
Bottom-Up Synthesis Strategies Enabling the Investigation of Metal Catalyst-Carbon Support Interactions

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Carbon Synthesis Using Hydrothermal Carbonization (HTC)
2.3. Carbon Synthesis Using Evaporation-Induced Self-Assembly (EISA)
2.4. Catalyst Synthesis
2.5. Characterizations
3. Results and Discussion
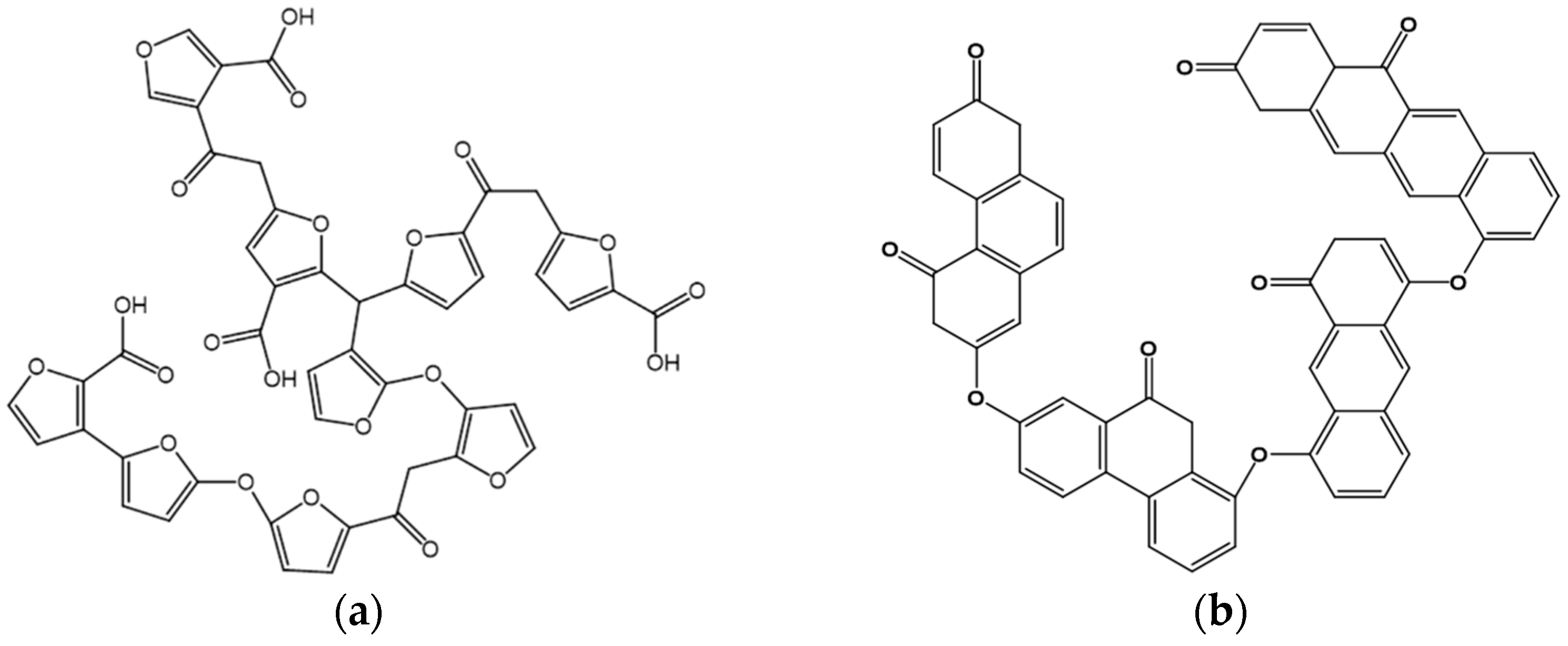
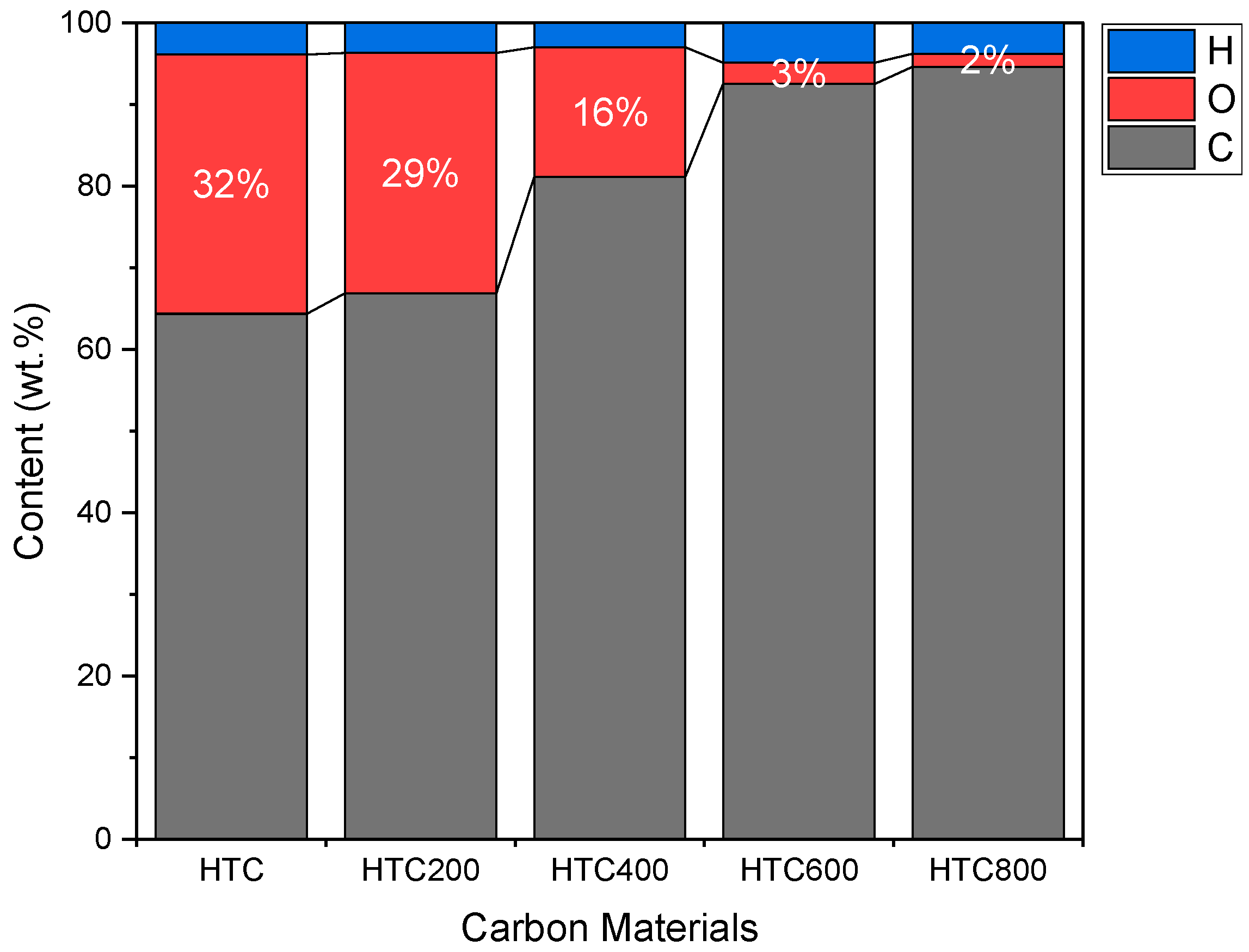
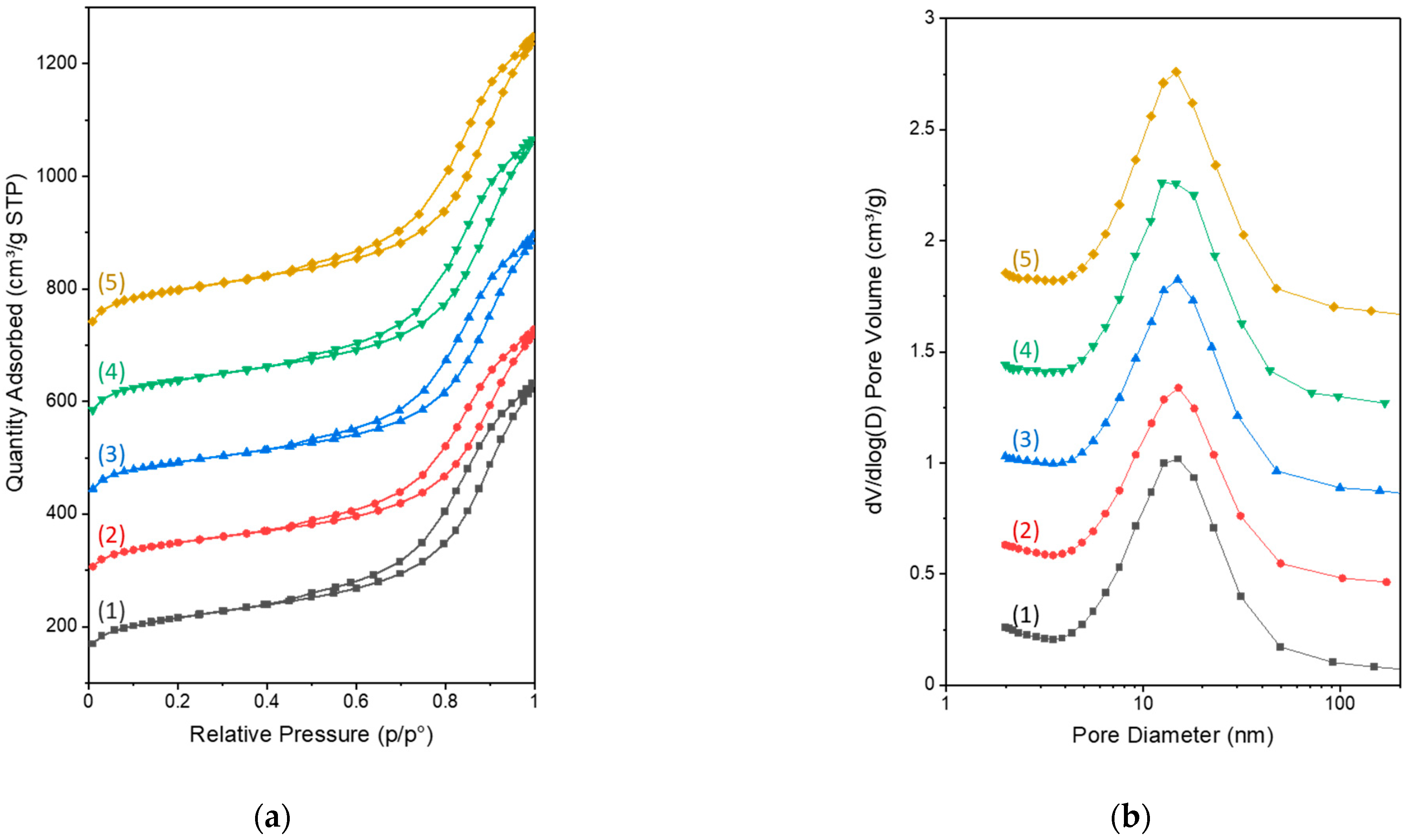
3.1. Hydrothermal Carbonization (HTC)
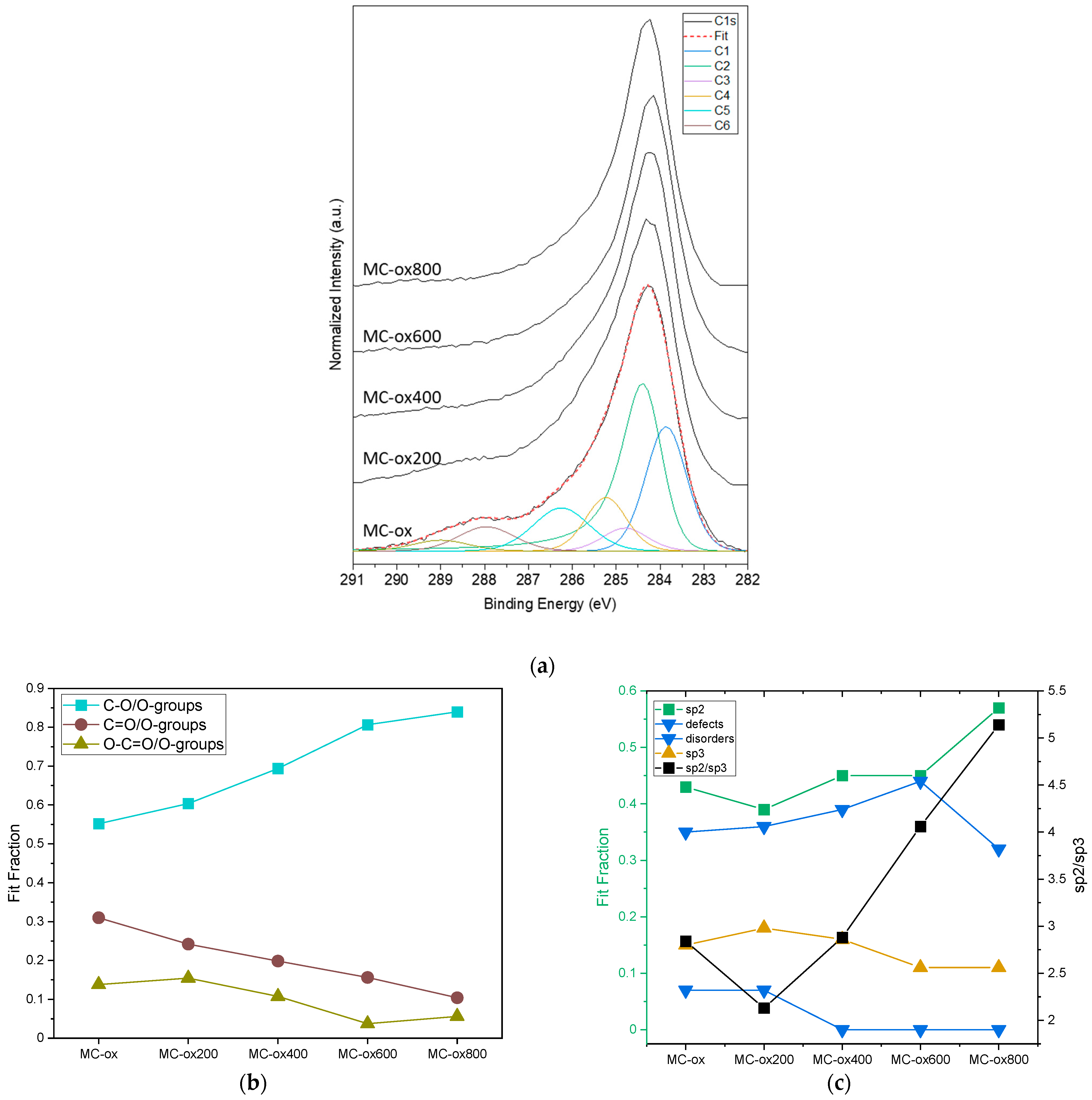
3.2. Evaporation-Induced Self-Assembly (EISA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Radovic, L.R. Physicochemical properties of carbon materials: A brief overview. In Carbon Materials for Catalysis; Serp, P., Figueiredo, J.L., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 1–44. [Google Scholar]
- Gerber, I.C.; Serp, P. A theory/experience description of support effects in carbon-supported catalysts. Chem. Rev. 2020, 120, 1250–1349. [Google Scholar] [CrossRef]
- Rodŕíguez-Reinoso, F.; Seṕulveda-Escribano, A. Carbon as catalyst support. In Carbon Materials for Catalysis; Serp, P., Figueiredo, J.L., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 131–155. [Google Scholar]
- Huo, J.; Tessonnier, J.-P.; Shanks, B.H. Improving hydrothermal stability of supported metal catalysts for biomass conversions: A review. ACS Catal. 2021, 11, 5248–5270. [Google Scholar] [CrossRef]
- Xiong, H.; Pham, H.N.; Datye, A.K. Hydrothermally stable heterogeneous catalysts for conversion of biorenewables. Green Chem. 2014, 16, 4627–4643. [Google Scholar] [CrossRef]
- Lou, Y.; Xu, J.; Zhang, Y.; Pan, C.; Dong, Y.; Zhu, Y. Metal-support interaction for heterogeneous catalysis: From nanoparticles to single atoms. Mater. Today Nano 2020, 12, 100093. [Google Scholar] [CrossRef]
- Ahmadi, M.; Mistry, H.; Roldan Cuenya, B. Tailoring the catalytic properties of metal nanoparticles via support interactions. J. Phys. Chem. Lett. 2016, 7, 3519–3533. [Google Scholar] [CrossRef] [PubMed]
- Bandosz, T.J. Surface chemistry of carbon materials. In Carbon Materials for Catalysis; Serp, P., Figueiredo, J.L., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 45–92. [Google Scholar]
- Jerigová, M.; Odziomek, M.; López-Salas, N. “We are here!” oxygen functional groups in carbons for electrochemical applications. ACS Omega 2022, 7, 11544–11554. [Google Scholar] [CrossRef]
- Rao, R.G.; Blume, R.; Hansen, T.W.; Fuentes, E.; Dreyer, K.; Moldovan, S.; Ersen, O.; Hibbitts, D.D.; Chabal, Y.J.; Schlogl, R.; et al. Interfacial charge distributions in carbon-supported palladium catalysts. Nat. Commun. 2017, 8, 340. [Google Scholar] [CrossRef] [PubMed]
- Habiba, K.; Makarov, V.I.; Weiner, B.R.; Morell, G. Chapter 10 fabrication of nanomaterials by pulsed laser synthesis. In Manufacturing Nanostructures; Ahmed, W., Ali, N., Eds.; One Central Press: Manchester, UK, 2014; pp. 263–292. [Google Scholar]
- Titirici, M.-M.; White, R.J.; Brun, N.; Budarin, V.L.; Su, D.S.; Del Monte, F.; Clark, J.H.; Maclachlan, M.J. Sustainable carbon materials. Chem. Soc. Rev. 2015, 44, 250–290. [Google Scholar] [CrossRef]
- Wang, Z.; Jin, K.W.M.; Melvin, G.J.H. Carbon materials from various sources for composite materials. In Composite Materials: Applications in Engineering, Biomedicine and Food Science; Siddiquee, S., Gan Jet Hong, M., Mizanur Rahman, M., Eds.; Springer: Cham, Switzerland, 2020; pp. 3–33. [Google Scholar]
- Inagaki, M.; Toyoda, M.; Soneda, Y.; Tsujimura, S.; Morishita, T. Templated mesoporous carbons: Synthesis and applications. Carbon 2016, 107, 448–473. [Google Scholar] [CrossRef]
- Xin, W.; Song, Y. Mesoporous carbons: Recent advances in synthesis and typical applications. RSC Adv. 2015, 5, 83239–83285. [Google Scholar] [CrossRef]
- Liang, C.; Li, Z.; Dai, S. Mesoporous carbon materials: Synthesis and modification. Angew. Chem. Int. Ed. 2008, 47, 3696–3717. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Pint, C.L.; Islam, A.E.; Weatherup, R.S.; Hofmann, S.; Meshot, E.R.; Wu, F.; Zhou, C.; Dee, N.; Amama, P.B.; et al. Carbon nanotubes and related nanomaterials: Critical advances and challenges for synthesis toward mainstream commercial applications. ACS Nano 2018, 12, 11756–11784. [Google Scholar] [CrossRef] [PubMed]
- Sakintuna, B.; Yürüm, Y. Templated porous carbons: A review article. Ind. Eng. Chem. Res. 2005, 44, 2893–2902. [Google Scholar] [CrossRef]
- Nicolae, S.A.; Au, H.; Modugno, P.; Luo, H.; Szego, A.E.; Qiao, M.; Li, L.; Yin, W.; Heeres, H.J.; Berge, N.; et al. Recent advances in hydrothermal carbonisation: From tailored carbon materials and biochemicals to applications and bioenergy. Green Chem. 2020, 22, 4747–4800. [Google Scholar] [CrossRef]
- Hu, B.; Wang, K.; Wu, L.; Yu, S.-H.; Antonietti, M.; Titirici, M.-M. Engineering carbon materials from the hydrothermal carbonization process of biomass. Adv. Mater. 2010, 22, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Liu, Y.; Zhang, J. Effect of solution pH on the carbon microsphere synthesized by hydrothermal carbonization. Procedia Environ. Sci. 2011, 11, 1322–1327. [Google Scholar] [CrossRef][Green Version]
- Zhao, Q.; Tao, S.; Miao, X.; Zhu, Y. A green, rapid, scalable and versatile hydrothermal strategy to fabricate monodisperse carbon spheres with tunable micrometer size and hierarchical porosity. Chem. Eng. J. 2019, 372, 1164–1173. [Google Scholar] [CrossRef]
- Titirici, M.-M.; Antonietti, M.; Baccile, N. Hydrothermal carbon from biomass: A comparison of the local structure from poly- to monosaccharides and pentoses/hexoses. Green Chem. 2008, 10, 1204–1212. [Google Scholar] [CrossRef]
- Sevilla, M.; Fuertes, A.B. Chemical and structural properties of carbonaceous products obtained by hydrothermal carbonization of saccharides. Chemistry 2009, 15, 4195–4203. [Google Scholar] [CrossRef]
- van Zandvoort, I.; Koers, E.J.; Weingarth, M.; Bruijnincx, P.C.A.; Baldus, M.; Weckhuysen, B.M. Structural characterization of 13C-enriched humins and alkali-treated 13C humins by 2D solid-state NMR. Green Chem. 2015, 17, 4383–4392. [Google Scholar] [CrossRef]
- Wang, Z.; Opembe, N.; Kobayashi, T.; Nelson, N.C.; Slowing, I.I.; Pruski, M. Quantitative atomic-scale structure characterization of ordered mesoporous carbon materials by solid state NMR. Carbon 2018, 131, 102–110. [Google Scholar] [CrossRef]
- Papaioannou, N.; Marinovic, A.; Yoshizawa, N.; Goode, A.E.; Fay, M.; Khlobystov, A.; Titirici, M.-M.; Sapelkin, A. Structure and solvents effects on the optical properties of sugar-derived carbon nanodots. Sci. Rep. 2018, 8, 6559. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Buenaposada, I.D.; Rey-Raap, N.; Calvo, E.G.; Angel Menéndez, J.; Arenillas, A. Effect of methanol content in commercial formaldehyde solutions on the porosity of RF carbon xerogels. J. Non-Cryst. Solids 2015, 426, 13–18. [Google Scholar] [CrossRef]
- Libbrecht, W.; Verberckmoes, A.; Thybaut, J.W.; Van Der Voort, P.; De Clercq, J. Tunable large pore mesoporous carbons for the enhanced adsorption of humic acid. Langmuir 2017, 33, 6769–6777. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Deng, Q.-F.; Agula, B.; Ren, T.-Z.; Liu, Y.-P.; Zhaorigetu, B.; Yuan, Z.-Y. Synthesis of ordered mesoporous carbon materials and their catalytic performance in dehydrogenation of propane to propylene. Catal. Today 2012, 186, 35–41. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Arrigo, R.; Blume, R.; Streibel, V.; Genovese, C.; Roldan, A.; Schuster, M.E.; Ampelli, C.; Perathoner, S.; Velasco Vélez, J.J.; Hävecker, M.; et al. Dynamics at polarized carbon dioxide–iron oxyhydroxide interfaces unveil the origin of multicarbon product formation. ACS Catal. 2021, 12, 411–430. [Google Scholar] [CrossRef]
- Blume, R.; Rosenthal, D.; Tessonnier, J.-P.; Li, H.; Knop-Gericke, A.; Schlögl, R. Characterizing graphitic carbon with X-ray photoelectron spectroscopy: A step-by-step approach. ChemCatChem 2015, 7, 2871–2881. [Google Scholar] [CrossRef]
- Nikolov, P.Y.; Yaylayan, V.A. Thermal decomposition of 5-(hydroxymethyl)-2-furaldehyde (HMF) and its further transformations in the presence of glycine. J. Agric. Food Chem. 2011, 59, 10104–10113. [Google Scholar] [CrossRef]
- Zhao, M.; Li, B.; Cai, J.-X.; Liu, C.; McAdam, K.G.; Zhang, K. Thermal & chemical analyses of hydrothermally derived carbon materials from corn starch. Fuel Process. Technol. 2016, 153, 43–49. [Google Scholar] [CrossRef]
- Gerber, I.; Oubenali, M.; Bacsa, R.; Durand, J.; Gonçalves, A.; Pereira, M.F.R.; Jolibois, F.; Perrin, L.; Poteau, R.; Serp, P. Theoretical and Experimental Studies on the Carbon-Nanotube Surface Oxidation by Nitric Acid: Interplay between Functionalization and Vacancy Enlargement. Chem.–Eur. J. 2011, 17, 11467–11477. [Google Scholar] [CrossRef] [PubMed]
- Gomezserrano, V.; Acedoramos, M.; Lopezpeinado, A.; Valenzuelacalahorro, C. Oxidation of activated carbon by hydrogen peroxide. Study of surface functional groups by FT-i.r. Fuel 1994, 73, 387–395. [Google Scholar] [CrossRef]
- Eigler, S.; Dotzer, C.; Hirsch, A.; Enzelberger, M.; Müller, P. Formation and Decomposition of CO2 Intercalated Graphene Oxide. Chem. Mater. 2012, 24, 1276–1282. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Annealing Temperature (°C) | Total Surface Area (m2/g) | Micropores Surface Area (m2/g) | Fraction of Mesoporous Surface Area (%) | Average Pore Size (nm) * |
|---|---|---|---|---|---|
| HTC | - | 4 | - | - | - |
| HTC200 | 200 | 4.5 | - | - | - |
| HTC400 | 400 | 351 | 263 | 25 | 4.1 |
| HTC600 | 600 | 448 | 395 | 11.7 | 5.1 |
| HTC800 | 800 | 438 | 393 | 10.2 | 5.5 |
| t-HTC800 | 800 | 531 | 440 | 17.0 | 8.0 |
| Catalyst | BET Surface Area (m2/g) | BJH Surface Area (m2/g) | BJH Pore Size (nm) | BJH Pore Volume (cm3/g) | Micropore Volume (cm3/g) |
|---|---|---|---|---|---|
| Formic Acid | 521 | 379 | 6.1 | 0.581 | 0.072 |
| Citric Acid | 501 | 383 | 6.2 | 0.589 | 0.059 |
| Acetic Acid | 236 | 112 | 13.9 | 0.389 | 0.041 |
| Nitric Acid | 455 | 276 | 5.4 | 0.372 | 0.076 |
| Hydrochloric Acid | 572 | 263 | 14.5 | 0.740 | 0.128 |
| Catalyst | Annealing Temperature (°C) | Elemental Composition (wt%) | Surface Area * (m2/g) | Average Pores Size (nm) * | |||
|---|---|---|---|---|---|---|---|
| N | C | H | O | ||||
| MC-ox | - | 0.7 | 71.9 | 1.0 | 26.4 | 322 | 9.9 |
| MC-ox200 | 200 | 0.7 | 73.0 | 0.7 | 25.6 | 290 | 10.0 |
| MC-ox400 | 400 | 0.6 | 78.7 | 0.3 | 20.4 | 303 | 10.1 |
| MC-ox600 | 600 | 0.5 | 83.7 | 0.4 | 15.4 | 325 | 10.1 |
| MC-ox800 | 800 | 0.7 | 93.9 | 0.7 | 4.7 | 340 | 10.1 |
| Catalyst | Average Pore Size (nm) a | Pd Loading (wt.%) b | Metal Dispersion (%) c | Metallic Surface Area (m2/g) c | Average Particle Size (nm) c |
|---|---|---|---|---|---|
| Pd/MC-ox200 | 9.9 | 4.76 | 33.8 | 7.177 | 3.31 |
| Pd/MC-ox400 | 10.0 | 4.80 | 33.4 | 7.141 | 3.37 |
| Pd/MC-ox600 | 9.9 | 4.82 | 32.4 | 6.946 | 3.47 |
| Pd/MC-ox800 | 9.9 | 4.55 | 31.6 | 6.403 | 3.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bateni, H.; Prabhu, P.T.; Gebur, H.E.; Tessonnier, J.-P. Bottom-Up Synthesis Strategies Enabling the Investigation of Metal Catalyst-Carbon Support Interactions. C 2022, 8, 37. https://doi.org/10.3390/c8030037
Bateni H, Prabhu PT, Gebur HE, Tessonnier J-P. Bottom-Up Synthesis Strategies Enabling the Investigation of Metal Catalyst-Carbon Support Interactions. C. 2022; 8(3):37. https://doi.org/10.3390/c8030037
Chicago/Turabian StyleBateni, Hamed, Prathamesh T. Prabhu, Hannah E. Gebur, and Jean-Philippe Tessonnier. 2022. "Bottom-Up Synthesis Strategies Enabling the Investigation of Metal Catalyst-Carbon Support Interactions" C 8, no. 3: 37. https://doi.org/10.3390/c8030037
APA StyleBateni, H., Prabhu, P. T., Gebur, H. E., & Tessonnier, J.-P. (2022). Bottom-Up Synthesis Strategies Enabling the Investigation of Metal Catalyst-Carbon Support Interactions. C, 8(3), 37. https://doi.org/10.3390/c8030037