
Theoretical Investigation of O2 and CO2 Adsorption on Small PdNi Clusters Supported on N-Doped Graphene Quantum Dots

 , and
, and
Abstract

1. Introduction
2. Computational Details
3. Results and Discussion
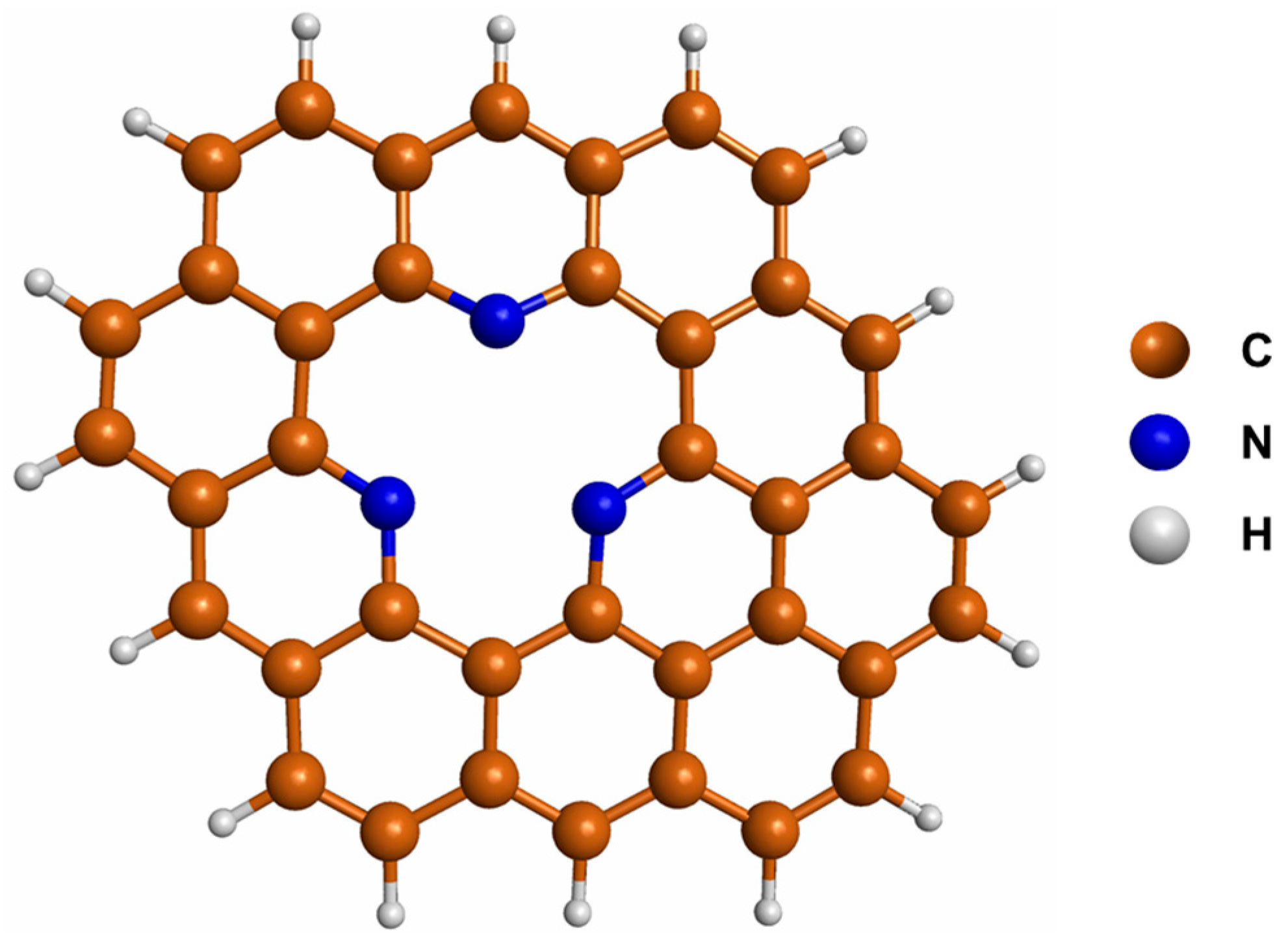
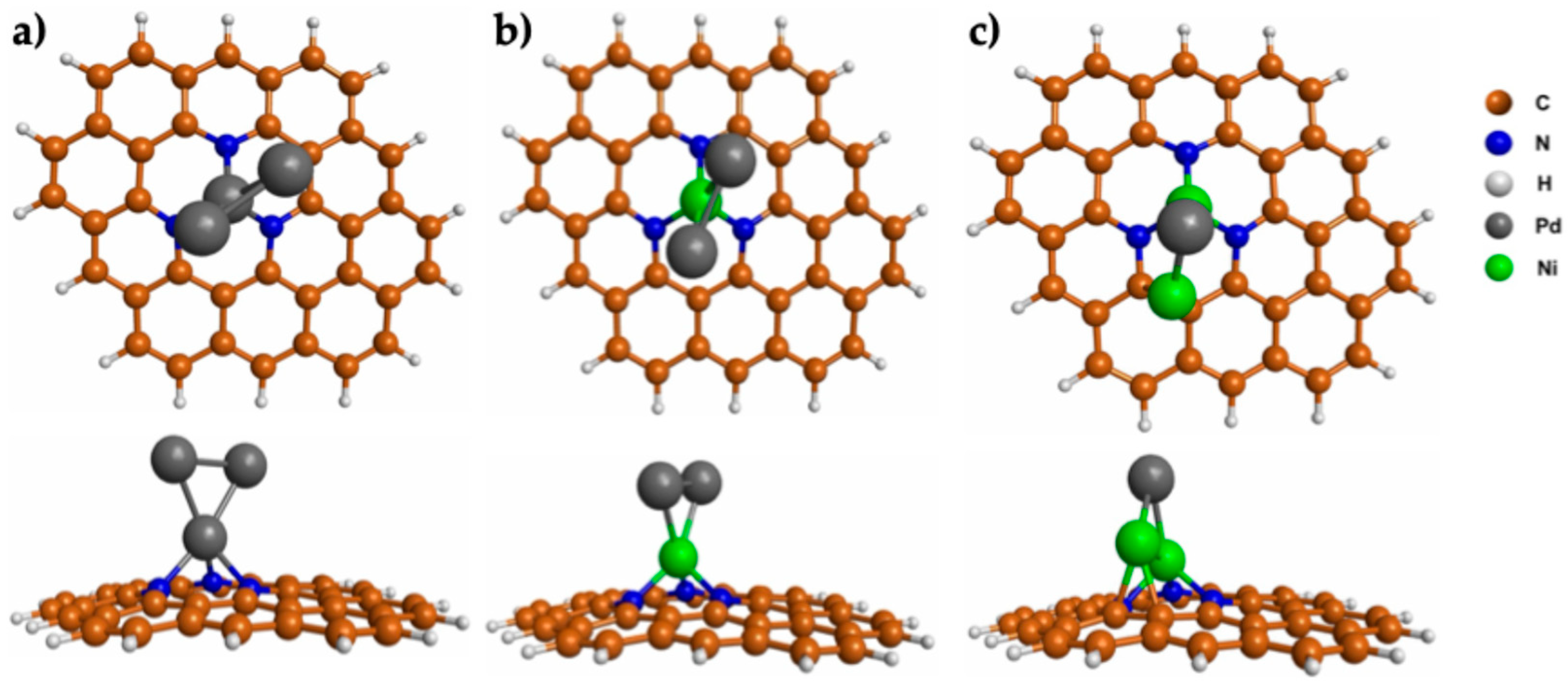
3.1. Properties of Pd3−nNin (n = 0–2) Clusters Deposited on N-GQDs
3.2. O2 Adsorption on Pd3−nNin (n = 0–2) Clusters Embedded in N-GQDs
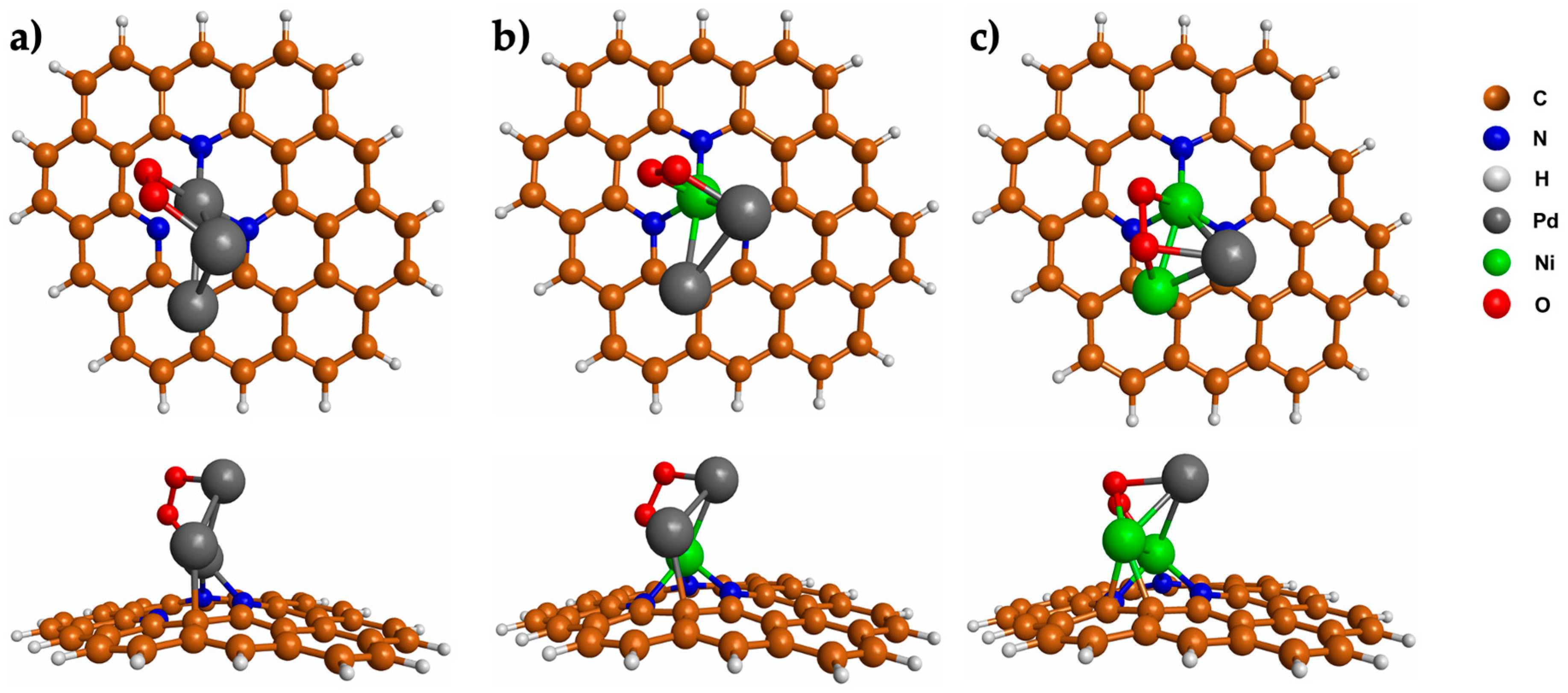
3.3. CO2 Adsorption on Pd3−nNin (n = 0–2) Clusters Embedded in N-GQDs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Holechek, J.L.; Geli, H.M.; Sawalhah, M.N.; Valdez, R. A global assessment: Can renewable energy replace fossil fuels by 2050? Sustainability 2022, 14, 4792. [Google Scholar] [CrossRef]
- Jeje, S.O.; Marazani, T.; Obiko, J.O.; Shongwe, M.B. Advancing the hydrogen production economy: A comprehensive review of technologies, sustainability, and future prospects. Int. J. Hydrogen Energy 2024, 78, 642–661. [Google Scholar] [CrossRef]
- Gomez-Sanchez, A.; Franco-Luján, V.A.; Alfaro-López, H.M.; Hernández-Sánchez, L.; Cruz-Martínez, H.; Medina, D.I. Carbon material-reinforced polymer composites for bipolar plates in polymer electrolyte membrane fuel cells. Polymers 2024, 16, 671. [Google Scholar] [CrossRef]
- Tellez-Cruz, M.M.; Escorihuela, J.; Solorza-Feria, O.; Compañ, V. Proton exchange membrane fuel cells (PEMFCs): Advances and challenges. Polymers 2021, 13, 3064. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Martínez, H.; Guerra-Cabrera, W.; Flores-Rojas, E.; Ruiz-Villalobos, D.; Rojas-Chávez, H.; Peña-Castañeda, Y.A.; Medina, D.I. Pt-free metal nanocatalysts for the oxygen reduction reaction combining experiment and theory: An overview. Molecules 2021, 26, 6689. [Google Scholar] [CrossRef]
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Martínez, H.; Rojas-Chávez, H.; Matadamas-Ortiz, P.T.; Ortiz-Herrera, J.C.; López-Chávez, E.; Solorza-Feria, O.; Medina, D.I. Current progress of Pt-based ORR electrocatalysts for PEMFCs: An integrated view combining theory and experiment. Mater. Today Phys. 2021, 19, 100406. [Google Scholar] [CrossRef]
- Jacob, T. The mechanism of forming H2O from H2 and O2 over a Pt catalyst via direct oxygen reduction. Fuel cells 2006, 6, 159–181. [Google Scholar] [CrossRef]
- Woldu, A.R.; Huang, Z.; Zhao, P.; Hu, L.; Astruc, D. Electrochemical CO2 reduction (CO2RR) to multi-carbon products over copper-based catalysts. Coord. Chem. Rev. 2022, 454, 214340. [Google Scholar] [CrossRef]
- Chang, B.; Pang, H.; Raziq, F.; Wang, S.; Huang, K.W.; Ye, J.; Zhang, H. Electrochemical reduction of carbon dioxide to multicarbon (C2+) products: Challenges and perspectives. Energy Environ. Sci. 2023, 16, 4714–4758. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Huang, L.; Wei, M.; Qi, R.; Dong, C.L.; Dang, D.; Yang, C.C.; Xia, C.; Chen, C.; Zaman, S.; Li, F.M.; et al. An integrated platinum-nanocarbon electrocatalyst for efficient oxygen reduction. Nat. Commun. 2022, 13, 6703. [Google Scholar] [CrossRef]
- Huang, L.; Zaman, S.; Tian, X.; Wang, Z.; Fang, W.; Xia, B.Y. Advanced platinum-based oxygen reduction electrocatalysts for fuel cells. Acc. Chem. Res. 2021, 54, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Todorova, T.K.; Schreiber, M.W.; Fontecave, M. Mechanistic understanding of CO2 reduction reaction (CO2RR) toward multicarbon products by heterogeneous copper-based catalysts. ACS Catal. 2019, 10, 1754–1768. [Google Scholar] [CrossRef]
- Xue, Y.; Guo, Y.; Cui, H.; Zhou, Z. Catalyst design for electrochemical reduction of CO2 to multicarbon products. Small Methods 2021, 5, 2100736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, J.; Ge, R.; Zhang, J.; Cairney, J.M.; Li, Y.; Zhu, M.; Li, S.; Li, W. The effect of coordination environment on the activity and selectivity of single-atom catalysts. Coord. Chem. Rev. 2022, 461, 214493. [Google Scholar] [CrossRef]
- An, Q.; Bo, S.; Jiang, J.; Gong, C.; Su, H.; Cheng, W.; Liu, Q. Atomic-level interface engineering for boosting oxygen electrocatalysis performance of single-atom catalysts: From metal active center to the first coordination sphere. Adv. Sci. 2023, 10, 2205031. [Google Scholar] [CrossRef]
- Karmodak, N.; Nørskov, J.K. Activity and stability of single-and di-atom catalysts for the O2 reduction reaction. Angew. Chem. Int. Ed. 2023, 62, e202311113. [Google Scholar] [CrossRef]
- Feng, M.; Wu, X.; Cheng, H.; Fan, Z.; Li, X.; Cui, F.; Fan, S.; Dai, Y.; Lei, G.; He, G. Well-defined Fe–Cu diatomic sites for efficient catalysis of CO2 electroreduction. J. Mater. Chem. A 2021, 9, 23817–23827. [Google Scholar] [CrossRef]
- Han, H.; Lee, S.; Im, J.; Lee, M.; Lee, T.; Hyun, S.T.; Hong, J.; Seok, T.; Choo, D. Large-scale CO2-to-CO electroconversion on highly efficient diatomic catalysts. Chem. Eng. J. 2024, 479, 147603. [Google Scholar] [CrossRef]
- Yan, X.; Liu, D.; Guo, P.; He, Y.; Wang, X.; Li, Z.; Pan, H.; Sun, D.; Fang, F.; Wu, R. Atomically Dispersed Co2MnN8 Triatomic Sites Anchored in N-Doped Carbon Enabling Efficient Oxygen Reduction Reaction. Adv. Mater. 2023, 35, 2210975. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Cao, S.; Wei, S.; Wang, Z.; Chen, H.; Lin, X.; Chen, X.; Liu, S.; Lu, X. Triple-atom catalysts 3TM-GYs (TM = Cu, Fe, and Co; GY = graphyne) for high-performance CO2 reduction reaction to C1 products. Appl. Mater. Today 2021, 25, 101245. [Google Scholar] [CrossRef]
- Liu, M.; Huang, J.; Hu, S.; Ma, Z.; Yang, Y.; Chen, Y.; Liu, Y. Mechanism study of trimetallic single-cluster catalysts loaded on graphdiyne for highly efficient oxygen-reduction reactions. Appl. Surf. Sci. 2024, 672, 160860. [Google Scholar] [CrossRef]
- Liu, J.; Xu, H.; Zhu, J.; Cheng, D. Understanding the pathway switch of the oxygen reduction reaction from single-to double-/triple-atom catalysts: A dual channel for electron acceptance–backdonation. JACS Au 2023, 3, 3031–3044. [Google Scholar] [CrossRef]
- Li, S.; Tong, L.; Peng, Z.; Zhang, B.; Fu, X. Efficient CO2 reduction for CO production using triatomic catalyst screening: A DFT study. Appl. Surf. Sci. 2024, 644, 158804. [Google Scholar] [CrossRef]
- Ren, Y.; Sun, X.; Jing, H.; Xie, Z.; Zhao, Z. Efficient Electrochemical Reduction of CO2 on g-C3N4 Monolayer-supported Metal Trimer Catalysts: A DFT Study. Chem. Asian J. 2023, 18, e202201232. [Google Scholar] [CrossRef]
- Xing, N.; Liu, Z.; Wang, Z.; Gao, Y.; Li, Q.; Wang, H. The reduction reaction of carbon dioxide on a precise number of Fe atoms anchored on two-dimensional biphenylene. Phys. Chem. Chem. Phys. 2022, 24, 27474–27482. [Google Scholar] [CrossRef]
- Geudtner, G.; Calaminici, P.; Carmona-Espindola, J.; del Campo, J.M.; Dominguez-Soria, V.D.; Moreno, R.F.; Gamboa, G.U.; Goursot, A.; Köster, A.M.; Reveles, J.U.; et al. DeMon2k. WIREs Comput. Mol. Sci. 2012, 2, 548–555. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, W. Comment on “Generalized gradient approximation made simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Mintmire, J.W.; Dunlap, B.I. Fitting the Coulomb potential variationally in linear-combination-of-atomic-orbitals density-functional calculations. Phys. Rev. A 1982, 25, 88. [Google Scholar] [CrossRef]
- Andrae, D.; Haeussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Calaminici, P.; Janetzko, F.; Köster, A.M.; Mejia-Olvera, R.; Zuniga-Gutierrez, B. Density functional theory optimized basis sets for gradient corrected functionals: 3d transition metal systems. J. Chem. Phys. 2007, 126, 044108. [Google Scholar] [CrossRef]
- Köster, A.M.; Reveles, J.U.; del Campo, J.M. Calculation of exchange-correlation potentials with auxiliary function densities. J. Chem. Phys. 2004, 121, 3417–3424. [Google Scholar] [CrossRef] [PubMed]
- Reveles, J.U.; Köster, A.M. Geometry optimization in density functional methods. J. Comput. Chem. 2004, 25, 1109–1116. [Google Scholar] [CrossRef]
- López-Sosa, L.; Cruz-Martínez, H.; Solorza-Feria, O.; Calaminici, P. Nickel and copper doped palladium clusters from a first-principles perspective. Int. J. Quantum Chem. 2019, 119, e26013. [Google Scholar] [CrossRef]
- Chaves, A.S.; Piotrowski, M.J.; Da Silva, J.L. Evolution of the structural, energetic, and electronic properties of the 3d, 4d, and 5d transition-metal clusters (30 TMn systems for n = 2–15): A density functional theory investigation. Phys. Chem. Chem. Phys. 2017, 19, 15484–15502. [Google Scholar] [CrossRef]
- Granja-DelRío, A.; Abdulhussein, H.A.; Johnston, R.L. DFT-based global optimization of sub-nanometer Ni–Pd clusters. J. Phys. Chem. C 2019, 123, 26583–26596. [Google Scholar] [CrossRef]
- Ferro, Y.; Teillet-Billy, D.; Rougeau, N.; Sidis, V.; Morisset, S.; Allouche, A. Stability and magnetism of hydrogen dimers on graphene. Phys. Rev. B 2008, 78, 085417. [Google Scholar] [CrossRef]
- Nieman, R.; Aquino, A.J.; Hardcastle, T.P.; Kotakoski, J.; Susi, T.; Lischka, H. Structure and electronic states of a graphene double vacancy with an embedded Si dopant. J. Chem. Phys. 2017, 147, 194702. [Google Scholar] [CrossRef]
- Domancich, N.F.; Ferullo, R.M.; Castellani, N.J. Interaction of aluminum dimer with defective graphene. Comput. Theor. Chem. 2015, 1059, 27–34. [Google Scholar] [CrossRef]
- Martínez-Espinosa, J.A.; Cruz-Martínez, H.; Calaminici, P.; Medina, D.I. Structures and properties of Co13−xCux (x = 0–13) nanoclusters and their interaction with pyridinic N3-doped graphene nanoflake. Phys. E 2021, 134, 114858. [Google Scholar] [CrossRef]
- Rangel, E.; Sansores, E. Theoretical study of hydrogen adsorption on nitrogen doped graphene decorated with palladium clusters. Int. J. Hydrogen Energy 2014, 39, 6558–6566. [Google Scholar] [CrossRef]
- Rangel, E.; Sansores, E.; Vallejo, E.; Hernández-Hernández, A.; López-Pérez, P.A. Study of the interplay between N-graphene defects and small Pd clusters for enhanced hydrogen storage via a spill-over mechanism. Phys. Chem. Chem. Phys. 2016, 18, 33158–33170. [Google Scholar] [CrossRef]
- Zhao, C.; Wu, H. Density functional investigation of mercury and arsenic adsorption on nitrogen doped graphene decorated with palladium clusters: A promising heavy metal sensing material in farmland. Appl. Surf. Sci. 2017, 399, 55–66. [Google Scholar] [CrossRef]
- García-Hilerio, B.; Santiago-Silva, L.; Vásquez-García, A.; Gomez-Sanchez, A.; Franco-Luján, V.A.; Cruz-Martínez, H. H2 adsorption on small Pd-Ni clusters deposited on N-doped graphene: A theoretical study. C 2024, 10, 73. [Google Scholar] [CrossRef]
- Martínez-Vargas, A.; Vásquez-López, A.; Antonio-Ruiz, C.D.; Cruz-Martínez, H.; Medina, D.I.; Montejo-Alvaro, F. Stability, energetic, and reactivity properties of NiPd alloy clusters deposited on graphene with defects: A density functional theory study. Materials 2022, 15, 4710. [Google Scholar] [CrossRef]
- Jin, C.; Cheng, L.; Feng, G.; Ye, R.; Lu, Z.H.; Zhang, R.; Yu, X. Adsorption of transition-metal clusters on graphene and N-doped graphene: A DFT study. Langmuir 2022, 38, 3694–3710. [Google Scholar] [CrossRef]
- Rêgo, C.R.; Tereshchuk, P.; Oliveira, L.N.; Da Silva, J.L. Graphene-supported small transition-metal clusters: A density functional theory investigation within van der Waals corrections. Phys. Rev. B 2017, 95, 235422. [Google Scholar] [CrossRef]
- Liu, C.S.; Liu, X.C.; Wang, G.C.; Liang, R.P.; Qiu, J.D. Preparation of nitrogen-doped graphene supporting Pt nanoparticles as a catalyst for oxygen reduction and methanol oxidation. J. Electroanal. Chem. 2014, 728, 41–50. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, J.; Liu, D.; Li, T.; Yan, Y.; Wei, X.; Yang, Y.; Yan, S.; Zou, Z. N-doped graphene-coated commercial Pt/C catalysts toward high-stability and antipoisoning in oxygen reduction reaction. J. Phys. Chem. Lett. 2022, 13, 2019–2026. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Shao, X.; Cao, D. Nitrogen-doped graphene as an excellent candidate for selective gas sensing. Sci. China Chem. 2014, 57, 911–917. [Google Scholar] [CrossRef]
- Yan, P.; Liu, J.; Yuan, S.; Liu, Y.; Cen, W.; Chen, Y. The promotion effects of graphitic and pyridinic N combinational doping on graphene for ORR. Appl. Surf. Sci. 2018, 445, 398–403. [Google Scholar] [CrossRef]
- Jian-feng, L.; Ge, S.; Ting, W.; Kai, N.; Bin-xia, Y.; Wei-guo, P. The oxygen reduction activity of nitrogen-doped graphene. Pol. J. Chem. Technol. 2022, 24, 29–34. [Google Scholar] [CrossRef]
- Ma, J.; Habrioux, A.; Luo, Y.; Ramos-Sanchez, G.; Calvillo, L.; Granozzi, G.; Balbuena, P.B.; Alonso-Vante, N. Electronic interaction between platinum nanoparticles and nitrogen-doped reduced graphene oxide: Effect on the oxygen reduction reaction. J. Mater. Chem. A 2015, 3, 11891–11904. [Google Scholar] [CrossRef]
- Liu, S.; Huang, S. Theoretical insights into the activation of O2 by Pt single atom and Pt4 nanocluster on functionalized graphene support: Critical role of Pt positive polarized charges. Carbon 2017, 115, 11–17. [Google Scholar] [CrossRef]
- Montejo-Alvaro, F.; Navarro-Ibarra, D.C.; Franco-Luján, V.A.; Alfaro-López, H.M.; Vásquez-García, A.; Medina, D.I.; Cruz-Martínez, H. CO2 adsorption on 3d transition metal-alloyed Pt clusters supported on pyridinic N-doped graphene. Inorg. Chim. Acta 2024, 573, 122339. [Google Scholar] [CrossRef]
- Montejo-Alvaro, F.; Martínez-Espinosa, J.A.; Rojas-Chávez, H.; Navarro-Ibarra, D.C.; Cruz-Martínez, H.; Medina, D.I. CO2 adsorption over 3d transition-metal nanoclusters supported on pyridinic N3-doped graphene: A DFT investigation. Materials 2022, 15, 6136. [Google Scholar] [CrossRef]
- del Castillo, R.M.; Calles, A.G.; Espejel-Morales, R.; Hernandez-Coronado, H. Adsorption of CO2 on graphene surface modified with defects. Comput. Condens. Matter 2018, 16, e00315. [Google Scholar] [CrossRef]
- Gálvez-González, L.E.; Juárez-Sánchez, J.O.; Pacheco-Contreras, R.; Garzón, I.L.; Paz-Borbón, L.O.; Posada-Amarillas, A. CO2 adsorption on gas-phase Cu4−xPtx (x = 0–4) clusters: A DFT study. Phys. Chem. Chem. Phys. 2018, 20, 17071–17080. [Google Scholar] [CrossRef]
- Batista, K.E.; Ocampo-Restrepo, V.K.; Soares, M.D.; Quiles, M.G.; Piotrowski, M.J.; Da Silva, J.L. Ab initio investigation of CO2 adsorption on 13-atom 4d clusters. J. Chem. Inf. Model. 2022, 60, 537–545. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pd3/N-GQDs | Pd2Ni1/N-GQDs | Pd1Ni2/N-GQDs | |
|---|---|---|---|
| Eint (eV) | −2.71 | −3.89 | −5.04 |
| Bader charges (e) | 0.27 | 0.37 | 0.70 |
| HOMO-LUMO gap (eV) | 0.17 | 0.20 | 0.58 |
| Pd3/N-GQDs | Pd2Ni/N-GQDs | PdNi2/N-GQDs | |
|---|---|---|---|
| Eads (eV) | −1.645 | −1.624 | −1.740 |
| O–O bond length (Å) | 1.379 | 1.420 | 1.490 |
| Average metal–O bond lengths (Å) | 1.982 | 1.905 | 1.964 |
| Bader charges (e) | −0.594 | −0.776 | −0.936 |
| Properties | Pd3/N-GQDs | Pd2Ni/N-GQDs | PdNi2/N-GQDs |
|---|---|---|---|
| Eads (eV) | −1.043 | −1.051 | −0.823 |
| Bending angle of CO2 (°) | 132.90 | 133.71 | 135.04 |
| Average C–O bond lengths (Å) | 1.265 | 1.264 | 1.261 |
| Metal–O bond lengths (Å) | 2.082 | 1.954 | 1.953 |
| Metal–C bond lengths (Å) | 1.986 | 1.980 | 1.971 |
| Bader charges (e) | −0.547 | −0.574 | −0.582 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Hilerio, B.; Santiago-Silva, L.; Matadamas-Ortiz, P.T.; Gomez-Sanchez, A.; Franco-Luján, V.A.; Cruz-Martínez, H. Theoretical Investigation of O2 and CO2 Adsorption on Small PdNi Clusters Supported on N-Doped Graphene Quantum Dots. C 2025, 11, 43. https://doi.org/10.3390/c11030043
García-Hilerio B, Santiago-Silva L, Matadamas-Ortiz PT, Gomez-Sanchez A, Franco-Luján VA, Cruz-Martínez H. Theoretical Investigation of O2 and CO2 Adsorption on Small PdNi Clusters Supported on N-Doped Graphene Quantum Dots. C. 2025; 11(3):43. https://doi.org/10.3390/c11030043
Chicago/Turabian StyleGarcía-Hilerio, Brenda, Lidia Santiago-Silva, Pastor T. Matadamas-Ortiz, Alejandro Gomez-Sanchez, Víctor A. Franco-Luján, and Heriberto Cruz-Martínez. 2025. "Theoretical Investigation of O2 and CO2 Adsorption on Small PdNi Clusters Supported on N-Doped Graphene Quantum Dots" C 11, no. 3: 43. https://doi.org/10.3390/c11030043
APA StyleGarcía-Hilerio, B., Santiago-Silva, L., Matadamas-Ortiz, P. T., Gomez-Sanchez, A., Franco-Luján, V. A., & Cruz-Martínez, H. (2025). Theoretical Investigation of O2 and CO2 Adsorption on Small PdNi Clusters Supported on N-Doped Graphene Quantum Dots. C, 11(3), 43. https://doi.org/10.3390/c11030043