
An Expanded Landscape of Unusually Short RNAs in 11 Samples from Six Eukaryotic Organisms

 , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
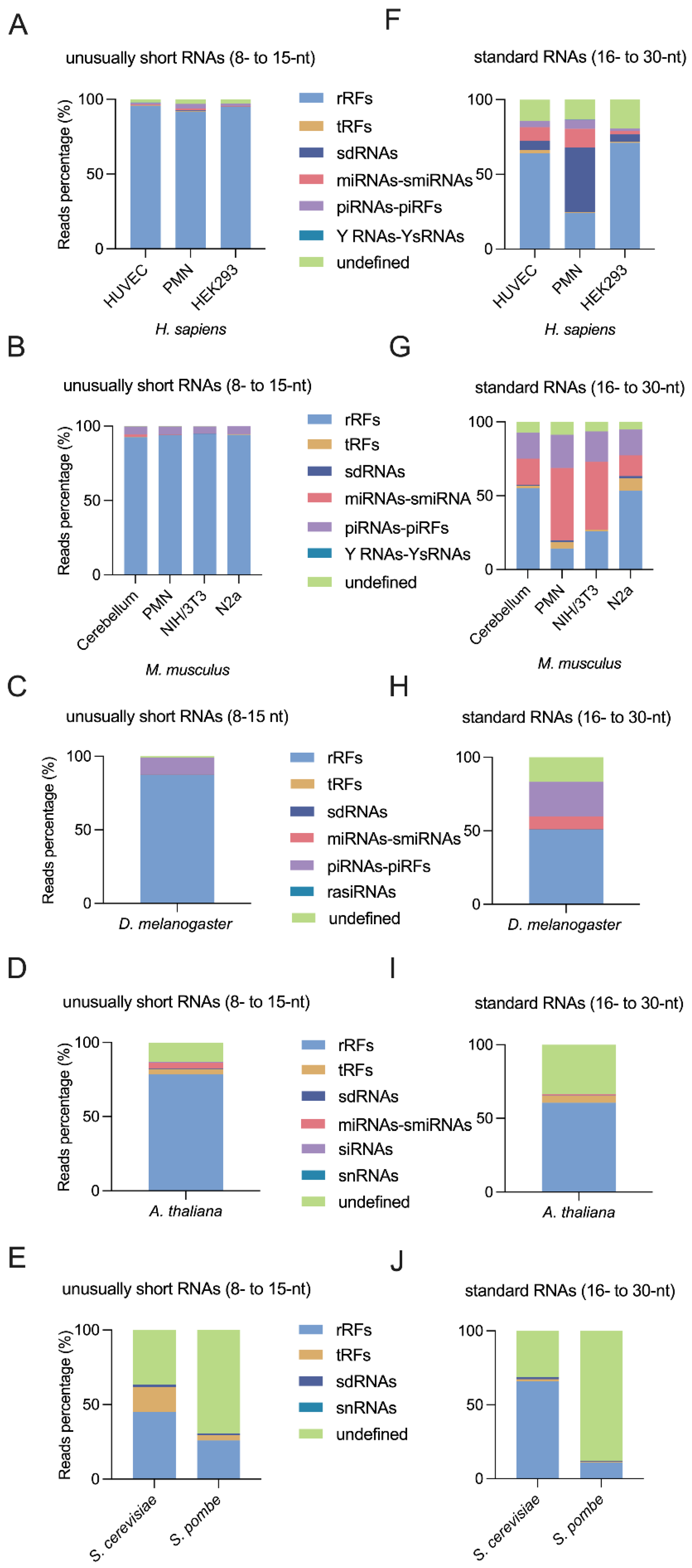
2.1. Identification of Highly Abundant Unusually Short RNAs
2.2. RNAs Shorter Than 16- nt Are More Abundant in Bilaterian Organisms
2.3. Small Non Coding RNA Distribution upon Biotypes
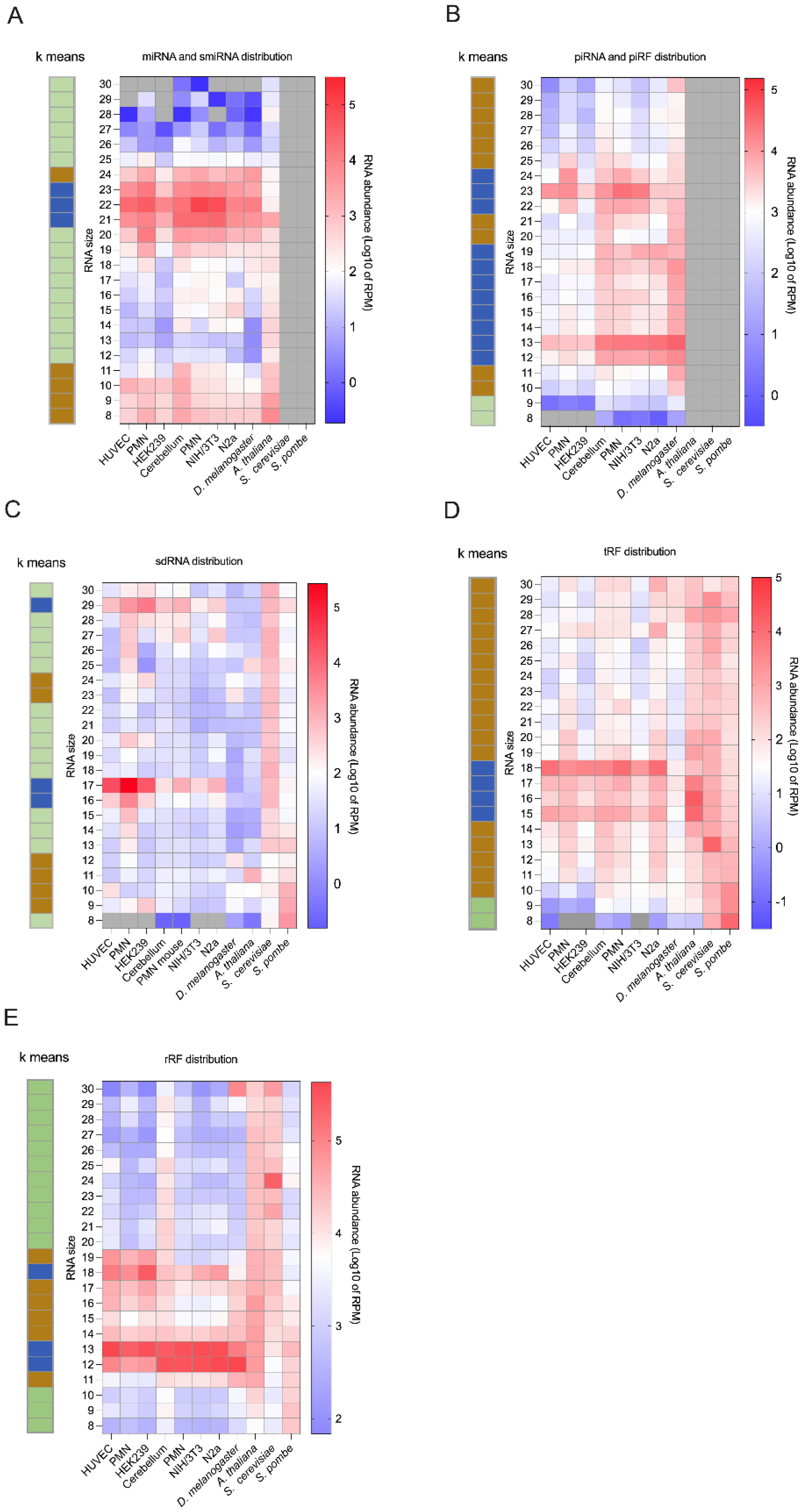
2.3.1. MicroRNAs Are Less Abundant Than Unusually Short RNAs
2.3.2. MicroRNA Fragments May Be Detected in the 8- to 15-nt Window
2.3.3. piRNA Fragments Are as Abundant and Diverse as piRNAs in Bilaterian Organisms
2.3.4. Discovery of Highly Abundant, Diverse, and Unusually Short sdRNAs
2.3.5. Detection of tRFs Shorter Than 16-nt
2.3.6. rRFs Are Overly Abundant in Bilaterian Organisms
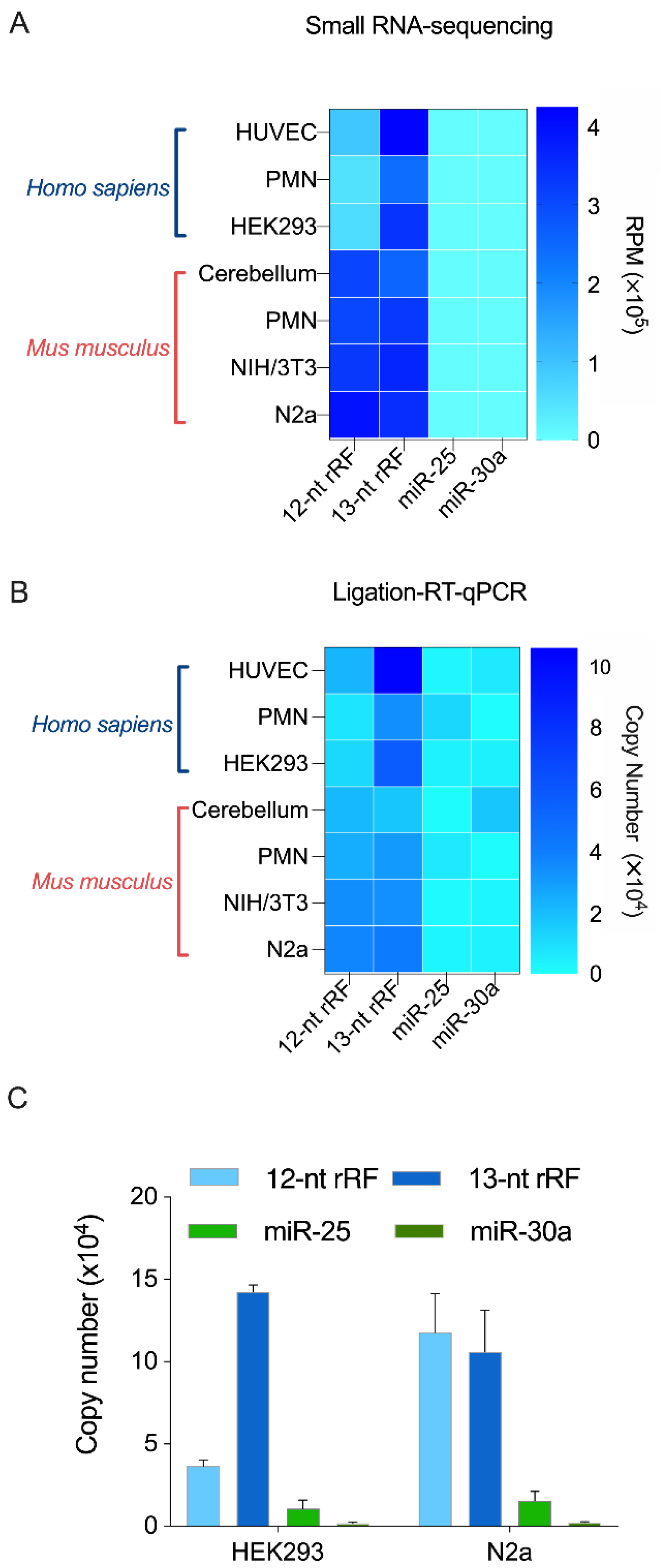
2.4. RT-qPCR Validation of Two Unusually Short rRFs of 12 and 13 nt
2.5. Conclusions
3. Materials and Methods
3.1. Ethical Statement
3.1.1. Human Blood Samples
3.1.2. Mouse Tissue Samples
3.2. Biological Samples
3.2.1. Primary and Cultured Human Cells
3.2.2. Primary and Cultured Mouse Cells and Tissues
3.2.3. Drosophila melanogaster
3.2.4. Arabidopsis thaliana
3.3. Total RNA Isolation
3.4. Small RNA Library and Sequencing
3.5. Analysis Workflow
3.6. Adapter-Ligated RT-qPCR Method
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | fetal bovine serum |
| HEK293 | human embryonic kidney 293 |
| HTS | high-throughput sequencing |
| HUVEC | human umbilical vein endothelial cells |
| mRNA | messenger RNA |
| nt | nucleotide |
| piRNA | PIWI-associated RNA |
| PMN | polymorphonuclear leukocyte |
| rRF | ribosomal RNA fragment |
| rRNA | ribosomal RNA |
| RT-qPCR | reverse transcription—quantitative polymerase chain reaction |
| sdRNA | small nucleolar RNA-derived RNA |
| sncRNA | small non-coding RNA |
| snoRNA | small nucleolar RNA |
| sRNA | small RNA |
| sRNA-Seq | small RNA sequencing |
| tiRNAs | tRNA halves derived from tRNA |
| tRF | transfer RNA (tRNA)-derived fragment |
| tRNA | transfer RNA |
| Y RNA | cytoplasmic RNA |
| YsRNA | Y RNA-derived small RNA |
| siRNA | small interfering RNA |
| rasiRNA | repeat-associated short interfering RNA |
| snRNA | small nuclear RNA |
| NIH/3T3 | mouse embryonic fibroblast cells |
| N2a | mouse neuroblastoma cells |
| miRNA | microRNA |
| smiRNA | semi-microRNA |
| piRFs | piRNA-derived fragments |
| siRNA | small interfering RNA |
| RPM | reads per million. |
References
- Reddy, R.; Henning, D.; Busch, H. Substitutions, insertions, and deletions in two highly conserved U3 RNA species. J. Biol. Chem. 1980, 255, 7029–7033. [Google Scholar] [CrossRef]
- Mizuno, T.; Chou, M.Y.; Inouye, M. A unique mechanism regulating gene expression: Translational inhibition by a complementary RNA transcript (micRNA). Proc. Natl. Acad. Sci. USA 1984, 81, 1966–1970. [Google Scholar] [CrossRef]
- Lambert, M.; Benmoussa, A.; Provost, P. Small Non-Coding RNAs Derived from Eukaryotic Ribosomal RNA. Non-Coding RNA 2019, 5, 16. [Google Scholar] [CrossRef]
- Castellano, L.; Stebbing, J. Deep sequencing of small RNAs identifies canonical and non-canonical miRNA and endogenous siRNAs in mammalian somatic tissues. Nucleic Acids Res. 2013, 41, 3339–3351. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Houwing, S.; Kamminga, L.M.; Berezikov, E.; Cronembold, D.; Girard, A.; van den Elst, H.; Filippov, D.V.; Blaser, H.; Raz, E.; Moens, C.B.; et al. A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell 2007, 129, 69–82. [Google Scholar] [CrossRef]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Guglas, K.; Kołodziejczak, I.; Kolenda, T.; Kopczyńska, M.; Teresiak, A.; Sobocińska, J.; Bliźniak, R.; Lamperska, K. YRNAs and YRNA-Derived Fragments as New Players in Cancer Research and Their Potential Role in Diagnostics. Int. J. Mol. Sci. 2020, 21, 5682. [Google Scholar] [CrossRef]
- Shigematsu, M.; Kirino, Y. tRNA-Derived Short Non-coding RNA as Interacting Partners of Argonaute Proteins. Gene Regul. Syst. Biol. 2015, 9, 27–33. [Google Scholar] [CrossRef]
- Lui, L.; Lowe, T. Small nucleolar RNAs and RNA-guided post-transcriptional modification. Essays Biochem. 2013, 54, 53–77. [Google Scholar] [CrossRef]
- Moyano, M.; Stefani, G. piRNA involvement in genome stability and human cancer. J. Hematol. Oncol. 2015, 8, 38. [Google Scholar] [CrossRef]
- Zheng, Y. Chapter 1—Introduction to Non-coding RNAs and High Throughput Sequencing. In Computational Non-Coding RNA Biology; Zheng, Y., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 3–31. [Google Scholar] [CrossRef]
- Chow, R.D.; Chen, S. Sno-derived RNAs are prevalent molecular markers of cancer immunity. Oncogene 2018, 37, 6442–6462. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Sun, Q.; Yao, Y. Characterization of Small RNAs Derived from tRNAs, rRNAs and snoRNAs and Their Response to Heat Stress in Wheat Seedlings. PLoS ONE 2016, 11, e0150933. [Google Scholar] [CrossRef]
- Abel, Y.; Rederstorff, M. SnoRNAs and the emerging class of sdRNAs: Multifaceted players in oncogenesis. Biochimie 2019, 164, 17–21. [Google Scholar] [CrossRef]
- Green, D.; Fraser, W.D.; Dalmay, T. Transfer RNA-derived small RNAs in the cancer transcriptome. Pflug. Arch. 2016, 468, 1041–1047. [Google Scholar] [CrossRef]
- Mleczko, A.M.; Machtel, P.; Walkowiak, M.; Wasilewska, A.; Pietras, P.J.; Bąkowska-Żywicka, K. Levels of sdRNAs in cytoplasm and their association with ribosomes are dependent upon stress conditions but independent from snoRNA expression. Sci. Rep. 2019, 9, 18397. [Google Scholar] [CrossRef]
- Guan, L.; Grigoriev, A. Computational meta-analysis of ribosomal RNA fragments: Potential targets and interaction mechanisms. Nucleic Acids Res. 2021, 49, 4085–4103. [Google Scholar] [CrossRef]
- Kowalski, M.P.; Krude, T. Functional roles of non-coding Y RNAs. Int. J. Biochem. Cell Biol. 2015, 66, 20–29. [Google Scholar] [CrossRef]
- Xie, Y.; Yao, L.; Yu, X.; Ruan, Y.; Li, Z.; Guo, J. Action mechanisms and research methods of tRNA-derived small RNAs. Signal Transduct. Target. Ther. 2020, 5, 109. [Google Scholar] [CrossRef]
- Gebetsberger, J.; Wyss, L.; Mleczko, A.M.; Reuther, J.; Polacek, N. A tRNA-derived fragment competes with mRNA for ribosome binding and regulates translation during stress. RNA Biol. 2017, 14, 1364–1373. [Google Scholar] [CrossRef]
- Kishore, S.; Khanna, A.; Zhang, Z.; Hui, J.; Balwierz, P.J.; Stefan, M.; Beach, C.; Nicholls, R.D.; Zavolan, M.; Stamm, S. The snoRNA MBII-52 (SNORD 115) is processed into smaller RNAs and regulates alternative splicing. Hum. Mol. Genet. 2010, 19, 1153–1164. [Google Scholar] [CrossRef]
- Ender, C.; Krek, A.; Friedländer, M.R.; Beitzinger, M.; Weinmann, L.; Chen, W.; Pfeffer, S.; Rajewsky, N.; Meister, G. A human snoRNA with microRNA-like functions. Mol. Cell 2008, 32, 519–528. [Google Scholar] [CrossRef]
- Plewka, P.; Szczesniak, M.W.; Stepien, A.; Zywicki, M.; Pacak, A.; Colombo, M.; Makalowska, I.; Ruepp, M.; Raczynska, K.D. FUS controls the processing of snoRNAs into smaller RNA fragments that can regulate gene expression. bioRxiv 2018. [Google Scholar] [CrossRef]
- Giraldez, M.D.; Spengler, R.M.; Etheridge, A.; Godoy, P.M.; Barczak, A.J.; Srinivasan, S.; De Hoff, P.L.; Tanriverdi, K.; Courtright, A.; Lu, S.; et al. Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nat. Biotechnol. 2018, 36, 746–757. [Google Scholar] [CrossRef]
- Raabe, C.A.; Tang, T.-H.; Brosius, J.; Rozhdestvensky, T.S. Biases in small RNA deep sequencing data. Nucleic Acids Res. 2014, 42, 1414–1426. [Google Scholar] [CrossRef]
- Olivares, D.; Perez-Hernandez, J.; Perez-Gil, D.; Chaves, F.J.; Redon, J.; Cortes, R. Optimization of small RNA library preparation protocol from human urinary exosomes. J. Transl. Med. 2020, 18, 132. [Google Scholar] [CrossRef]
- Plante, I.; Plé, H.; Landry, P.; Gunaratne, P.H.; Provost, P. Modulation of microRNA Activity by Semi-microRNAs. Front. Genet. 2012, 3, 99. [Google Scholar] [CrossRef]
- Lambert, M.; Benmoussa, A.; Diallo, I.; Ouellet-Boutin, K.; Dorval, V.; Majeau, N.; Joly-Beauparlant, C.; Droit, A.; Bergeron, A.; Têtu, B.; et al. Identification of Abundant and Functional dodecaRNAs (doRNAs) Derived from Ribosomal RNA. Int. J. Mol. Sci. 2021, 22, 9757. [Google Scholar] [CrossRef]
- Taft, R.J.; Glazov, E.A.; Lassmann, T.; Hayashizaki, Y.; Carninci, P.; Mattick, J.S. Small RNAs derived from snoRNAs. RNA 2009, 15, 1233–1240. [Google Scholar] [CrossRef]
- He, X.; Chen, X.; Zhang, X.; Duan, X.; Pan, T.; Hu, Q.; Zhang, Y.; Zhong, F.; Liu, J.; Zhang, H.; et al. An Lnc RNA (GAS5)/SnoRNA-derived piRNA induces activation of TRAIL gene by site-specifically recruiting MLL/COMPASS-like complexes. Nucleic Acids Res. 2015, 43, 3712–3725. [Google Scholar] [CrossRef]
- Feng, L.; Zhang, F.; Zhang, H.; Zhao, Y.; Meyers, B.C.; Zhai, J. An Online Database for Exploring Over 2,000 Arabidopsis Small RNA Libraries. Plant Physiol. 2020, 182, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.P.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J. ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef]
- Qian, F.-H.; Deng, X.; Zhuang, Q.-X.; Wei, B.; Zheng, D.-D. miR-625-5p suppresses inflammatory responses by targeting AKT2 in human bronchial epithelial cells. Mol. Med. Rep. 2019, 19, 1951–1957. [Google Scholar] [CrossRef]
- Yuan, J.; Xiao, C.; Lu, H.; Yu, H.; Hong, H.; Guo, C.; Wu, Z. Effect of miR-12136 on Drug Sensitivity of Drug-Resistant Cell Line Michigan Cancer Foundation-7/Doxorubicin by Regulating ATP Binding Cassette Subfamily B Member 1. J. Biomater. Tissue Eng. 2020, 10, 1431–1435. [Google Scholar] [CrossRef]
- Jadideslam, G.; Ansarin, K.; Sakhinia, E.; Babaloo, Z.; Abhari, A.; Ghahremanzadeh, K.; Khalili, M.; Radmehr, R.; Kabbazi, A. Diagnostic biomarker and therapeutic target applications of miR-326 in cancers: A systematic review. J. Cell. Physiol. 2019, 234, 21560–21574. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. miR-31: A crucial overseer of tumor metastasis and other emerging roles. Cell Cycle 2010, 9, 2124–2129. [Google Scholar] [CrossRef]
- Gainetdinov, I.; Skvortsova, Y.; Kondratieva, S.; Funikov, S.; Azhikina, T. Two modes of targeting transposable elements by piRNA pathway in human testis. RNA 2017, 23, 1614–1625. [Google Scholar] [CrossRef]
- Wu, X.; Pan, Y.; Fang, Y.; Zhang, J.; Xie, M.; Yang, F.; Yu, T.; Ma, P.; Li, W.; Shu, Y. The Biogenesis and Functions of piRNAs in Human Diseases. Mol. Ther. Nucleic Acids 2020, 21, 108–120. [Google Scholar] [CrossRef]
- Grimson, A.; Srivastava, M.; Fahey, B.; Woodcroft, B.; Chiang, H.R.; King, N.; Degnan, B.; Rokhsar, D.; Bartel, D.P. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature 2008, 455, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Perera, B.P.U.; Tsai, Z.T.-Y.; Colwell, M.; Jones, T.R.; Goodrich, J.M.; Wang, K.; Sartor, M.A.; Faulk, C.; Dolinoy, D.C. Somatic expression of piRNA and associated machinery in the mouse identifies short, tissue-specific piRNA. Epigenetics 2019, 14, 504–521. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Tan, W.; Zhou, Y. Transfer RNA-derived small RNAs: Potential applications as novel biomarkers for disease diagnosis and prognosis. Ann. Transl. Med. 2020, 8, 1092. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, T.; Suresh, P.S.; Tsutsumi, R. tRFs: miRNAs in disguise. Gene 2016, 579, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Hu, H.; Jiang, X.; Maierhofer, V.; Neb, E.; He, L.; Hu, Y.; Hu, H.; Li, N.; Chen, W.; et al. Widespread expression of piRNA-like molecules in somatic tissues. Nucleic Acids Res. 2011, 39, 6596–6607. [Google Scholar] [CrossRef]
- Pundhir, S.; Gorodkin, J. Differential and coherent processing patterns from small RNAs. Sci. Rep. 2015, 5, 12062. [Google Scholar] [CrossRef][Green Version]
- Scott, M.S.; Ono, M.; Yamada, K.; Endo, A.; Barton, G.J.; Lamond, A.I. Human box C/D snoRNA processing conservation across multiple cell types. Nucleic Acids Res. 2012, 40, 3676–3688. [Google Scholar] [CrossRef]
- López-Corral, L.; Mateos, M.V.; Corchete, L.A.; Sarasquete, M.E.; de la Rubia, J.; De Arriba, F.; Lahuerta, J.-J.; Garcia-Sanz, R.; Miguel, J.S.; Gutierrez, N.; et al. Genomic analysis of high-risk smoldering multiple myeloma. Haematologica 2012, 97, 1439–1443. [Google Scholar] [CrossRef]
- Chuang, T.-D.; Xie, Y.; Yan, W.; Khorram, O. Next-generation sequencing reveals differentially expressed small noncoding RNAs in uterine leiomyoma. Fertil. Steril. 2018, 109, 919–929. [Google Scholar] [CrossRef]
- Ma, X.; Liu, C.; Kong, X.; Liu, J.; Zhang, S.; Liang, S.; Luan, W.; Cao, X. Extensive profiling of the expressions of tRNAs and tRNA-derived fragments (tRFs) reveals the complexities of tRNA and tRF populations in plants. Sci. China Life Sci. 2021, 64, 495–511. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, M.; Hu, C. Exosomal transfer of miR-214 mediates gefitinib resistance in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2018, 507, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Magee, R.; Rigoutsos, I. On the expanding roles of tRNA fragments in modulating cell behavior. Nucleic Acids Res. 2020, 48, 9433–9448. [Google Scholar] [CrossRef] [PubMed]
- Cherlin, T.; Magee, R.; Jing, Y.; Pliatsika, V.; Loher, P.; Rigoutsos, I. Ribosomal RNA fragmentation into short RNAs (rRFs) is modulated in a sex- and population of origin-specific manner. BMC Biol. 2020, 18, 38. [Google Scholar] [CrossRef] [PubMed]
- Fowler, E.K.; Mohorianu, I.; Smith, D.T.; Dalmay, T.; Chapman, T. Small RNA populations revealed by blocking rRNA fragments in Drosophila melanogaster reproductive tissues. PLoS ONE 2018, 13, e0191966. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L. Molecular Cell Biology. In Processing of rRNA and tRNA, 4th ed.; WH Freeman: New York, NY, USA, 2000. [Google Scholar]
- Jackowiak, P.; Nowacka, M.; Strozycki, P.M.; Figlerowicz, M. RNA degradome—Its biogenesis and functions. Nucleic Acids Res. 2011, 39, 7361–7370. [Google Scholar] [CrossRef]
- Lambert, M.; Benmoussa, A.; Provost, P. A New Specific and Sensitive RT-qPCR Method Based on Splinted 5’ Ligation for the Quantitative Detection of RNA Species Shorter than microRNAs. Non-Coding RNA 2021, 7, 59. [Google Scholar] [CrossRef]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef]
- Wright, C.; Rajpurohit, A.; Burke, E.E.; Williams, C.; Collado-Torres, L.; Kimos, M.; Brandon, N.J.; Cross, A.J.; Jaffe, A.E.; Weinberger, D.R.; et al. Comprehensive assessment of multiple biases in small RNA sequencing reveals significant differences in the performance of widely used methods. BMC Genom. 2019, 20, 513. [Google Scholar] [CrossRef]
- Li, Z.; Kim, S.W.; Lin, Y.; Moore, P.S.; Chang, Y.; John, B. Characterization of Viral and Human RNAs Smaller than Canonical MicroRNAs. J. Virol. 2009, 83, 12751–12758. [Google Scholar] [CrossRef]
- Laffont, B.; Corduan, A.; Rousseau, M.; Duchez, A.C.; Lee, C.H.C.; Boilard, E.; Provost, P. Platelet microparticles reprogram macrophage gene expression and function. Thromb. Haemost. 2016, 115, 311–323. [Google Scholar] [CrossRef]
- Duchez, A.-C.; Boudreau, L.H.; Naika, G.S.; Bollinger, J.; Belleannée, C.; Cloutier, N.; Laffont, B.; Mendoza-Villarroel, R.E.; Levesque, T.; Rollet-Labelle, E.; et al. Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc. Natl. Acad. Sci. USA 2015, 112, E3564–E3573. [Google Scholar] [CrossRef] [PubMed]
- Assefa, A.T.; Vandesompele, J.; Thas, O. On the utility of RNA sample pooling to optimize cost and statistical power in RNA sequencing experiments. BMC Genom. 2020, 21, 312. [Google Scholar] [CrossRef] [PubMed]
- Benmoussa, A.; Laugier, J.; Beauparlant, C.J.; Lambert, M.; Droit, A.; Provost, P. Complexity of the microRNA transcriptome of cow milk and milk-derived extracellular vesicles isolated via differential ultracentrifugation. J. Dairy Sci. 2020, 103, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.P.; Young, P.E.; McCorkindale, A.L.; Dang, T.H.; Clancy, J.L.; Humphreys, D.; Preiss, T.; Hutvagner, G.; Martin, D.I.; Cropley, J.E.; et al. The human Piwi protein Hiwi2 associates with tRNA-derived piRNAs in somatic cells. Nucleic Acids Res. 2014, 42, 8984–8995. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length (nt) | Sequence | Reads * | Origin | ||
|---|---|---|---|---|---|
| H. sapiens | HUVEC | 17 | GTTTGTGATGACTTACA | 99.9 | 5′ end of SNORD30 |
| 29 | TTGCTGTGATGACTATCTTAGGACACCTT | 94.4 | 5′ end of SNORD58C | ||
| 29 | CTGCAGTGATGACTTTCTTAGGACACCTT | 4.4 | 5′ end of SNORD58A | ||
| PMN | 17 | GTTTGTGATGACTTACA | 99.9 | 5′ end of SNORD30 | |
| 29 | TTGCTGTGATGACTATCTTAGGACACCTT | 54.2 | 5′ end of SNORD58C | ||
| 29 | CTGCAGTGATGACTTTCTTAGGACACCTT | 29.7 | 5′ end of SNORD58A | ||
| HEK293 | 17 | GTTTGTGATGACTTACA | 99.9 | 5′ end of SNORD30 | |
| 29 | TTGCTGTGATGACTATCTTAGGACACCTT | 94.6 | 5′ end of SNORD58C | ||
| 29 | CTGCAGTGATGACTTTCTTAGGACACCTT | 4.7 | 5′ end of SNORD58A | ||
| M. musculus | Cerebellum | 17 | GTTCTGTGATGAGGCTC | 96 | 5′ end of SNORD83B, without the 3 first nt |
| 29 | TTGCTGTGATGACTATCTTAGGACACCTT | 64 | 5′ end of SNORD58, without the 3 first nt | ||
| 29 | CTGCAGTGATGACTATCTTAGGACACCTT | 17 | 5′ end of SNORD58, without the 3 first nt | ||
| PMN | 17 | GTTCTGTGATGAGGCTC | 98 | 5′ end of SNORD83B, without the 3 first nt | |
| 29 | TTGCTGTGATGACTATCTTAGGACACCTT | 73 | 5′ end of SNORD58, without the 3 first nt | ||
| 29 | CTGCAGTGATGACTATCTTAGGACACCTT | 13 | 5′ end of SNORD58, without the 3 first nt | ||
| NIH | 17 | GTTCTGTGATGAGGCTC | 99 | 5′ end of SNORD83B, without the 3 first nt | |
| 29 | TTGCTGTGATGACTATCTTAGGACACCTT | 47 | 5′ end of SNORD58, without the 3 first nt | ||
| 29 | CTGCAGTGATGACTATCTTAGGACACCTT | 35 | 5′ end of SNORD58, without the 3 first nt | ||
| N2a | 17 | GTTCTGTGATGAGGCTC | 99 | 5′ end of SNORD83B, without the 3 first nt | |
| 29 | TTGCTGTGATGACTATCTTAGGACACCTT | 45 | 5′ end of SNORD58, without the 3 first nt | ||
| 29 | CTGCAGTGATGACTATCTTAGGACACCTT | 26 | 5′ end of SNORD58, without the 3 first nt |
| Length (nt) | Sequence | Origin (piRBase Name) | ||
|---|---|---|---|---|
| H. sapiens | HUVEC | 15 | GACCAATGATGTGAA | piR-hsa-4433698 5′ end |
| 23 | TCCTGTACTGAGCTGCCCCGAGA | piR-hsa-145507 | ||
| 23 | TCCTGTACTGAGCTGCCCCGAGT | piR-hsa-145507 | ||
| PMN | 15 | TACAACTTTTGGCAA | piR-hsa-7695930 3′ end | |
| 14 | ACAACTTTTGGCAA | piR-hsa-7695930 3′ end | ||
| 23 | TATTGCACTTGTCCCGGCCTGTA | piR-hsa-137098 | ||
| HEK293 | 14 | GATGGGTGACCGCC | piR-hsa-741077 fragment | |
| 13 | ATGGGTGACCGCC | piR-hsa-741077 fragment | ||
| 23 | TCCTGTACTGAGCTGCCCCGAGA | piR-hsa-145507 | ||
| M. musculus | Cerebellum | 15 | GCATTGGTGGTTCAG | piR-mmu-10912946 5′ end |
| 18 | GCATTGGTGGTTCAGTGG | piR-mmu-10912946 5′ end | ||
| 23 | AACCCGTAGATCCGAACTTGTGA | piR-mmu-29307247 5′ end | ||
| 23 | TCCTGTACTGAGCTGCCCCGAGA | piR-mmu-25873647 5′ end | ||
| PMN | 15 | GCATTGGTGGTTCAG | piR-mmu-10912946 5′ end | |
| 16 | AGCGGAGTAGAGCAGT | piR-mmu-23655655 5′ end | ||
| 23 | TCCTGTACTGAGCTGCCCCGAGA | piR-mmu-25873647 5′ end | ||
| 23 | TCCTGTACTGAGCTGCCCCGAGT | piR-mmu-25873647 5′ end | ||
| 22 | CCTGTACTGAGCTGCCCCGAGA | piR-mmu-25873647 5′ end | ||
| 23 | GTACCCTGTAGATCCGAATTTGT | piR-mmu-11542414 | ||
| NIH/3T3 | 16 | AGCGGAGTAGAGCAGT | piR-mmu-23655655 5′ end | |
| 22 | CCTGTACTGAGCTGCCCCGAGA | piR-mmu-25873647 5′ end | ||
| 23 | TCCTGTACTGAGCTGCCCCGAGA | piR-mmu-25873647 5′ end | ||
| 24 | GTCCTGTACTGAGCTGCCCCGAGA | piR-mmu-25873647 5′ end | ||
| N2a | 12 | TCGCTGTGATGA | piR-mmu-24106721 | |
| 23 | CACCCGTAGAACCGACCTTGCGT | piR-mmu-31228201 5′ end | ||
| 27 | GGCTCTGTGGCGCAATGGATAGCGCAT | piR-mmu-5102689 | ||
| 28 | TGGCCAAGGATGAGAACTCTAACCTGAC | piR-mmu-7884931 | ||
| D. melanogaster | 13 | GAGGAAACTCTGG | piR-dme-108681 5′ end | |
| 15 | AAGGGAAGGGTATTG | piR-dme-5048778 5′ end | ||
| 16 | AAAGGGAAGGGTATTG | piR-dme-5048778 5′ end | ||
| 18 | CTGGGTCGGCCGGGGCGC | piR-dme-34359551 fragment | ||
| 20 | TAGGGACGGTCGGGGGCATC | piR-dme-40694119 3′ end | ||
| 21 | ATAGGGACGGTCGGGGGCATC | piR-dme-40694119 3′ end |
| Length (nt) | Sequence | Reads * | Origin | ||
|---|---|---|---|---|---|
| H. sapiens | HUVEC | 9 | GGCTAATGA | 97.9 | 5′ end of SNORD-like-snoRNA,alias:ZL45, ID:snoID_0724, without the 3 first nt |
| 12 | TCGCTATGATGA | 36.9 | 5′ end of SNORD14B | ||
| 10 | GGACCAATGA | 96.0 | 5′ end of SNORD114-12 | ||
| PMN | 9 | GGCTAATGA | 99.7 | 5′ end of SNORD-like-snoRNA,alias:ZL45, ID:snoID_0724, without the 3 first nt | |
| 12 | TCGCTATGATGA | 23.9 | 5′ end of SNORD14B | ||
| 11 | CCCGTCTGACC | 22.0 | 3′ end of SNORD13 | ||
| HEK293 | 9 | GGCTAATGA | 100,0 | 5′ end of SNORD-like-snoRNA,alias:ZL45, ID:snoID_0724, without the 3 first nt | |
| 10 | TGGCTAATGA | 45,2 | 5′ end of SNORD-like-snoRNA,alias:ZL45, ID:snoID_0724, without the 2 first nt | ||
| 11 | GTAAGTATATT | 41,4 | Middle of SNORA24L2 | ||
| M. musculus | Cerebellum | 11 | CGCTGTGATGA | 32.6 | 5′ end of SNORD14C, without the first nt |
| 9 | ATTGAGGAC | 7.9 | CD_40-1_ (chr16) 20684238,20684314 | ||
| 12 | AATTGTGGTAAC | 13.6 | Middle of SCARNA10 | ||
| PMN | 11 | CGCTGTGATGA | 37.9 | 5′ end of SNORD14C, without the first nt | |
| 12 | AATTGTGGTAAC | 8.3 | Middle of SCARNA10 | ||
| 11 | ATTGTGGTAAC | 11.3 | Middle of SCARNA10 | ||
| NIH/3T3 | 11 | CGCTGTGATGA | 19.5 | 5′ end of SNORD14C, without the first nt | |
| 11 | AGAGAGGTGAG | 18.1 | Middle of SNORA17 | ||
| 12 | TGCTGTGATGAC | 39.6 | 5′ end of SNORD58C, without the first nt | ||
| N2a | 11 | CGCTGTGATGA | 80.4 | 5′ end of SNORD14C, without the first nt | |
| 12 | AGGGATTGTGGG | 28.2 | 5′ end of SNORA71 | ||
| 10 | GCGGGTGTGG | 24.1 | SNORA74B | ||
| D. melanogaster | 12 | GTGGAGGTAAAG | 98.0 | 5′ end snoRNA:Psi18S-525f | |
| 9 | ATAGGGACG | 71.3 | snoRNA:Psi18S-525k (Dmel_CR34569) | ||
| 10 | TTATAAACTG | 43.7 | PsiU2-38.40.42 (scaRNA:PsiU2-38.40.42) | ||
| A. thaliana | 11 | AGATATGATGA | 95.1 | 5′ end of SnoR18a | |
| 10 | AATATTGAAA | 31.4 | Middle of SnoR96 | ||
| 11 | TAATATTGAAA | 1.4 | Middle of SnoR96 | ||
| S. cerevisae | 10 | CCTTCTGAAA | 22.1 | SnoRNA86 | |
| 11 | TCCTTCTGAAA | 26.6 | SnoRNA86 | ||
| 10 | TCGGGGCTGA | 11.4 | SnoRNA86 | ||
| S. pombe | 9 | TCAACTGTA | 28.0 | SnR70 | |
| 10 | TGTTCTGATG | 35.5 | SnR81 | ||
| 9 | TGTCTGATC | 6.7 | Snr41 |
| Length (nt) | Sequence | Origin | ||
|---|---|---|---|---|
| H. sapiens | HUVEC | 18 | GCATTGGTGGTTCAGTGG | 5′ end of tRNA-Gly-GCC-3-1 |
| 18 | GCATGGGTGGTTCAGTGG | 5′ end of tRNA-Gly-GCC-3-1 | ||
| 15 | GCATTGGTGGTTCAG | 5′ end of tRNA-Gly-GCC-3-1 | ||
| PMN | 18 | GCATTGGTGGTTCAGTGG | 5′ end of tRNA-Gly-GCC-3-1 | |
| 15 | GCATTGGTGGTTCAG | 5′ end of tRNA-Gly-GCC-3-1 | ||
| 14 | TAGAATTCTCGCCT | Middle of tRNA-Gly-CCC-1-1 | ||
| HEK293 | 15 | GCATTGGTGGTTCAG | 5′ end of tRNA-Gly-GCC-3-1 | |
| 18 | GCATTGGTGGTTCAGTGG | 5′ end of tRNA-Gly-GCC-3-1 | ||
| 18 | GCATGGGTGGTTCAGTGG | 5′ end of tRNA-Gly-GCC-3-1 | ||
| M. musculus | Cerebellum | 18 | GCATTGGTGGTTCAGTGG | 5′ end of tRNA-Gly-GCC-3-1 |
| 15 | GCATTGGTGGTTCAG | 5′ end of tRNA-Gly-GCC-3-1 | ||
| 14 | CTTCGTGGTCGCCA | Partial 3035a trf-3 | ||
| PMN | 18 | GCATTGGTGGTTCAGTGG | 5′ end of tRNA-Gly-GCC-3-1 | |
| 17 | CATTGGTGGTTCAGTGG | 5′ end of tRNA-Gly-GCC-3-1 | ||
| 15 | GCATTGGTGGTTCAG | 5′ end of tRNA-Gly-GCC-3-1 | ||
| NIH/3T3 | 18 | GCATTGGTGGTTCAGTGG | 5′ end of tRNA-Gly-GCC-3-1 | |
| 15 | GCATTGGTGGTTCAG | 5′ end of tRNA-Gly-GCC-3-1 | ||
| N2a | 18 | GCATTGGTGGTTCAGTGG | 5′ end of tRNA-Gly-GCC-3-1 | |
| 15 | GCATTGGTGGTTCAG | 5′ end of tRNA-Gly-GCC-3-1 | ||
| D. melanogaster | 30 | CATCGGTGGTTCAGTGGTAGAATGCTCGCC | 5′ end of tRNA-Gly-GCC-3-1 | |
| 28 | GCATCGGTGGTTCAGTGGTAGAATGCTC | 5′ end of tRNA-Gly-GCC-3-1 | ||
| 17 | CCCGGGTTTCGGCACCA | 3023 trf-3 | ||
| A. thaliana | 15 | GGCTAGGTAACATAA | PT-261581 tRF-5 | |
| 16 | GGGGATGTAGCTCATA | 5′ end of tRNA-Ala-CGC-2-1 | ||
| 16 | GGCGGATGTAGCCAAG | PT-218828 tRF-5 | ||
| S. cerevisae | 13 | GCGGATTTAGCTC | trna9-PheGAA | |
| 13 | GCTTCAGTAGCTC | trna19-MetCAT | ||
| 28 | TCCTTAGTTCGATCCTGAGTGCGAGCTC | tRNA-Cys-GCA-1-1 | ||
| 29 | TCCGTGATAGTTTAATGGTCAGAATGGGC | trna1-AspGTC | ||
| S. pombe | 8 | GCTTCAGT | trna49-LeuCAG | |
| 8 | GCGGATTT | trna17-SerGCT | ||
| 10 | CCCTGGGTTC | trna15-AlaTGC |
| Length (nt) | Sequence | ||
|---|---|---|---|
| H. sapiens | HUVEC | 18 | TCGTACGACTCTTAGCGG |
| 19 | CTCGTACGACTCTTAGCGG | ||
| 18 | TCGTACGACTCTTAGCGG | ||
| 12 | GACTCTTAGCGG | ||
| 13 | CGACTCTTAGCGG | ||
| PMN | 12 | GACTCTTAGCGG | |
| 13 | CGACTCTTAGCGG | ||
| 18 | TCGTACGACTCTTAGCGG | ||
| HEK293 | 12 | GACTCTTAGCGG | |
| 13 | CGACTCTTAGCGG | ||
| 18 | TCGTACGACTCTTAGCGG | ||
| M. musculus | Cerebellum | 12 | GACTCTTAGCGG |
| 13 | CGACTCTTAGCGG | ||
| 25 | CAAACGAGAACTTTGAAGGCCGAAG | ||
| PMN | 12 | GACTCTTAGCGG | |
| 13 | CGACTCTTAGCGG | ||
| 18 | CGATACGACTCTTAGCGG | ||
| NIH | 12 | GACTCTTAGCGG | |
| 13 | CGACTCTTAGCGG | ||
| 18 | CGATACGACTCTTAGCGG | ||
| N2a | 12 | GACTCTTAGCGG | |
| 13 | CGACTCTTAGCGG | ||
| 18 | CGATACGACTCTTAGCGG | ||
| D. melanogaster | 11 | ACTCTAAGCGG | |
| 12 | AACTCTAAGCGG | ||
| 30 | TGCTTGGACTACATATGGTTGAGGGTTGTA | ||
| A. thaliana | 12 | GAGTCTGGTAAT | |
| 14 | GGGATGGGTCGGCC | ||
| 18 | TAGGATAGTGGCCTACCA | ||
| S. cerevisae | 13 | TTGACCTCAAATC | |
| 18 | TATCTGGTTGATCCTGCC | ||
| 19 | GCGGCTGTCTGATCAGGCA | ||
| S. pombe | 13 | TAAAACTTTCAGC | |
| 13 | TTGACCTCAAATC | ||
| 24 | TTTGACCTCAAATCAGGTAGGACT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lambert, M.; Guellal, S.; Ho, J.; Benmoussa, A.; Laffont, B.; Bélanger, R.; Provost, P. An Expanded Landscape of Unusually Short RNAs in 11 Samples from Six Eukaryotic Organisms. Non-Coding RNA 2022, 8, 34. https://doi.org/10.3390/ncrna8030034
Lambert M, Guellal S, Ho J, Benmoussa A, Laffont B, Bélanger R, Provost P. An Expanded Landscape of Unusually Short RNAs in 11 Samples from Six Eukaryotic Organisms. Non-Coding RNA. 2022; 8(3):34. https://doi.org/10.3390/ncrna8030034
Chicago/Turabian StyleLambert, Marine, Sara Guellal, Jeffrey Ho, Abderrahim Benmoussa, Benoit Laffont, Richard Bélanger, and Patrick Provost. 2022. "An Expanded Landscape of Unusually Short RNAs in 11 Samples from Six Eukaryotic Organisms" Non-Coding RNA 8, no. 3: 34. https://doi.org/10.3390/ncrna8030034
APA StyleLambert, M., Guellal, S., Ho, J., Benmoussa, A., Laffont, B., Bélanger, R., & Provost, P. (2022). An Expanded Landscape of Unusually Short RNAs in 11 Samples from Six Eukaryotic Organisms. Non-Coding RNA, 8(3), 34. https://doi.org/10.3390/ncrna8030034