54, 949–964. [Google Scholar] [CrossRef]
- Kieliszek, M.; Bierla, K.; Jiménez-Lamana, J.; Kot, A.M.; Alcántara-Durán, J.; Piwowarek, K.; Błazejak, S.; Szpunar, J. Metabolic response of the yeast Candida utilis during enrichment in selenium. Int. J. Mol. Sci. 2020, 21, 5287. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zhou, Y.; Yang, H.; Liao, Y.; Ma, T.; Wang, F. Enhanced selenocysteine biosynthesis for seleno-methylselenocysteine production in Bacillus subtilis. Appl. Microbiol. Biotechnol. 2023, 107, 2843–2854. [Google Scholar] [CrossRef]
- Wang, Z.X.; Zhou, X.Z.; Meng, H.M.; Liu, Y.J.; Zhou, Q.; Huang, B. Comparative transcriptomic analysis of the heat stress response in the filamentous fungus Metarhizium anisopliae using RNA-Seq. Appl. Microbiol. Biotechnol. 2014, 98, 5589–5597. [Google Scholar] [CrossRef]
- Li, X.L.; Yan, L.J.; Li, Q.; Tan, H.; Zhou, J.; Miao, R.Y.; Ye, L.; Peng, W.H.; Zhang, X.P.; Tan, W.; et al. Transcriptional profiling of Auricularia cornea in selenium accumulation. Sci. Rep. 2019, 9, 5641. [Google Scholar] [CrossRef]
- Yang, X.Y.; Dai, X.F.; Jin, H.N.; Lin, G.G.; Wang, Z.H.; Song, Y.; Zhang, W.; Man, C.X.; Jiang, Y.J. Physicochemical and transcriptomic responses of Lactobacillus brevis JLD715 to sodium selenite. J. Sci. Food Agr. 2021, 101, 4332–4341. [Google Scholar] [CrossRef] [PubMed]
- Barker, M.B.; Kroll, K.; Vödisch, M.; Mazurie, A.; Kniemeyer, O.; Cramer, R.A. Transcriptomic and proteomic analyses of the Aspergillus fumigatus hypoxia response using an oxygencontrolled fermenter. BMC Genom. 2012, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Higgins, V.J.; Beckhouse, A.G.; Oliver, A.D.; Rogers, P.J.; Dawes, I.W. Yeast genome-wide expression analysis identifies a strong ergosterol and oxidative stress response during the initial stages of an industrial lager fermentation. Appl. Environ. Microbiol. 2003, 69, 4777–4787. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Freitas, D.F.; da Rocha, I.M.; Vieira-da-Motta, O.; Santos, C.P. The role of melanin in the biology and ecology of nematophagous fungi. J. Chem. Ecol. 2021, 47, 597–613. [Google Scholar] [CrossRef]
- Liu, J.; Shi, L.; Ma, X.X.; Jiang, S.J.; Hou, X.Y.; Li, P.; Cheng, Y.; Lv, J.; Li, S.R.; Ma, T.Y.; et al. Characterization and anti-infammatory effect of selenium-enriched probiotic Bacillus amyloliquefaciens C-1, a potential postbiotics. Sci. Rep. 2023, 13, 14302. [Google Scholar]
- Oide, S.; Krasnoff, S.B.; Gibson, D.M.; Turgeon, B.G. Intracellular siderophores are essential for ascomycete sexual development in heterothallic Cochliobolus heterostrophus and homothallic Gibberella zeae. Eukaryot. Cell 2007, 6, 1339–1353. [Google Scholar] [CrossRef]
- Haas, H.; Eisendle, M.; Turgeon, B.G. Siderophores in fungal physiology and virulence. Annu. Rev. Phytopathol. 2008, 46, 149–187. [Google Scholar] [CrossRef]
- Koulman, A.; Lee, V.T.; Fraser, K.; Johnson, L.; Arcus, V.; Lott, S.J.; Rasmussen, S. Identification of extracellular siderophores and a related peptide from the endophytic fungus Epichloë festucae in culture and endophyte-infected Lolium perenne. Phytochemistry 2012, 75, 128–139. [Google Scholar] [CrossRef]
- Rashmi, V.; ShylajaNaciyar, M.; Rajalakshmi, R.; D’Souza, S.F.; Prabaharan, D.; Uma, L. Siderophore mediated uranium sequestration by marine cyanobacterium Synechococcus elongatus BDU 130911. Bioresour. Technol. 2013, 130, 204–210. [Google Scholar] [CrossRef]
- Song, Y.; Wu, X.; Li, Z.; Ma, Q.Q.; Bao, R. Molecular mechanism of siderophore regulation by the Pseudomonas aeruginosa BfmRS two-component system in response to osmotic stress. Commun. Biol. 2024, 7, 295. [Google Scholar] [CrossRef]
- Dimkpa, C.O.; Merten, D.; Svatos, A.; Buechel, G.; Kothe, E. Siderophores mediate reduced and increased uptake of cadmium by Streptomyces tendae F4 and sunflower (Helianthus annuus), respectively. J. Appl. Microbiol. 2009, 107, 1687–1696. [Google Scholar] [CrossRef] [PubMed]
- Kragl, C.; Schrettl, M.; Abt, B.; Sarg, B.; Lindner, H.H.; Haas, H. EstB-mediated hydrolysis of the siderophore triacetylfusarinine C optimizes iron uptake of Aspergillus fumigatus. Eukaryot. Cell 2007, 6, 1278–1285. [Google Scholar] [CrossRef]
- Wallner, A.; Blatzer, M.; Schrettl, M.; Sarg, B.; Lindner, H.; Haas, H. Ferricrocin, a siderophore involved in intra- and transcellular iron distribution in Aspergillus fumigates. Appl. Environ. Microbiol. 2009, 75, 4194–4196. [Google Scholar] [CrossRef] [PubMed]
- Eisendle, M.; Oberegger, H.; Zadra, I.; Haas, H. The intracellular siderophore ferricrocin is involved in iron storage, oxidative-stress resistance, germination, and sexual development in Aspergillus nidulans. Eukaryot. Cell 2006, 5, 1596–1603. [Google Scholar] [CrossRef]
- Hof, C.; Eisfeld, K.; Kai, W.; Antelo, L.; Foster, A.J.; Anke, H. Ferricrocin synthesis in Magnaporthe grisea and its role in pathogenicity in rice. Mol. Plant Pathol. 2007, 8, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Jirakkakul, J.; Wichienchote, N.; Likhitrattanapisal, S.; Supawadee Ingsriswang, S.; Yoocha, T.; Tangphatsornruang, S.; Wasuwan, R.; Cheevadhanarak, S.; Morakot Tanticharoen, M.; Amnuaykanjanasin, A. Iron homeostasis in the absence of ferricrocin and its consequences in fungal development and insect virulence in Beauveria bassiana. Sci. Rep. 2021, 11, 19624. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).