
Truncated Isoforms of lncRNA ANRIL Are Overexpressed in Bladder Cancer, But Do Not Contribute to Repression of INK4 Tumor Suppressors

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
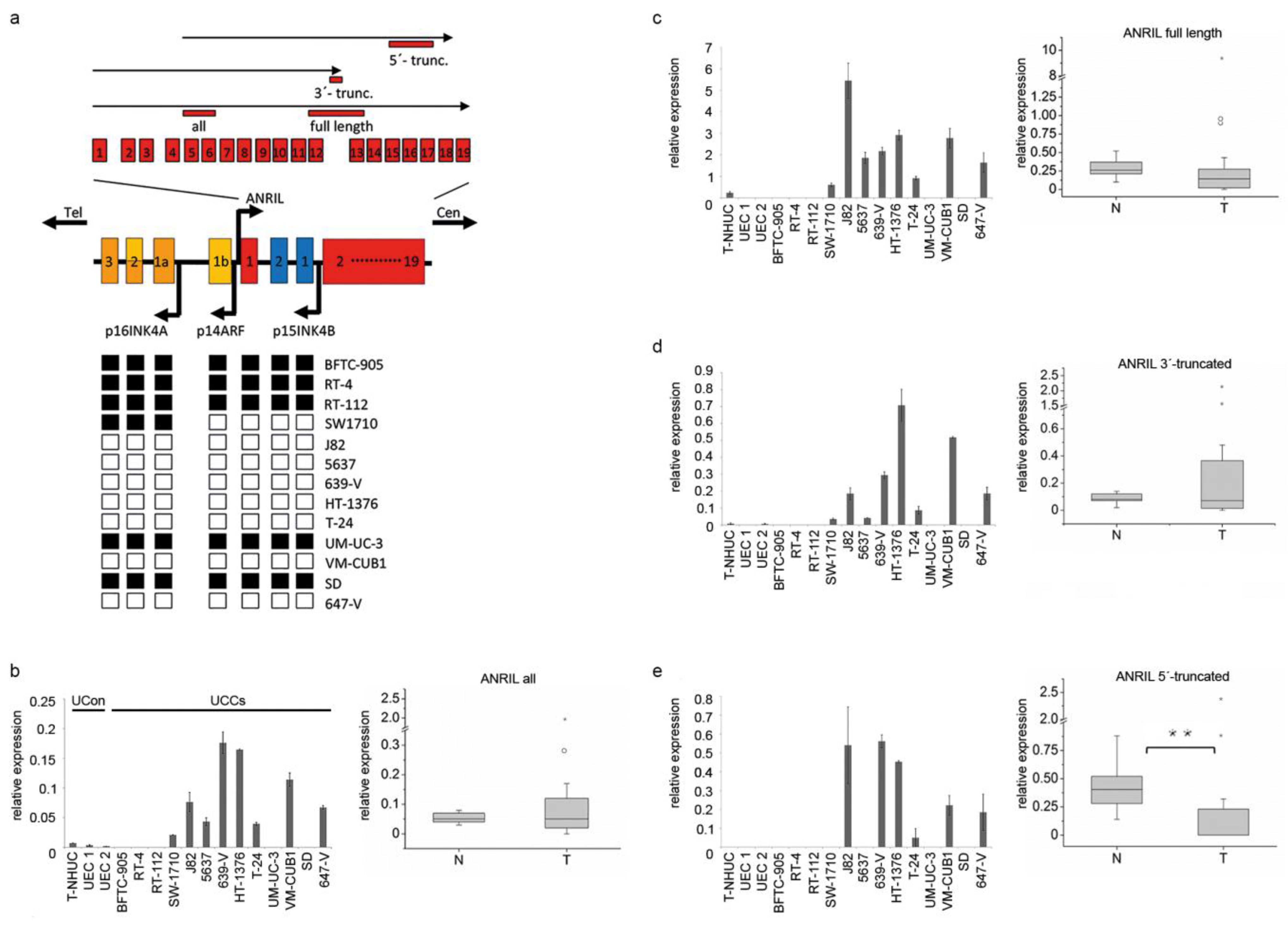
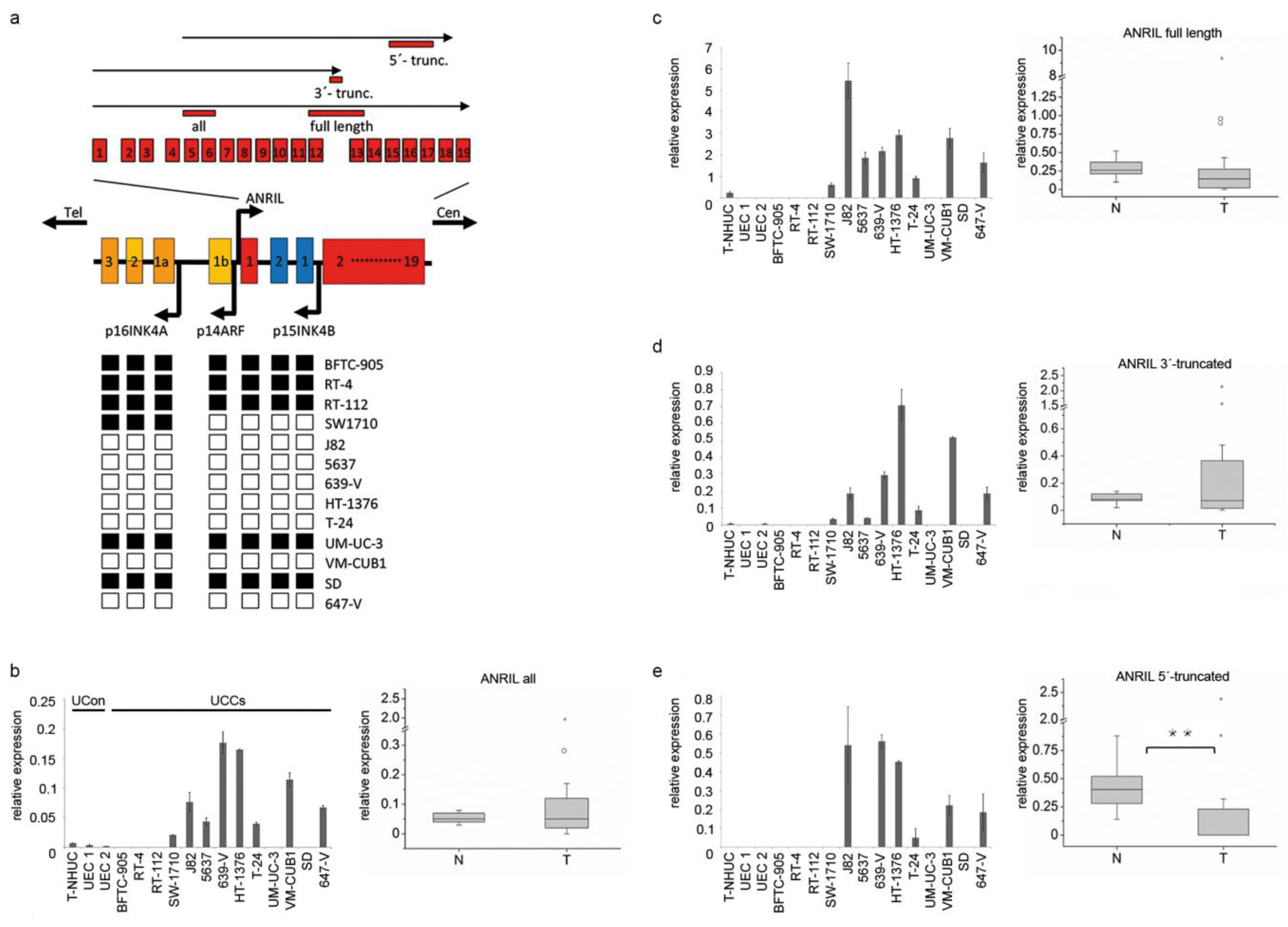
2.1. 3’-Truncated Isoforms of ANRIL Are Overexpressed in UC
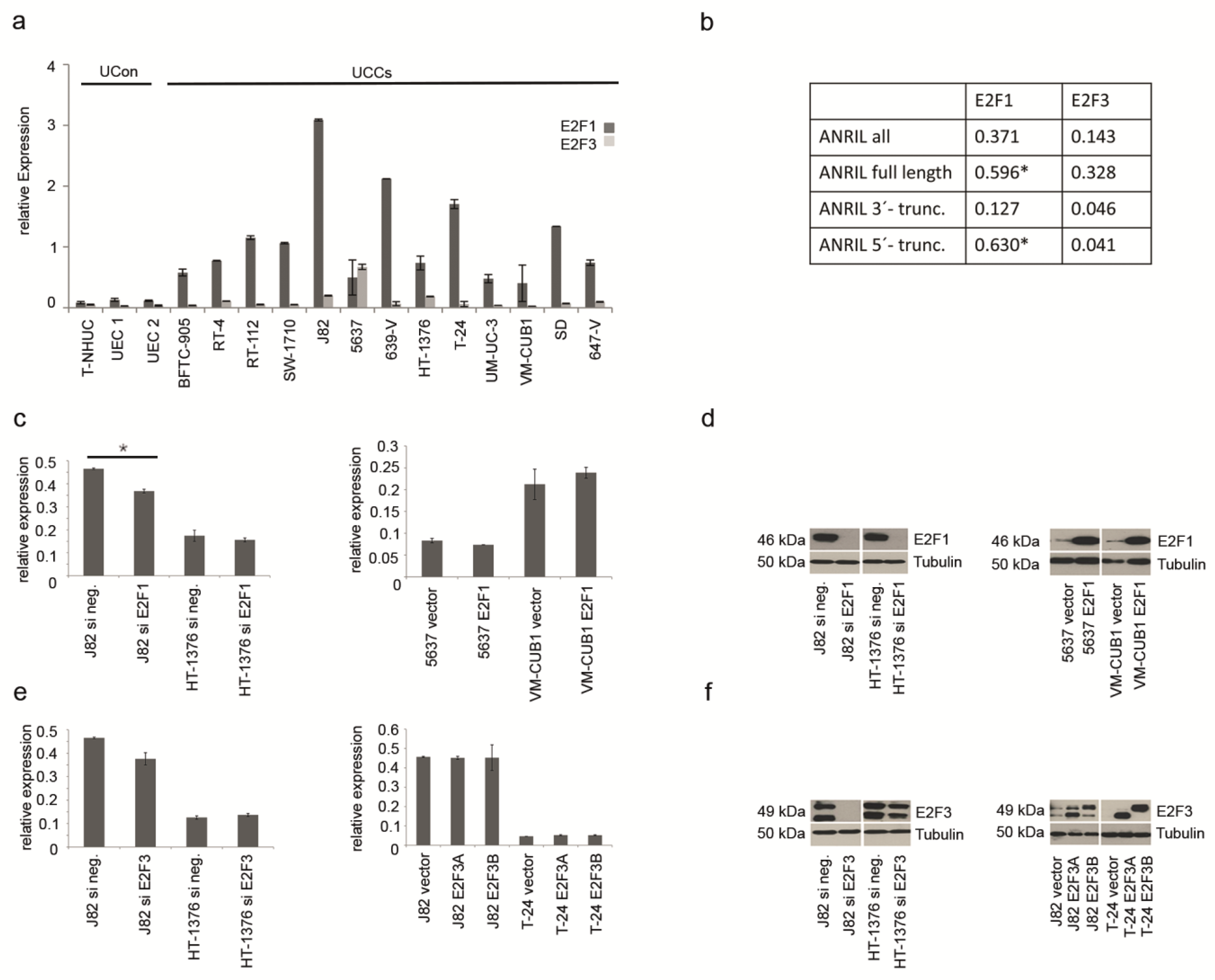
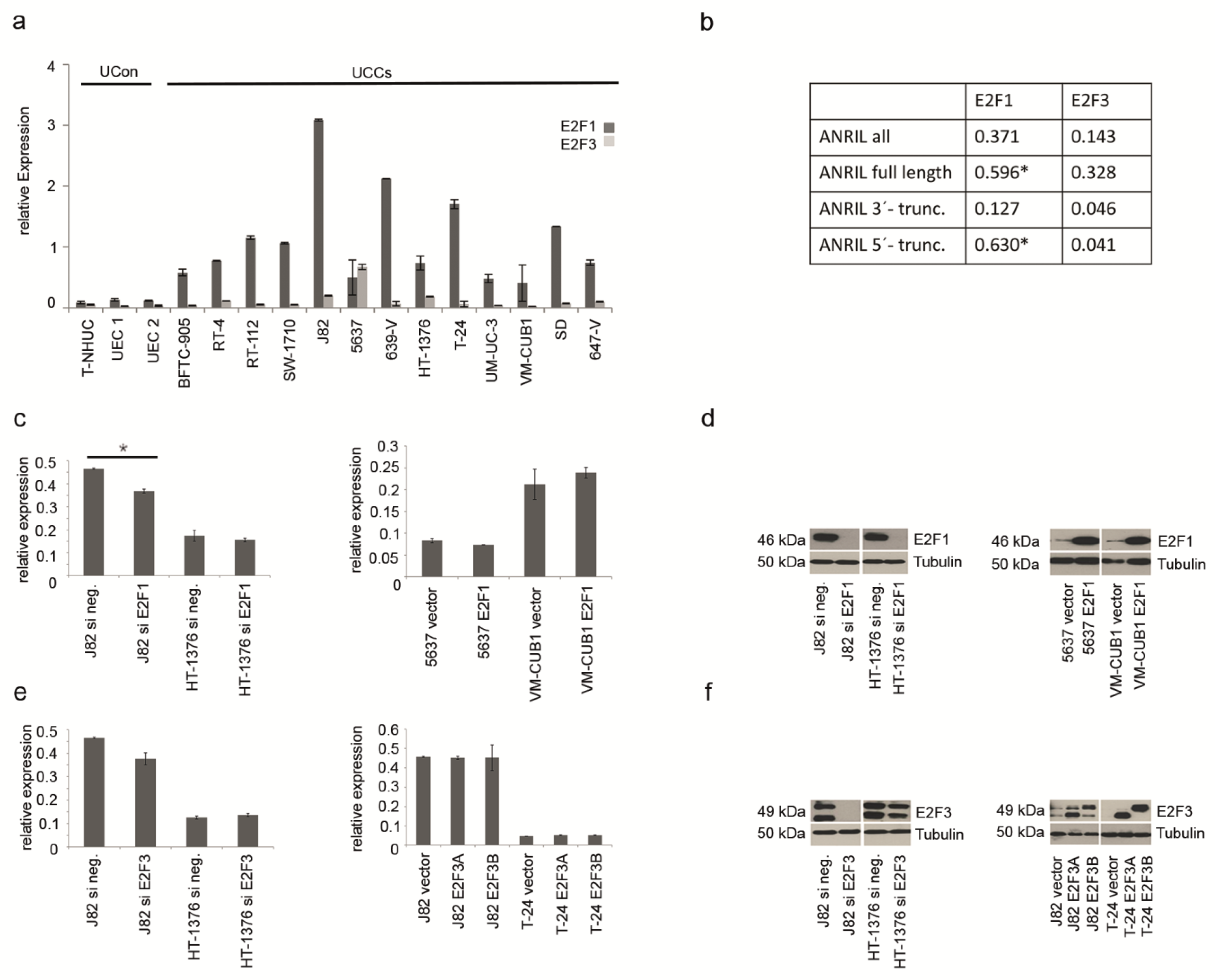
2.2. ANRIL Expression in UC Can Be Stimulated by E2F1, But Not by E2F3
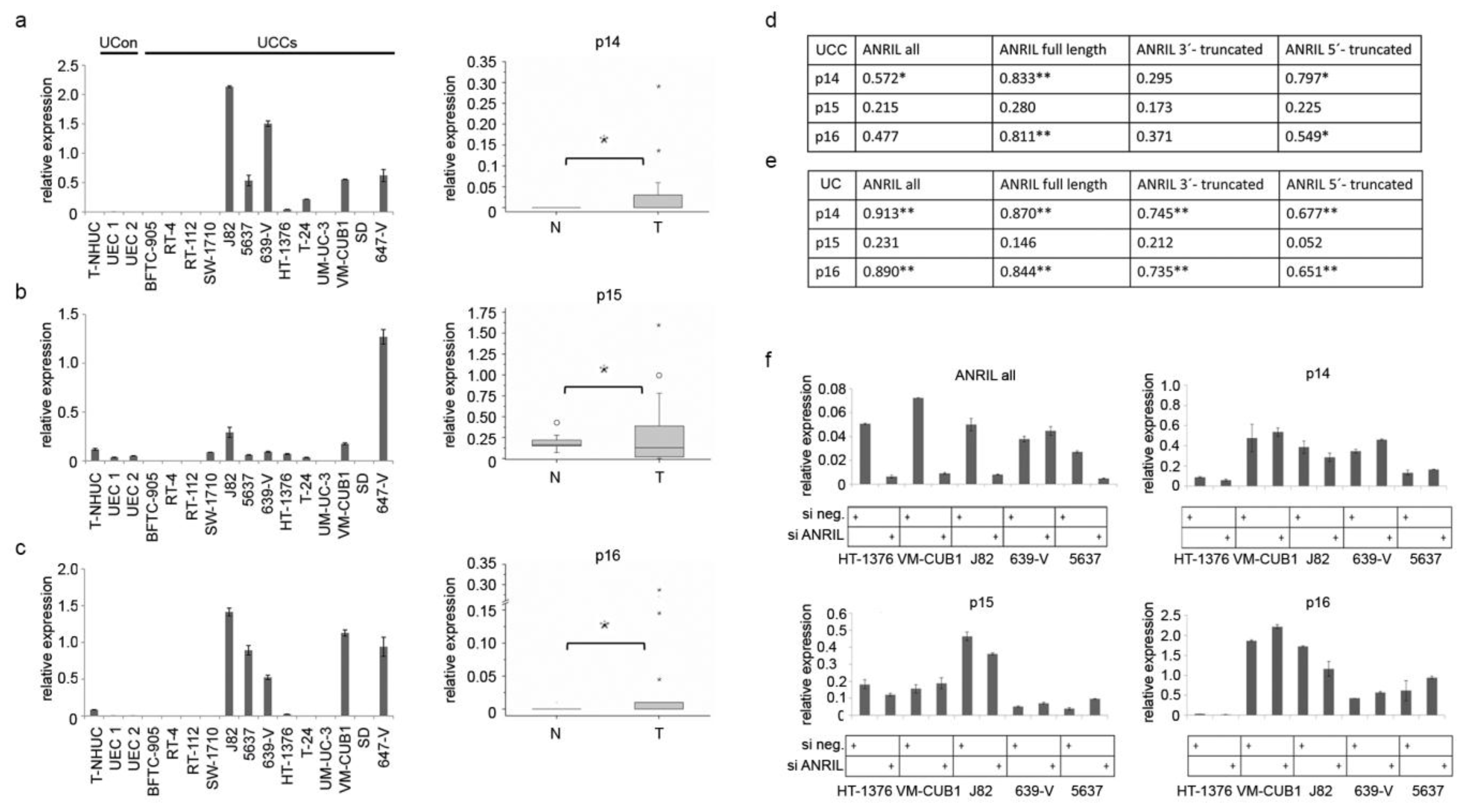
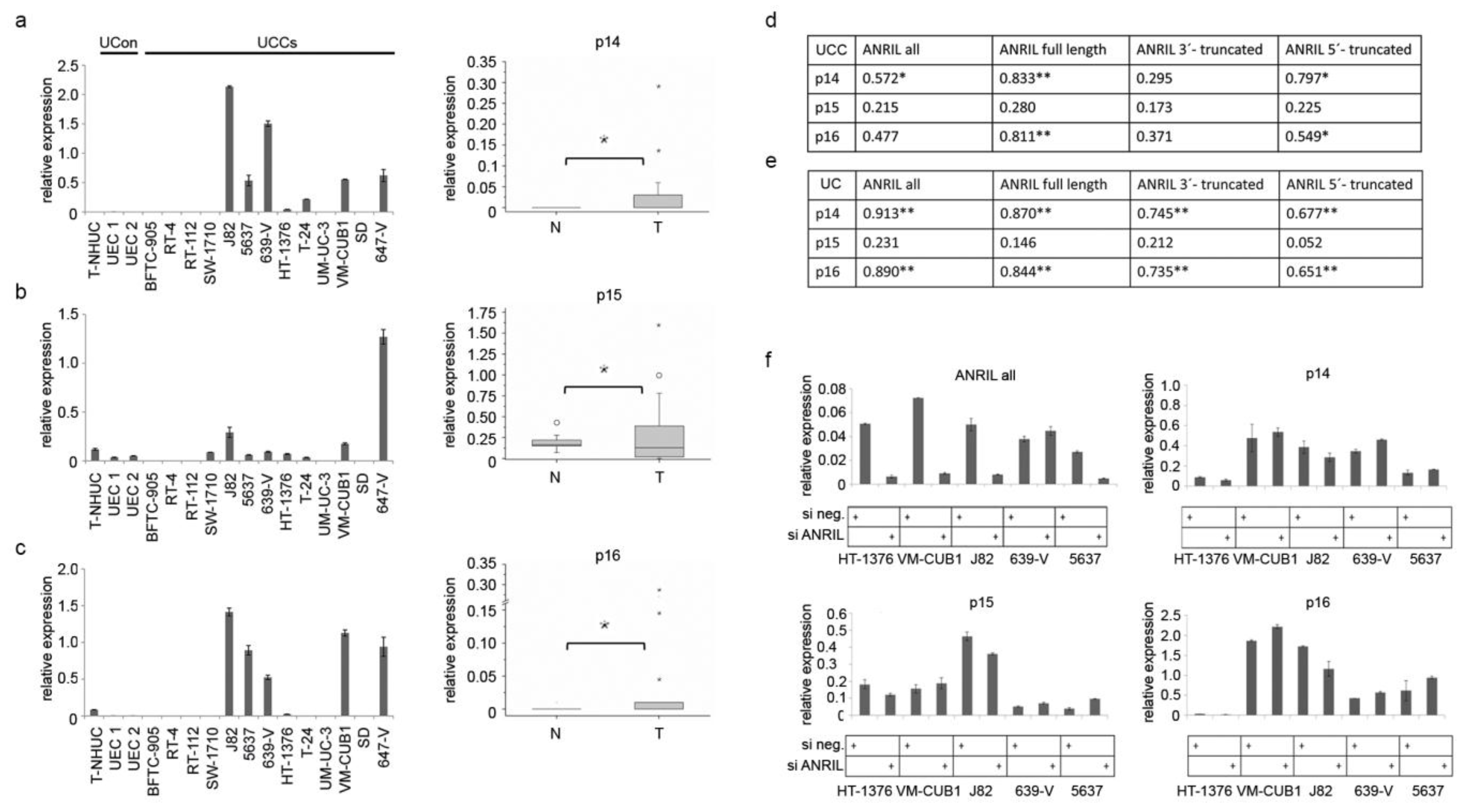
2.3. ANRIL Is Not Involved in Epigenetic Silencing of the INK4 Tumor Suppressor Locus in UC
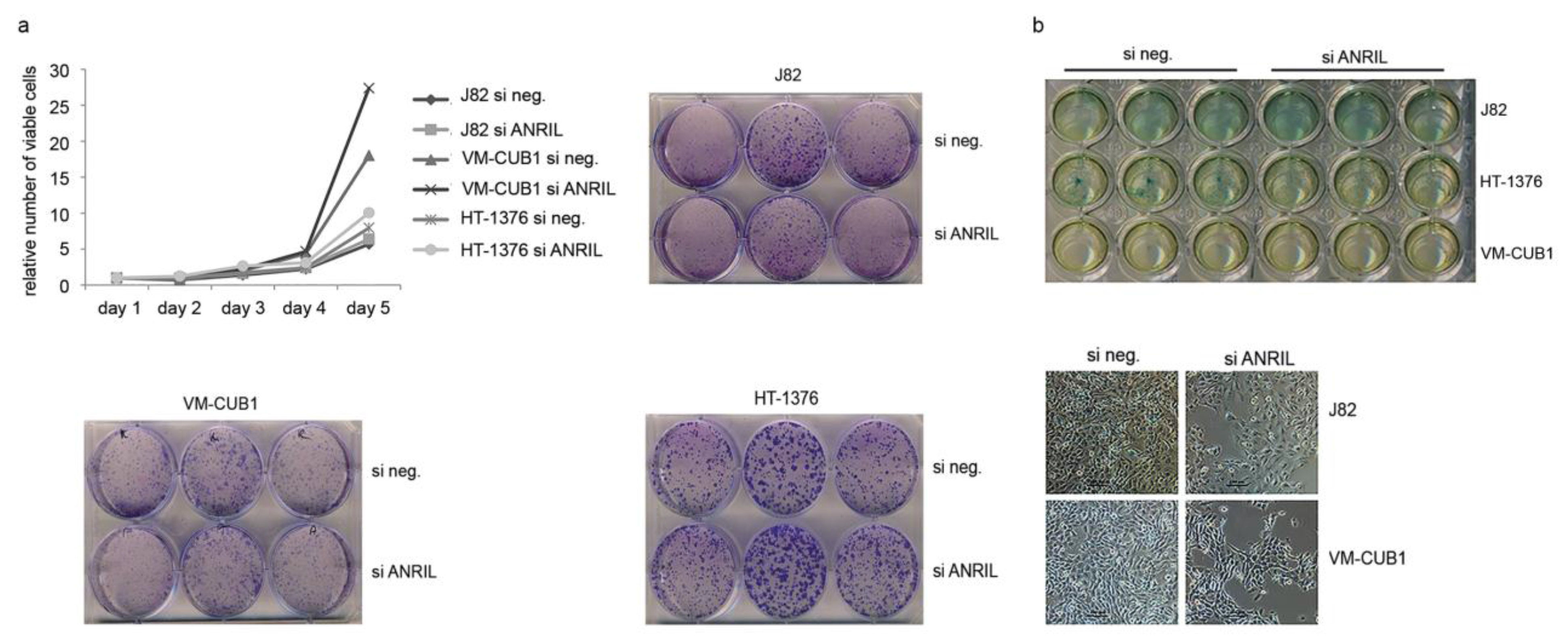
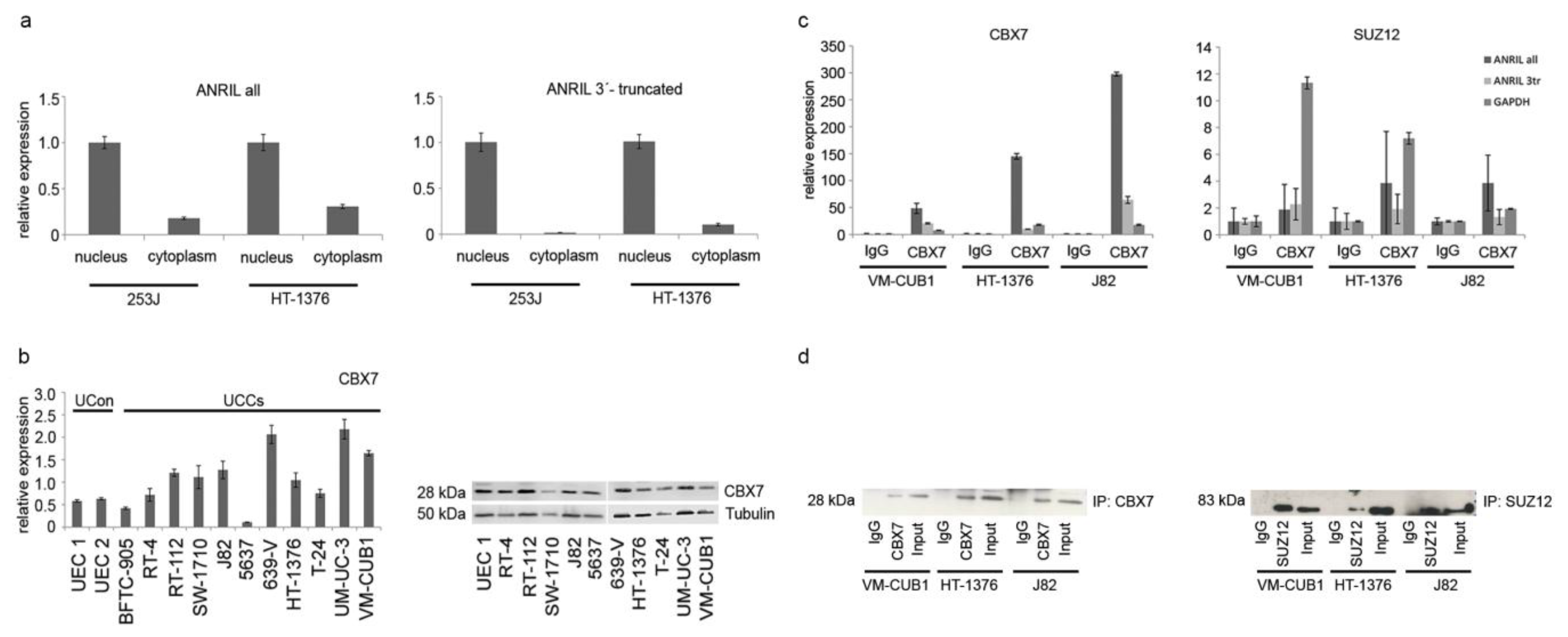
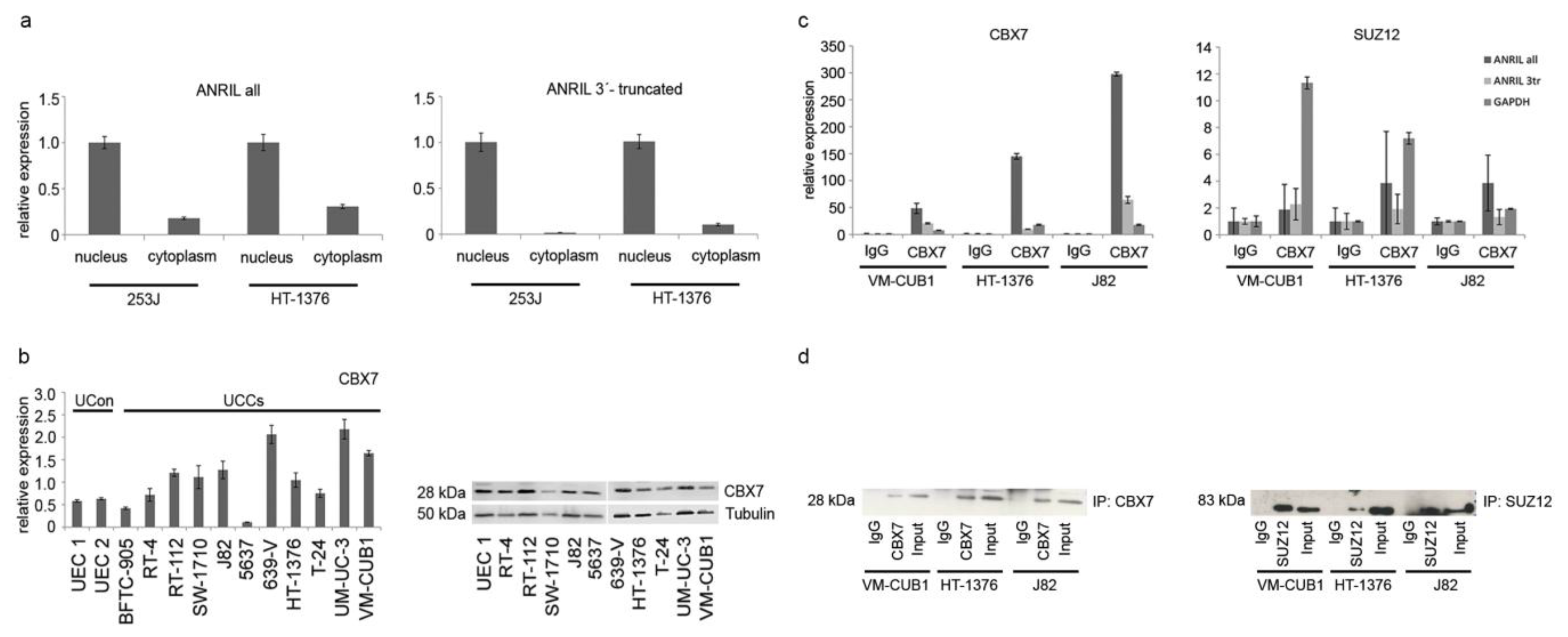
2.4. ANRIL Isoforms Interact with PcG Protein CBX7 in UC Cells, But Not with SUZ12
3. Discussion
4. Conclusions
5. Material and Methods
5.1. Patients and Tissues
5.2. Cell Culture and Transfection Experiments
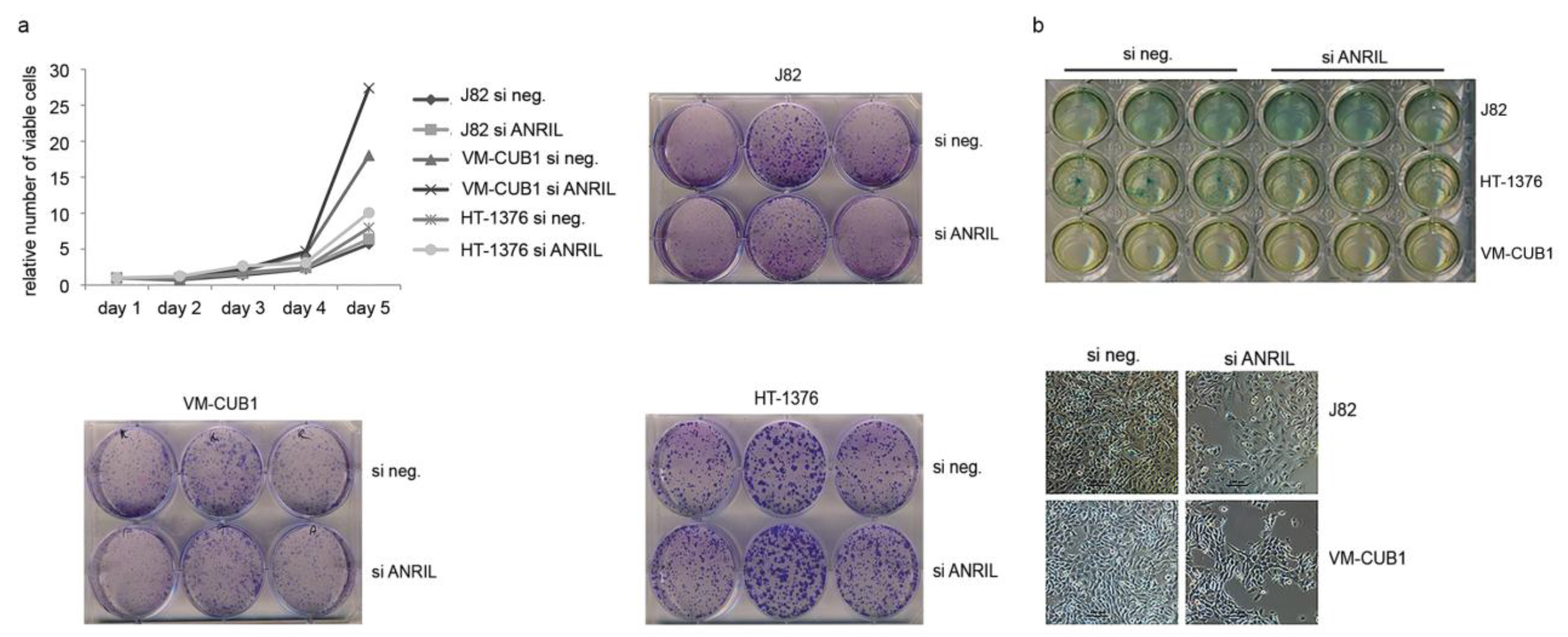
5.3. Assays for Cell Viability, Clonogenicity and Senescence
5.4. RNA Expression Analysis
5.5. RNA Immunoprecipitation
5.6. Western Blot Analysis
5.7. Statistical Analysis
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gil, J.; Peters, G. Regulation of the INK4B-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. INK4-ARF locus in cancer and aging. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Berggren de Verdier, P.J.; Kumar, R.; Adolfsson, J.; Larsson, P.; Norming, U.; Onelov, E.; Wijkstrom, H.; Steineck, G.; Hemminki, K. Prognostic significance of homozygous deletions and multiple duplications at the CDKN2A (P16INK4A)/ARF (P14ARF) locus in urinary bladder cancer. Scand. J. Urol. Nephrol. 2006, 40, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Florl, A.R.; Schulz, W.A. Peculiar structure and location of 9p21 homozygous deletion breakpoints in human cancer cells. Genes Chrom. Cancer 2003, 37, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, K.; Enokida, H.; Gotanda, T.; Kubo, H.; Nishiyama, K.; Kawahara, M.; Nakagawa, M. P16INK4A and P14ARF methylation as a potential biomarker for human bladder cancer. Biochem. Biophys. Res. Commun. 2006, 339, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Le Frere-Belda, M.A.; Cappellen, D.; Daher, A.; Gil-Diez-de-Medina, S.; Besse, F.; Abbou, C.C.; Thiery, J.P.; Zafrani, E.S.; Chopin, D.K.; Radvanyi, F. P15(INK4B) in bladder carcinomas: Decreased expression in superficial tumours. Br. J. Cancer 2001, 85, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Merlo, A.; Mao, L.; Lapidus, R.G.; Issa, J.P.; Davidson, N.E.; Sidransky, D.; Baylin, S.B. Inactivation of the CDKN2/P16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995, 55, 4525–4530. [Google Scholar] [PubMed]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Bernard, D.; Martinez, D.; Beach, D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat. Cell Biol. 2004, 6, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and polycomb-group gene BMI-1 regulates cell proliferation and senescence through the INK4A locus. Nature 1999, 397, 164–168. [Google Scholar] [PubMed]
- Maertens, G.N.; El Messaoudi-Aubert, S.; Racek, T.; Stock, J.K.; Nicholls, J.; Rodriguez-Niedenfuhr, M.; Gil, J.; Peters, G. Several distinct polycomb complexes regulate and co-localize on the INK4A tumor suppressor locus. PLoS ONE 2009, 4, e6380. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, Y.B.; Pirrotta, V. A new world of polycombs: Unexpected partnerships and emerging functions. Nat. Rev. Genetics 2013, 14, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Peschansky, V.J.; Wahlestedt, C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics 2014, 9, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Li, S.; Munoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4A. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of P15(INK4B) tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Holdt, L.M.; Hoffmann, S.; Sass, K.; Langenberger, D.; Scholz, M.; Krohn, K.; Finstermeier, K.; Stahringer, A.; Wilfert, W.; Beutner, F.; et al. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet. 2013, 9, e1003588. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Laurendeau, I.; Heron, D.; Vidaud, M.; Vidaud, D.; Bieche, I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: Identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007, 67, 3963–3969. [Google Scholar] [CrossRef] [PubMed]
- Holdt, L.M.; Beutner, F.; Scholz, M.; Gielen, S.; Gabel, G.; Bergert, H.; Schuler, G.; Thiery, J.; Teupser, D. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscl. Throm. Vasc. Biol. 2010, 30, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, Z.; Mao, C.; Zhou, Y.; Yu, L.; Yin, Y.; Wu, S.; Mou, X.; Zhu, Y. ANRIL inhibits P15(INK4B) through the TGFBETA1 signaling pathway in human esophageal squamous cell carcinoma. Cell. Immunol. 2014, 289, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.; Wang, C.Y.; Yao, K.H.; Chen, J.T.; Zhang, J.J.; Ma, W.L. High expression of long non-coding RNA ANRIL is associated with poor prognosis in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 3076–3082. [Google Scholar] [PubMed]
- Lin, L.; Gu, Z.T.; Chen, W.H.; Cao, K.J. Increased expression of the long non-coding RNA ANRIL promotes lung cancer cell metastasis and correlates with poor prognosis. Diagn. Pathol. 2015, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.B.; Kong, R.; Yin, D.D.; You, L.H.; Sun, M.; Han, L.; Xu, T.P.; Xia, R.; Yang, J.S.; de, W.; et al. Long noncoding RNA ANRIL indicates a poor prognosis of gastric cancer and promotes tumor growth by epigenetically silencing of miR-99a/miR-449a. Oncotarget 2014, 5, 2276–2292. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Jeck, W.R.; Liu, Y.; Sanoff, H.K.; Wang, Z.; Sharpless, N.E. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010, 6, e1001233. [Google Scholar] [CrossRef] [PubMed]
- Folkersen, L.; Kyriakou, T.; Goel, A.; Peden, J.; Malarstig, A.; Paulsson-Berne, G.; Hamsten, A.; Hugh, W.; Franco-Cereceda, A.; Gabrielsen, A.; et al. Relationship between CAD risk genotype in the chromosome 9p21 locus and gene expression. Identification of eight new ANRIL splice variants. PLoS ONE 2009, 4, e7677. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Mathur, R.; Hu, X.; Liu, Y.; Zhang, X.; Peng, G.; Lu, X. Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cell. Signal. 2013, 25, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Nakagawa, H.; Tajima, A.; Yoshida, K.; Inoue, I. ANRIL is implicated in the regulation of nucleus and potential transcriptional target of E2F1. Oncol. Rep. 2010, 24, 701–707. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar]
- Lee, J.S.; Leem, S.H.; Lee, S.Y.; Kim, S.C.; Park, E.S.; Kim, S.B.; Kim, S.K.; Kim, Y.J.; Kim, W.J.; Chu, I.S. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J. Clin. Oncol. 2010, 28, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Oeggerli, M.; Schraml, P.; Ruiz, C.; Bloch, M.; Novotny, H.; Mirlacher, M.; Sauter, G.; Simon, R. E2F3 is the main target gene of the 6p22 amplicon with high specificity for human bladder cancer. Oncogene 2006, 25, 6538–6543. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Hoffmann, M.J.; Hartmann, F.H.; Schulz, W.A. Amplification and overexpression of the ID4 gene at 6p22.3 in bladder cancer. Mol. Cancer 2005, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Hurst, C.D.; Tomlinson, D.C.; Williams, S.V.; Platt, F.M.; Knowles, M.A. Inactivation of the RB pathway and overexpression of both isoforms of E2F3 are obligate events in bladder tumours with 6p22 amplification. Oncogene 2008, 27, 2716–2727. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Zhang, E.B.; Yin, D.D.; You, L.H.; Xu, T.P.; Chen, W.M.; Xia, R.; Wan, L.; Sun, M.; Wang, Z.X.; et al. Long noncoding RNA PVT1 indicates a poor prognosis of gastric cancer and promotes cell proliferation through epigenetically regulating p15 and p16. Mol. Cancer 2015, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, F.; Pandey, R.R.; Nagano, T.; Chakalova, L.; Mondal, T.; Fraser, P.; Kanduri, C. KCNQ1OT1/LIT1 noncoding RNA mediates transcriptional silencing by targeting to the perinucleolar region. Mol. Cell. Biol. 2008, 28, 3713–3728. [Google Scholar] [CrossRef] [PubMed]
- Heubach, J.; Monsior, J.; Deenen, R.; Niegisch, G.; Szarvas, T.; Niedworok, C.; Schulz, W.A.; Hoffmann, M.J. The long noncoding RNA HOTAIR has tissue and cell type-dependent effects on HOX gene expression and phenotype of urothelial cancer cells. Mol. Cancer 2015, 14, 108. [Google Scholar] [CrossRef] [PubMed]
- Aguilo, F.; Zhou, M.M.; Walsh, M.J. Long noncoding RNA, polycomb, and the ghosts haunting INK4b-ARF-INK4a expression. Cancer Res. 2011, 71, 5365–5369. [Google Scholar] [CrossRef] [PubMed]
- Nie, F.Q.; Sun, M.; Yang, J.S.; Xie, M.; Xu, T.P.; Xia, R.; Liu, Y.W.; Liu, X.H.; Zhang, E.B.; Lu, K.H.; et al. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and p21 expression. Mol. Cancer Therap. 2015, 14, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, M.; Feber, A.; Dueñas, M.; Segovia, C.; Rubio, C.; Fernandez, M.; Villacampa, F.; Duarte, J.; López-Calderón, F.F.; Gómez-Rodriguez, M.J.; et al. Analysis of the Polycomb-related lncRNAs HOTAIR and ANRIL in bladder cancer. Clin. Epigenet. 2015, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, X.; Song, Y.; Zhang, P.; Xiao, Y.; Xing, Y. Long non-coding RNA ANRIL is up-regulated in bladder cancer and regulates bladder cancer cell proliferation and apoptosis through the intrinsic pathway. Biochem. Biophys. Res. Commun. 2015, 467, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Zacharatos, P.; Kotsinas, A.; Evangelou, K.; Karakaidos, P.; Vassiliou, L.V.; Rezaei, N.; Kyroudi, A.; Kittas, C.; Patsouris, E.; Papavassiliou, A.G.; et al. Distinct expression patterns of the transcription factor E2F1 in relation to tumour growth parameters in common human carcinomas. J. Pathol. 2004, 203, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Popov, N.; Gil, J. Epigenetic regulation of the INK4B-ARF-INK4A locus: In sickness and in health. Epigenetics 2010, 5, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Le Frere-Belda, M.A.; Gil Diez de Medina, S.; Daher, A.; Martin, N.; Albaud, B.; Heudes, D.; Abbou, C.C.; Thiery, J.P.; Zafrani, E.S.; Radvanyi, F.; et al. Profiles of the 2 INK4A gene products, p16 and p14ARF, in human reference urothelium and bladder carcinomas, according to pRB and p53 protein status. Hum. Pathol. 2004, 35, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.D.; Chen, W.M.; Qi, F.Z.; Xia, R.; Sun, M.; Xu, T.P.; Yin, L.; Zhang, E.B.; De, W.; Shu, Y.Q. Long non-coding RNA ANRIL is upregulated in hepatocellular carcinoma and regulates cell apoptosis by epigenetic silencing of KLF2. J. Hematol.Oncol. 2015, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.; Kempkensteffen, C.; Christoph, F.; Krause, H.; Schrader, M.; Schostak, M.; Miller, K.; Weikert, S. Expression parameters of the polycomb group proteins BMI1, SUZ12, RING1 and CBX7 in urothelial carcinoma of the bladder and their prognostic relevance. Tumour Biol. 2008, 29, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.J.; Florl, A.R.; Seifert, H.H.; Schulz, W.A. Multiple mechanisms downregulate CDKN1C in human bladder cancer. Int. J. Cancer 2005, 114, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Swiatkowski, S.; Seifert, H.H.; Steinhoff, C.; Prior, A.; Thievessen, I.; Schliess, F.; Schulz, W.A. Activities of MAP-kinase pathways in normal uroepithelial cells and urothelial carcinoma cell lines. Exp. Cell Res. 2003, 282, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Petersen, B.O.; Holm, K.; Bartek, J.; Helin, K. Deregulated expression of E2F family members induces S-phase entry and overcomes p16INK4A-mediated growth suppression. Mol. Cell. Biol. 1996, 16, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.A.; Saito, M.; Vidal, M.; Valentine, M.; Look, T.; Harlow, E.; Dyson, N.; Helin, K. The retinoblastoma protein binds to a family of E2F transcription factors. Mol. Cell. Biol. 1993, 13, 7813–7825. [Google Scholar] [CrossRef] [PubMed]
- Moran, V.A.; Niland, C.N.; Khalil, A.M. Co-immunoprecipitation of long noncoding RNAs. Meth. Mol. Biol. 2012, 925, 219–228. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, M.J.; Dehn, J.; Droop, J.; Niegisch, G.; Niedworok, C.; Szarvas, T.; Schulz, W.A. Truncated Isoforms of lncRNA ANRIL Are Overexpressed in Bladder Cancer, But Do Not Contribute to Repression of INK4 Tumor Suppressors. Non-Coding RNA 2015, 1, 266-284. https://doi.org/10.3390/ncrna1030266
Hoffmann MJ, Dehn J, Droop J, Niegisch G, Niedworok C, Szarvas T, Schulz WA. Truncated Isoforms of lncRNA ANRIL Are Overexpressed in Bladder Cancer, But Do Not Contribute to Repression of INK4 Tumor Suppressors. Non-Coding RNA. 2015; 1(3):266-284. https://doi.org/10.3390/ncrna1030266
Chicago/Turabian StyleHoffmann, Michèle J., Judith Dehn, Johanna Droop, Günter Niegisch, Christian Niedworok, Tibor Szarvas, and Wolfgang A. Schulz. 2015. "Truncated Isoforms of lncRNA ANRIL Are Overexpressed in Bladder Cancer, But Do Not Contribute to Repression of INK4 Tumor Suppressors" Non-Coding RNA 1, no. 3: 266-284. https://doi.org/10.3390/ncrna1030266
APA StyleHoffmann, M. J., Dehn, J., Droop, J., Niegisch, G., Niedworok, C., Szarvas, T., & Schulz, W. A. (2015). Truncated Isoforms of lncRNA ANRIL Are Overexpressed in Bladder Cancer, But Do Not Contribute to Repression of INK4 Tumor Suppressors. Non-Coding RNA, 1(3), 266-284. https://doi.org/10.3390/ncrna1030266