

Nanomaterial-Doped Xerogels for Biosensing Measurements of Xanthine in Clinical and Industrial Applications


Abstract
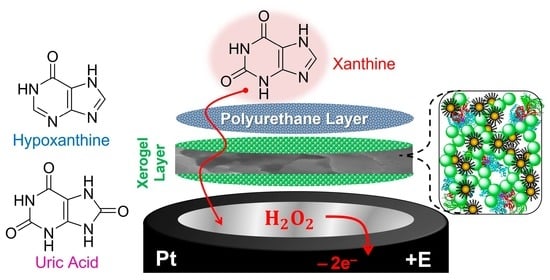
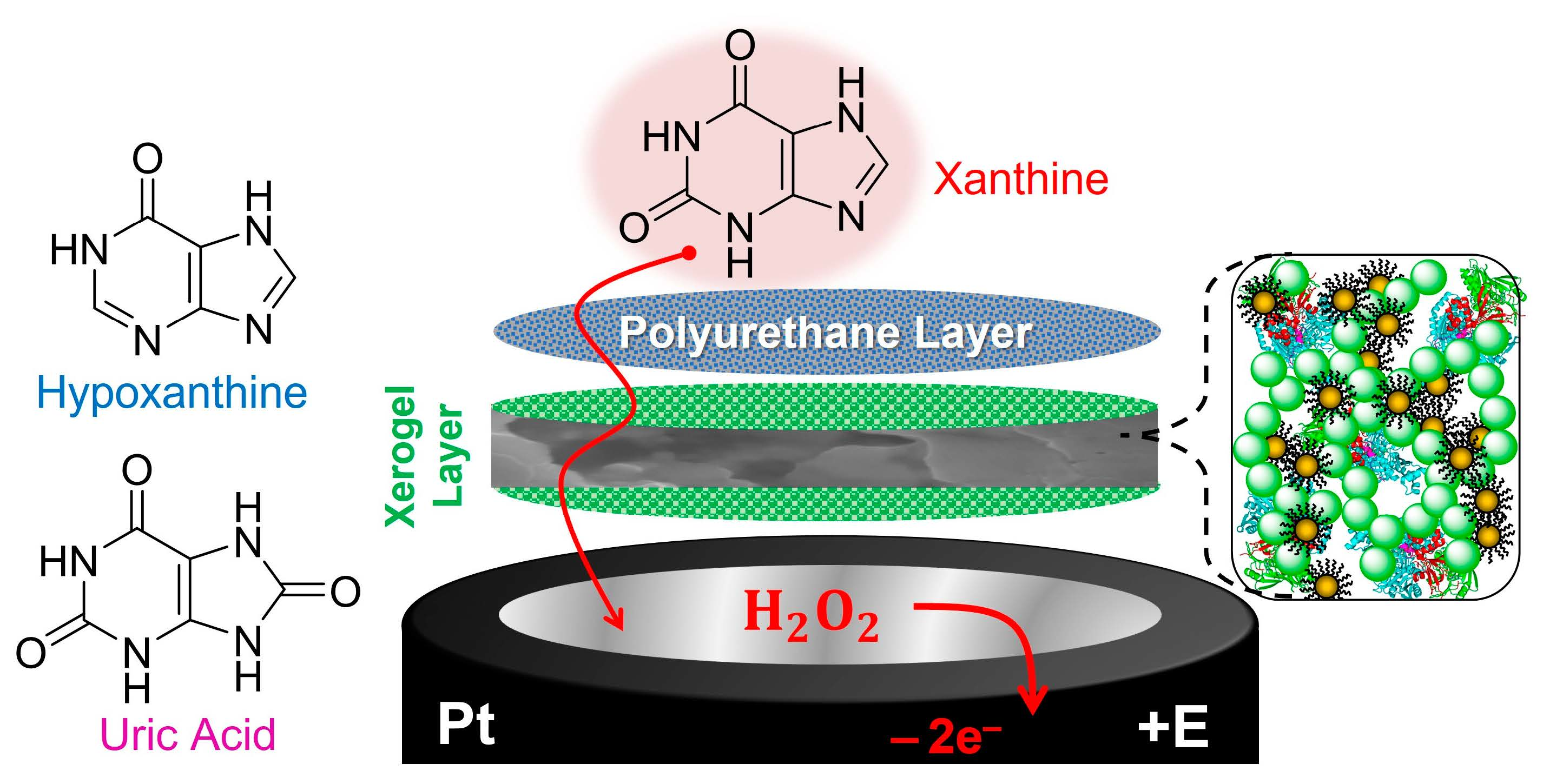
1. Introduction and Background
2. Results and Discussion
2.1. Layer-by-Layer Optimization
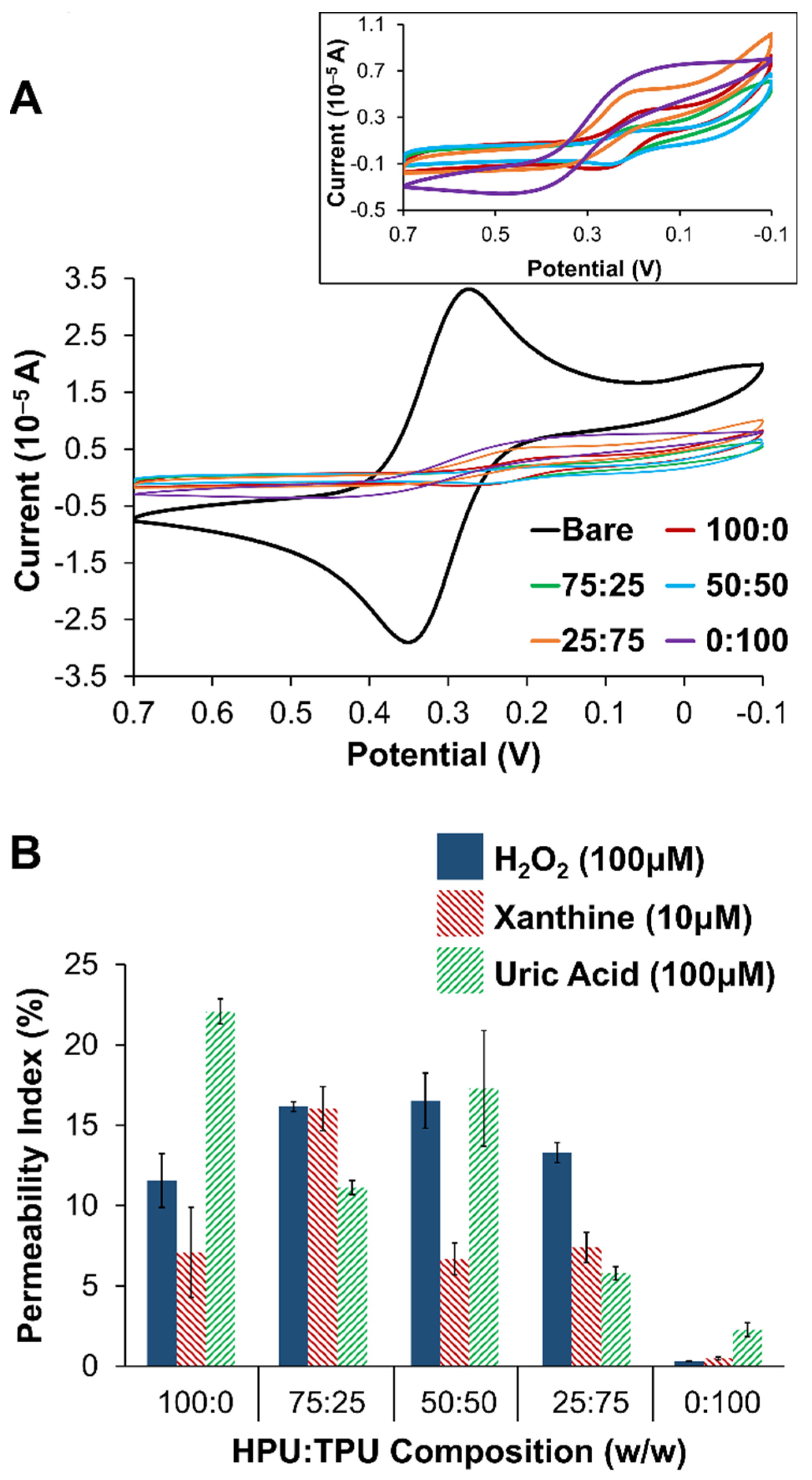
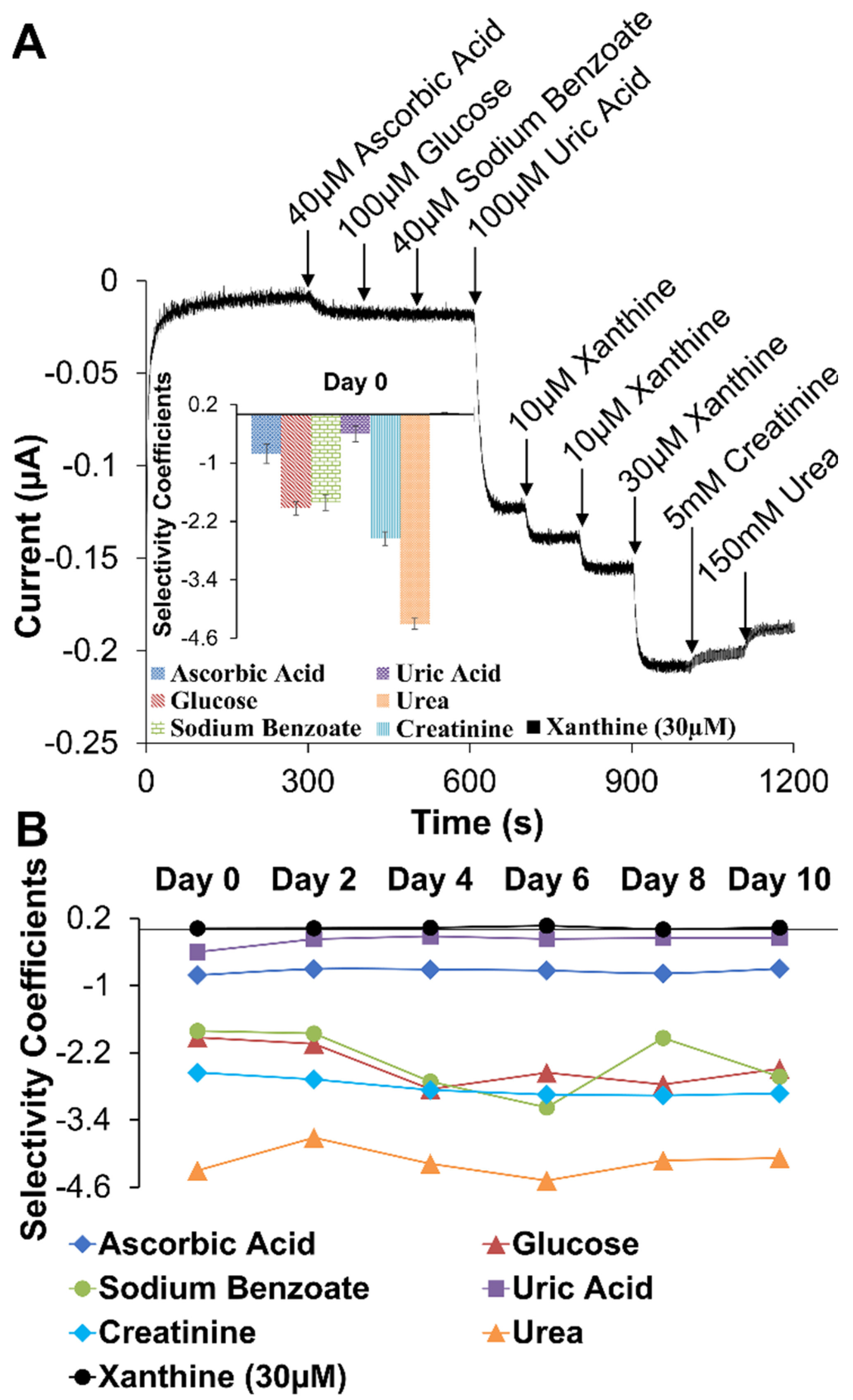
2.1.1. Outer Selective Polyurethane (PU) Layer
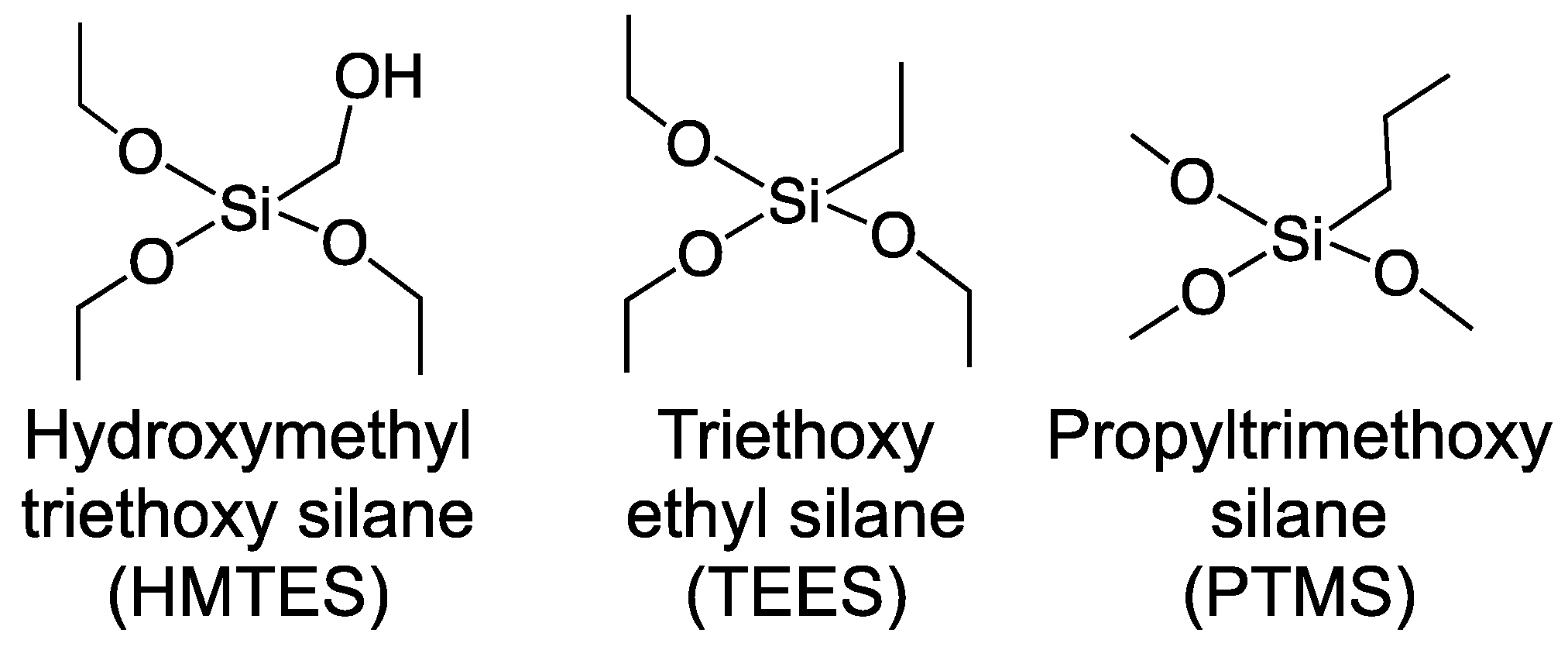
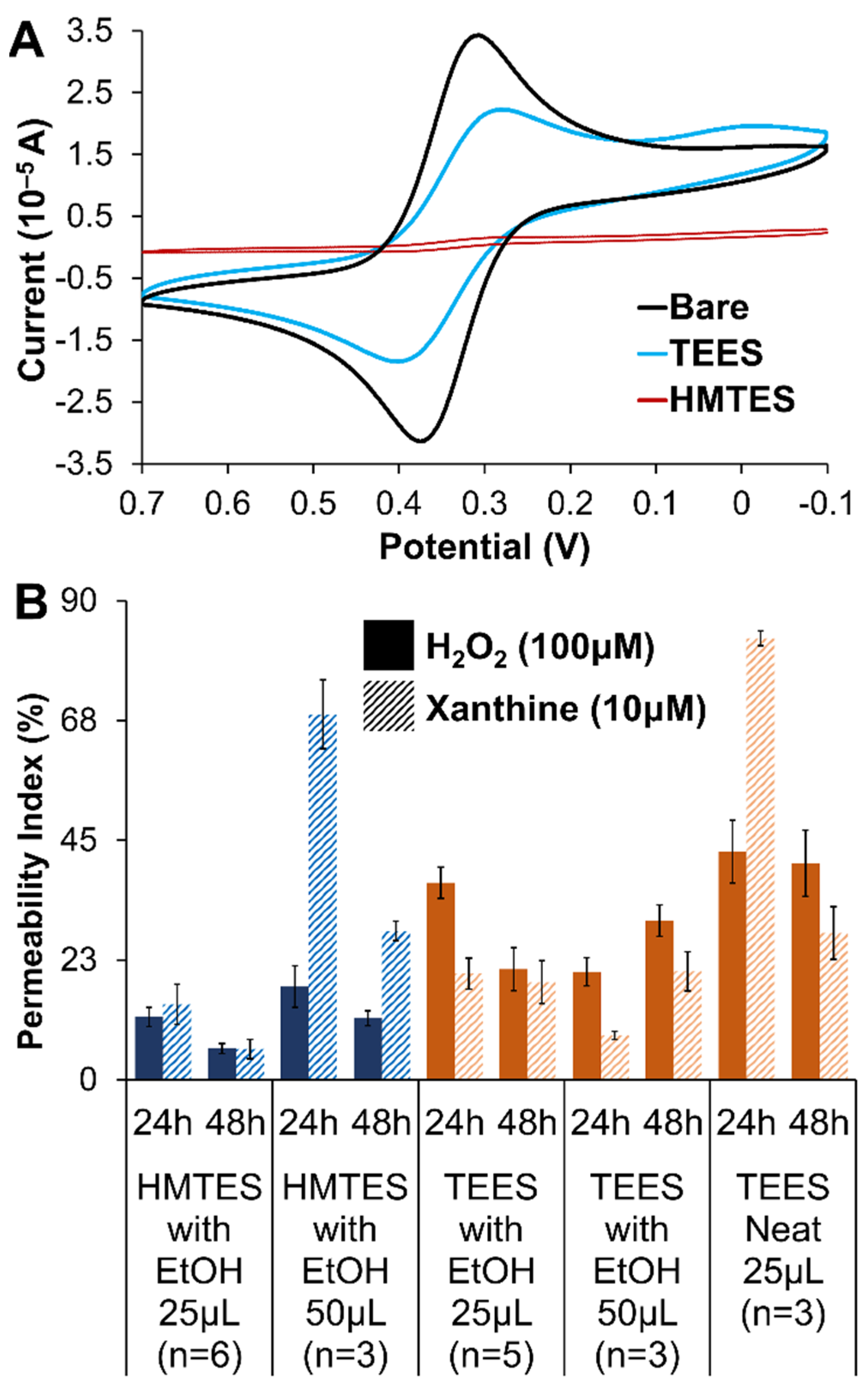
2.1.2. Xerogel Optimization—Silane Precursors, Aging, Enzyme Loading, and Multi-Layers
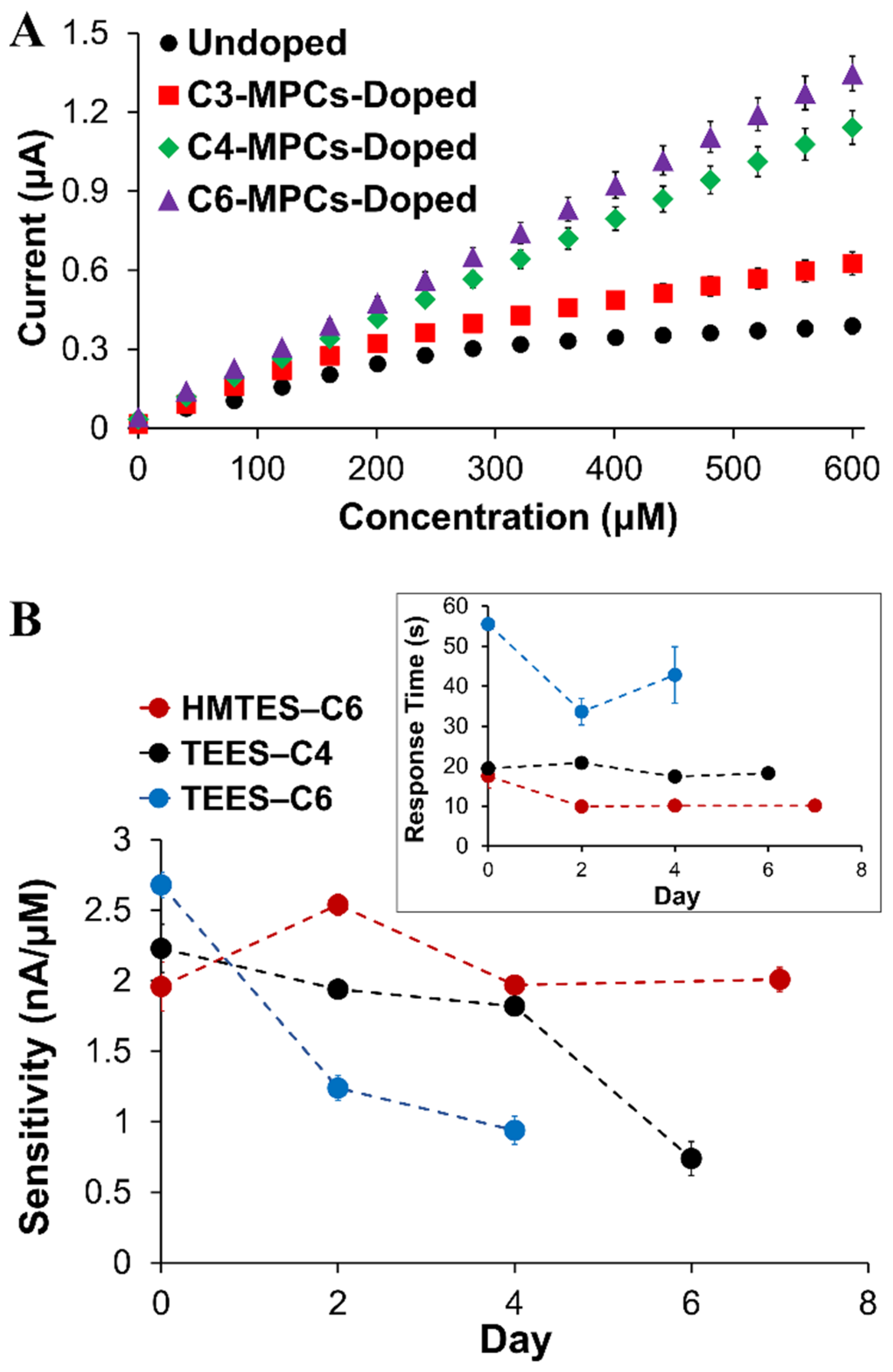
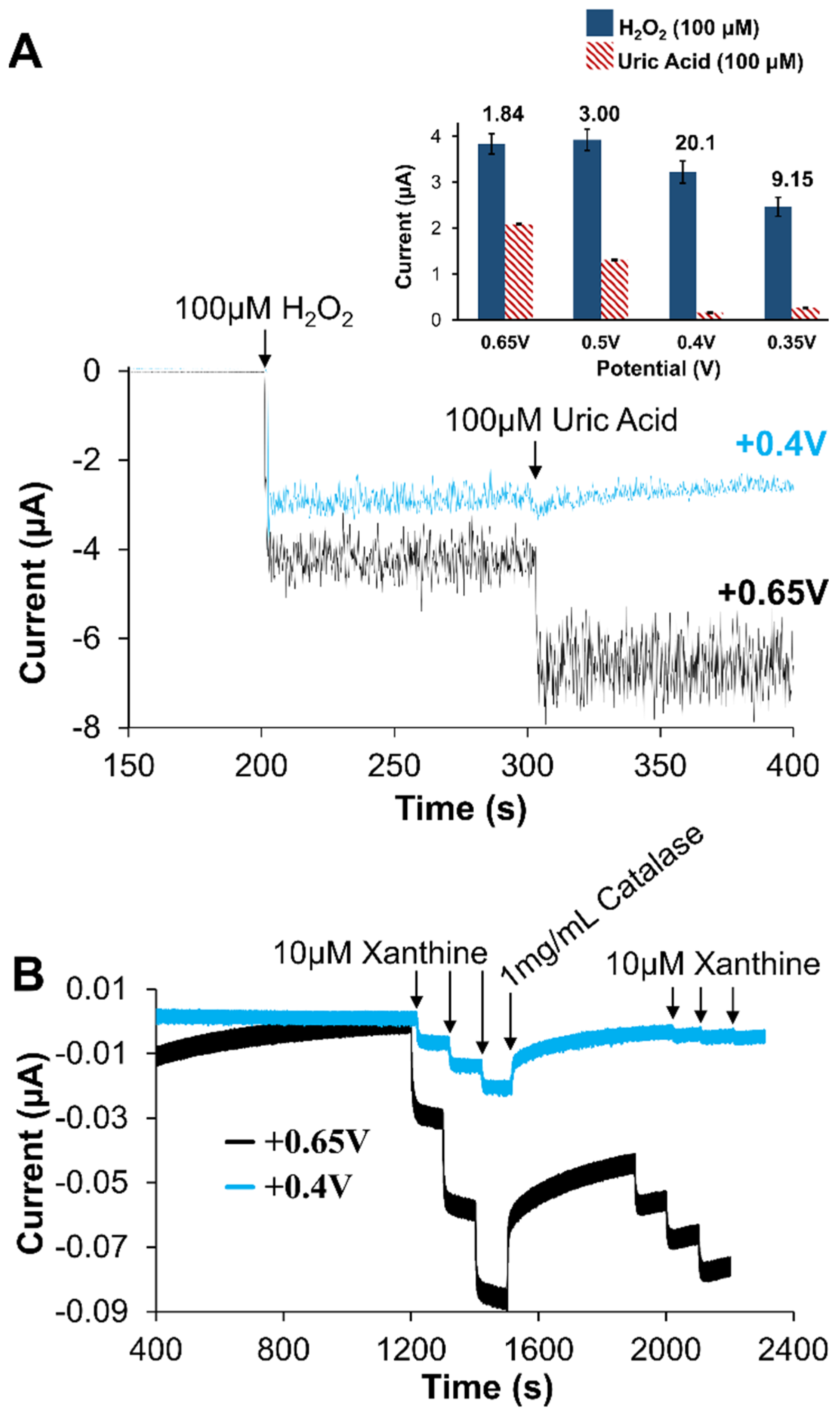
2.1.3. Nanoparticle Doping of Xerogels
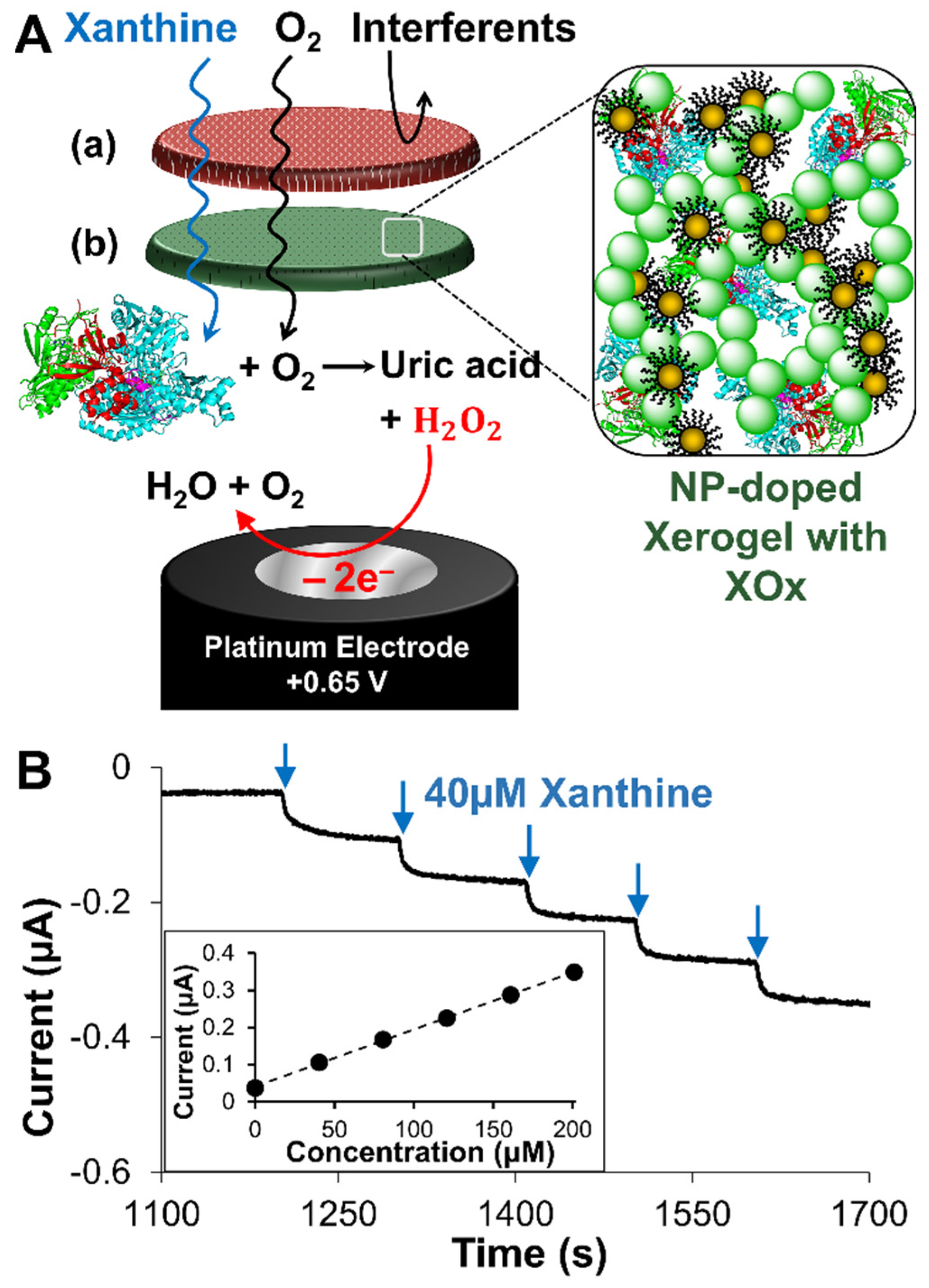
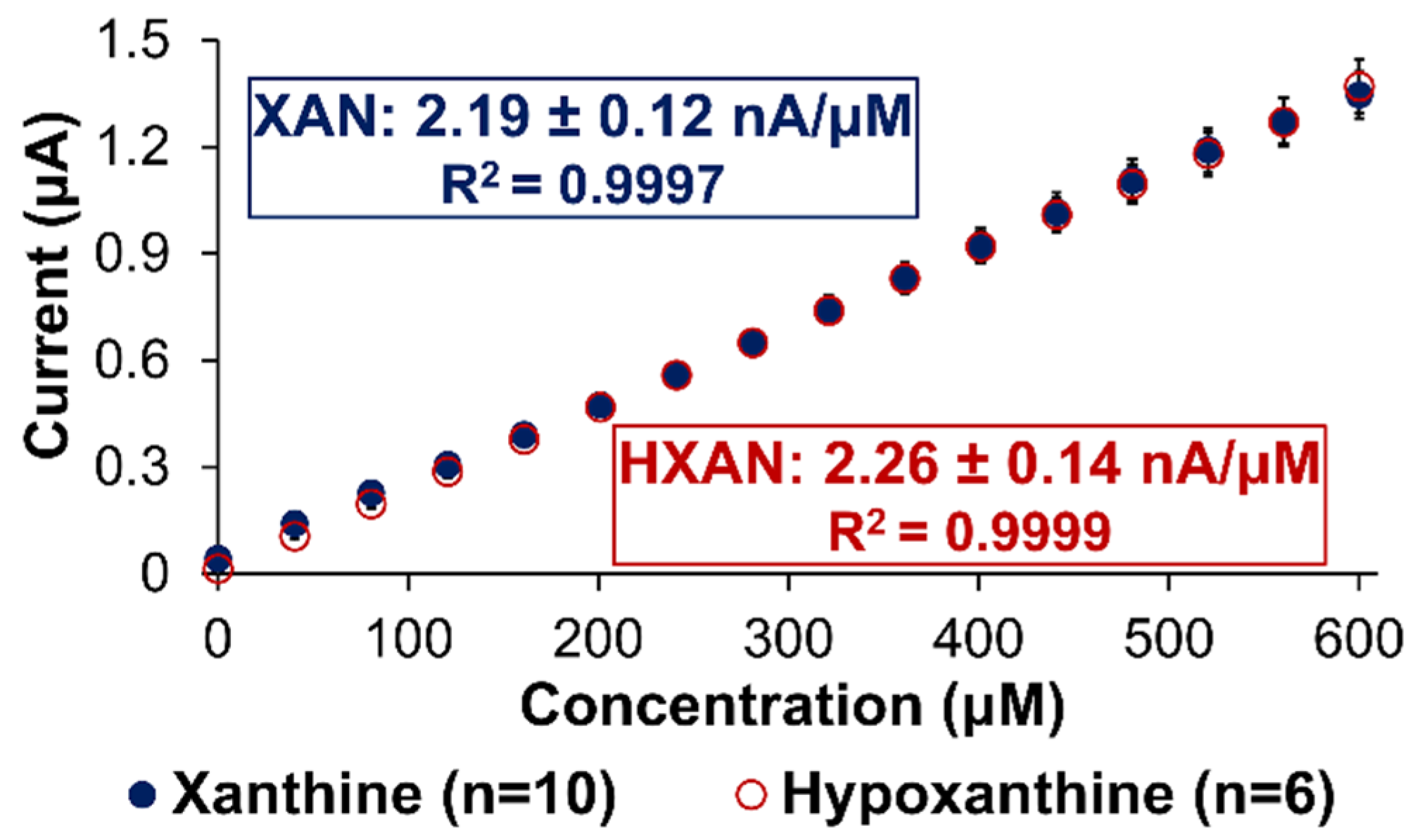
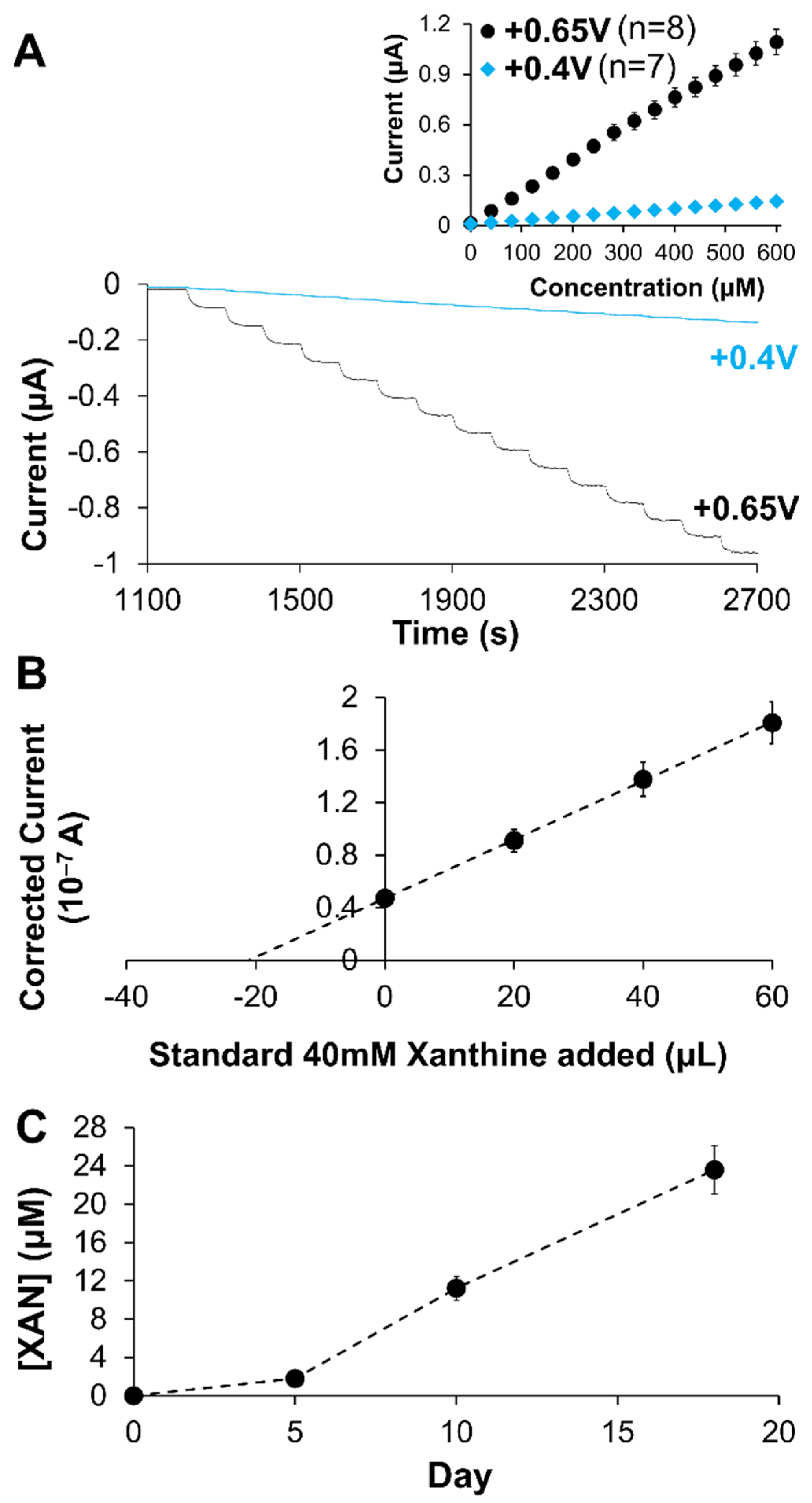
2.2. Analytical Performance of Optimized Xanthine Biosensing System
2.3. Signal Differentiation of XAN Biosensor Scheme
2.4. Clinical and Industrial Application of XAN Biosensors
3. Conclusions
4. Materials and Methods
4.1. Materials and Instrumentation
4.2. Nanoparticle Synthesis
4.3. Preparation of Amperometric Biosensors
4.4. Evaluation of Xanthine Biosensors
4.5. Evaluation of Xanthine Biosensors in Simulated Clinical Samples (Urine)
4.6. Evaluation of Xanthine Biosensors in Real Samples (Fish Meat)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Development Perspective of Bioelectrocatalysis-Based Biosensors. Sensors 2020, 20, 4826. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Gorton, L. Enzyme based amperometric biosensors. Curr. Opin. Electrochem. 2018, 10, 157–173. [Google Scholar] [CrossRef]
- Bollella, P.; Katz, E. Enzyme-Based Biosensors: Tackling Electron Transfer Issues. Sensors 2020, 20, 3517. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.H.; Lee, S.H.; Lee, U.J.; Fermin, C.D.; Kim, M. Immobilized Enzymes in Biosensor Applications. Materials 2019, 12, 121. [Google Scholar] [CrossRef]
- Kim, E.R.; Joe, C.; Mitchell, R.J.; Gu, M.B. Biosensors for healthcare: Current and future perspectives. Trends Biotechnol. 2022, 41, 374–395. [Google Scholar] [CrossRef]
- Kurbanoglu, S.; Erkmen, C.; Uslu, B. Frontiers in electrochemical enzyme based biosensors for food and drug analysis. TrAC Trends Anal. Chem. 2020, 124, 115809. [Google Scholar] [CrossRef]
- Pinyou, P.; Blay, V.; Muresan, L.M.; Noguer, T. Enzyme-modified electrodes for biosensors and biofuel cells. Mater. Horiz. 2019, 6, 1336–1358. [Google Scholar] [CrossRef]
- Huang, L.; Liu, G.; Fu, Y. Recent developments in biosensing strategies for the detection of small molecular contaminants to ensure food safety in aquaculture and fisheries. Trends Food Sci. Technol. 2023, 133, 15–27. [Google Scholar] [CrossRef]
- Chen, X.; Yao, C.; Li, Z. Microarray-based chemical sensors and biosensors: Fundamentals and food safety applications. TrAC Trends Anal. Chem. 2023, 158, 116785. [Google Scholar] [CrossRef]
- Wang, C.; Liu, M.; Wang, Z.; Li, S.; Deng, Y.; He, N. Point-of-care diagnostics for infectious diseases: From methods to devices. Nano Today 2021, 37, 101092. [Google Scholar] [CrossRef]
- Shrivastava, S.; Trung, T.Q.; Lee, N.-E. Recent progress, challenges, and prospects of fully integrated mobile and wearable point-of-care testing systems for self-testing. Chem. Soc. Rev. 2020, 49, 1812–1866. [Google Scholar] [CrossRef]
- Gupta, U.; Gupta, V.; Arun, R.K.; Chanda, N. Recent advances in enzymatic biosensors for point-of-care detection of biomolecules. Biotechnol. Bioeng. 2022, 119, 3393–3407. [Google Scholar] [CrossRef]
- Sempionatto, J.R.; Jeerapan, I.; Krishnan, S.; Wang, J. Wearable Chemical Sensors: Emerging Systems for On-Body Analytical Chemistry. Anal. Chem. 2019, 92, 378–396. [Google Scholar] [CrossRef]
- Teymourian, H.; Tehrani, F.; Mahato, K.; Wang, J. Lab under the Skin: Microneedle Based Wearable Devices. Adv. Heal. Mater. 2021, 10, e2002255. [Google Scholar] [CrossRef]
- Mahato, K.; Wang, J. Electrochemical sensors: From the bench to the skin. Sensors Actuators B: Chem. 2021, 344, 130178. [Google Scholar] [CrossRef]
- Tasca, F.; Tortolini, C.; Bollella, P.; Antiochia, R. Microneedle-based electrochemical devices for transdermal biosensing: A review. Curr. Opin. Electrochem. 2019, 16, 42–49. [Google Scholar] [CrossRef]
- Goud, K.Y.; Mahato, K.; Teymourian, H.; Longardner, K.; Litvan, I.; Wang, J. Wearable electrochemical microneedle sensing platform for real-time continuous interstitial fluid monitoring of apomorphine: Toward Parkinson management. Sensors Actuators B: Chem. 2021, 354, 131234. [Google Scholar] [CrossRef]
- Hughes, L.B.; Labban, N.; Conway, G.E.; Pollock, J.A.; Leopold, M.C. Adaptable Xerogel-Layered Amperometric Biosensor Platforms on Wire Electrodes for Clinically Relevant Measurements. Sensors 2019, 19, 2584. [Google Scholar] [CrossRef]
- Perdomo, S.A.; Marmolejo-Tejada, J.M.; Jaramillo-Botero, A. Review—Bio-Nanosensors: Fundamentals and Recent Applications. J. Electrochem. Soc. 2021, 168, 107506. [Google Scholar] [CrossRef]
- Putzbach, W.; Ronkainen, N.J. Immobilization Techniques in the Fabrication of Nanomaterial-Based Electrochemical Biosensors: A Review. Sensors 2013, 13, 4811–4840. [Google Scholar] [CrossRef]
- Zhu, C.; Yang, G.; Li, H.; Du, D.; Lin, Y. Electrochemical Sensors and Biosensors Based on Nanomaterials and Nanostructures. Anal. Chem. 2015, 87, 230–249. [Google Scholar] [CrossRef] [PubMed]
- Olloqui-Sariego, J.L.; Calvente, J.J.; Andreu, R. Immobilizing redox enzymes at mesoporous and nanostructured electrodes. Curr. Opin. Electrochem. 2020, 26, 100658. [Google Scholar] [CrossRef]
- Theyagarajan, K.; Kim, Y.-J. Recent Developments in the Design and Fabrication of Electrochemical Biosensors Using Functional Materials and Molecules. Biosensors 2023, 13, 424. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Bonilla-Cruz, J. Review on Healthcare Biosensing Nanomaterials. ACS Appl. Nano Mater. 2023, 6, 5042–5074. [Google Scholar] [CrossRef]
- Oliveira, T.M.B.F.; Morais, S. New Generation of Electrochemical Sensors Based on Multi-Walled Carbon Nanotubes. Appl. Sci. 2018, 8, 1925. [Google Scholar] [CrossRef]
- Labban, N.; Wayu, M.B.; Steele, C.M.; Munoz, T.S.; Pollock, J.A.; Case, W.S.; Leopold, M.C. First Generation Amperometric Biosensing of Galactose with Xerogel-Carbon Nanotube Layer-By-Layer Assemblies. Nanomaterials 2018, 9, 42. [Google Scholar] [CrossRef]
- Kumar, H.; Kumari, N.; Sharma, R. Nanocomposites (conducting polymer and nanoparticles) based electrochemical biosensor for the detection of environment pollutant: Its issues and challenges. Environ. Impact Assess. Rev. 2020, 85, 106438. [Google Scholar] [CrossRef]
- Heydari-Badrooei, E.; Ensafi, A.A. Nanomaterials-based biosensing strategies for biomarkers diagnosis, a review. Biosens. Bioelectron. X 2022, 13, 100245. [Google Scholar] [CrossRef]
- Ramya, M.; Kumar, P.S.; Rangasamy, G.; Shankar, V.U.; Rajesh, G.; Nirmala, K.; Saravanan, A.; Krishnapandi, A. A recent advancement on the applications of nanomaterials in electrochemical sensors and biosensors. Chemosphere 2022, 308, 136416. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, P.; Pournara, A.; Vellingiri, K.; Kim, K.-H. Nanomaterials for the sensing of narcotics: Challenges and opportunities. TrAC Trends Anal. Chem. 2018, 106, 84–115. [Google Scholar] [CrossRef]
- Guo, S.; Dong, S. Biomolecule-nanoparticle hybrids for electrochemical biosensors. TrAC Trends Anal. Chem. 2009, 28, 96–109. [Google Scholar] [CrossRef]
- Walcarius, A.; Collinson, M.M. Analytical Chemistry with Silica Sol-Gels: Traditional Routes to New Materials for Chemical Analysis. Annu. Rev. Anal. Chem. 2009, 2, 121–143. [Google Scholar] [CrossRef]
- Khalil, H.P.S.A.; Yahya, E.B.; Tajarudin, H.A.; Balakrishnan, V.; Nasution, H. Insights into the Role of Biopolymer-Based Xerogels in Biomedical Applications. Gels 2022, 8, 334. [Google Scholar] [CrossRef]
- Idumah, C.I. Emerging advancements in xerogel polymeric bionanoarchitectures and applications. JCIS Open 2023, 9, 100073. [Google Scholar] [CrossRef]
- Idumah, C.I.; Low, J.H.; Emmanuel, E.O. Recently emerging trends in xerogel polymeric nanoarchitectures and multifunctional applications. Polym. Bull. 2022, 1–31. [Google Scholar] [CrossRef]
- Narang, U.; Prasad, P.N.; Bright, F.V.; Ramanathan, K.; Kumar, N.D.; Malhotra, B.D.; Kamalasanan, M.N.; Chandra, S. Glucose Biosensor Based on a Sol-Gel-Derived Platform. Anal. Chem. 1994, 66, 3139–3144. [Google Scholar] [CrossRef]
- DiPasquale, L.T.; Poulos, N.G.; Hall, J.R.; Minocha, A.; Bui, T.A.; Leopold, M.C. Structure–function relationships affecting the sensing mechanism of monolayer-protected cluster doped xerogel amperometric glucose biosensors. J. Colloid Interface Sci. 2015, 450, 202–212. [Google Scholar] [CrossRef]
- Pundir, C.S.; Devi, R. Biosensing methods for xanthine determination: A review. Enzym. Microb. Technol. 2014, 57, 55–62. [Google Scholar] [CrossRef]
- Wayu, M.B.; Pannell, M.J.; Leopold, M.C. Layered Xerogel Films Incorporating Monolayer-Protected Cluster Networks on Platinum-Black-Modified Electrodes for Enhanced Sensitivity in First-Generation Uric Acid Biosensing. Chemelectrochem 2016, 3, 1245–1252. [Google Scholar] [CrossRef]
- Pannell, M.J.; Doll, E.E.; Labban, N.; Wayu, M.B.; Pollock, J.A.; Leopold, M.C. Versatile sarcosine and creatinine biosensing schemes utilizing layer-by-layer construction of carbon nanotube-chitosan composite films. J. Electroanal. Chem. 2018, 814, 20–30. [Google Scholar] [CrossRef]
- Freeman, M.H.; Hall, J.R.; Leopold, M.C. Monolayer-Protected Nanoparticle Doped Xerogels as Functional Components of Amperometric Glucose Biosensors. Anal. Chem. 2013, 85, 4057–4065. [Google Scholar] [CrossRef] [PubMed]
- Dervisevic, M.; Dervisevic, E.; Şenel, M. Recent progress in nanomaterial-based electrochemical and optical sensors for hypoxanthine and xanthine. A review. Microchim. Acta 2019, 186, 749. [Google Scholar] [CrossRef] [PubMed]
- Dervisevic, M.; Dervisevic, E.; Cevik, E.; Şenel, M. Novel electrochemical xanthine biosensor based on chitosan–polypyrrole–gold nanoparticles hybrid bio-nanocomposite platform. J. Food Drug Anal. 2017, 25, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, P.; Leimkühler, S.; Bernhardt, P.V. Low-Potential Amperometric Enzyme Biosensor for Xanthine and Hypoxanthine. Anal. Chem. 2012, 84, 10359–10365. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Atkin, D.; Lee, K.; Sell, M.; Chandra, S. Determining meat freshness using electrochemistry: Are we ready for the fast and furious? Meat Sci. 2018, 150, 40–46. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Guo, C.; Zhang, X.; Wang, Y.; Lv, L.; Wang, X.; Wei, M. A pattern-free paper enzyme biosensor for one-step detection of fish freshness indicator hypoxanthine with a microfluidic aggregation effect. Food Chem. 2023, 405, 134811. [Google Scholar] [CrossRef]
- Yazdanparast, S.; Benvidi, A.; Abbasi, S.; Rezaeinasab, M. Enzyme-based ultrasensitive electrochemical biosensor using poly(l-aspartic acid)/MWCNT bio-nanocomposite for xanthine detection: A meat freshness marker. Microchem. J. 2019, 149, 104000. [Google Scholar] [CrossRef]
- Sun, Y.; Fei, J.; Wu, K.; Hu, S. Simultaneous electrochemical determination of xanthine and uric acid at a nanoparticle film electrode. Anal. Bioanal. Chem. 2003, 375, 544–549. [Google Scholar] [CrossRef]
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free. Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef]
- Yu, H.; Chen, X.; Guo, X.; Chen, D.; Jiang, L.; Qi, Y.; Shao, J.; Tao, L.; Hang, J.; Lu, G.; et al. The clinical value of serum xanthine oxidase levels in patients with acute ischemic stroke. Redox Biol. 2023, 60, 102623. [Google Scholar] [CrossRef]
- Pundir, C.; Devi, R.; Narang, J.; Singh, S.; Nehra, J.; Chaudhry, S. FABRICATION OF AN AMPEROMETRIC XANTHINE BIOSENSOR BASED ON POLYVINYLCHLORIDE MEMBRANE. J. Food Biochem. 2011, 36, 21–27. [Google Scholar] [CrossRef]
- Khan, M.Z.H.; Ahommed, M.S.; Daizy, M. Detection of xanthine in food samples with an electrochemical biosensor based on PEDOT:PSS and functionalized gold nanoparticles. RSC Adv. 2020, 10, 36147–36154. [Google Scholar] [CrossRef]
- Liu, Y.; Li, W.; Wei, C.; Lü, L. Preparation of a Xanthine Sensor Based on the Immobilization of Xanthine Oxidase on a Chitosan Modified Electrode by Cross-linking. Chin. J. Chem. 2012, 30, 1601–1604. [Google Scholar] [CrossRef]
- Devi, R.; Yadav, S.; Nehra, R.; Yadav, S.; Pundir, C. Electrochemical biosensor based on gold coated iron nanoparticles/chitosan composite bound xanthine oxidase for detection of xanthine in fish meat. J. Food Eng. 2012, 115, 207–214. [Google Scholar] [CrossRef]
- Devi, R.; Batra, B.; Lata, S.; Yadav, S.; Pundir, C. A method for determination of xanthine in meat by amperometric biosensor based on silver nanoparticles/cysteine modified Au electrode. Process. Biochem. 2013, 48, 242–249. [Google Scholar] [CrossRef]
- Sahyar, B.Y.; Kaplan, M.; Ozsoz, M.; Celik, E.; Otles, S. Electrochemical xanthine detection by enzymatic method based on Ag doped ZnO nanoparticles by using polypyrrole. Bioelectrochemistry 2019, 130, 107327. [Google Scholar] [CrossRef]
- Pierini, G.D.; Robledo, S.N.; Zon, M.A.; Di Nezio, M.S.; Granero, A.M.; Fernández, H. Development of an electroanalytical method to control quality in fish samples based on an edge plane pyrolytic graphite electrode. Simultaneous determination of hypoxanthine, xanthine and uric acid. Microchem. J. 2018, 138, 58–64. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, L.; Zhang, C.; Deng, D.; Wang, D.; Luo, L. Simultaneous Determination of Xanthine and Hypoxanthine Using Polyglycine/rGO-Modified Glassy Carbon Electrode. Molecules 2023, 28, 1458. [Google Scholar] [CrossRef]
- Dervisevic, M.; Dervisevic, E.; Azak, H.; Çevik, E.; Şenel, M.; Yildiz, H.B. Novel amperometric xanthine biosensor based on xanthine oxidase immobilized on electrochemically polymerized 10-[4H-dithieno(3,2-b:2′,3′-d)pyrrole-4-yl]decane-1-amine film. Sensors Actuators B: Chem. 2016, 225, 181–187. [Google Scholar] [CrossRef]
- Rahman, A.; Won, M.-S.; Shim, Y.-B. Xanthine Sensors Based on Anodic and Cathodic Detection of Enzymatically Generated Hydrogen Peroxide. Electroanalysis 2007, 19, 631–637. [Google Scholar] [CrossRef]
- Dimcheva, N.; Horozova, E.; Jordanova, Z. An Amperometric Xanthine Oxidase Enzyme Electrode Based on Hydrogen Peroxide Electroreduction. Z. Für Nat. C 2002, 57, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of thiol-derivatised gold nanoparticles in a two-phase Liquid–Liquid system. J. Chem. Soc. Chem. Commun. 1994, 7, 801–802. [Google Scholar] [CrossRef]
- Conway, G.E.; Lambertson, R.H.; Schwarzmann, M.A.; Pannell, M.J.; Kerins, H.W.; Rubenstein, K.J.; Dattelbaum, J.D.; Leopold, M.C. Layer-by-layer design and optimization of xerogel-based amperometric first generation biosensors for uric acid. J. Electroanal. Chem. 2016, 775, 135–145. [Google Scholar] [CrossRef]
- Wayu, M.B.; Schwarzmann, M.A.; Gillespie, S.D.; Leopold, M.C. Enzyme-free uric acid electrochemical sensors using β-cyclodextrin-modified carboxylic acid-functionalized carbon nanotubes. J. Mater. Sci. 2017, 52, 6050–6062. [Google Scholar] [CrossRef]
- Templeton, A.C.; Wuelfing, M.P.; Murray, R.W. Monolayer protected cluster molecules. Acc. Chem. Res. 2000, 33, 27–36. [Google Scholar] [CrossRef]
- Sadeghi, S.; Fooladi, E.; Malekaneh, M. A nanocomposite/crude extract enzyme-based xanthine biosensor. Anal. Biochem. 2014, 464, 51–59. [Google Scholar] [CrossRef]
- Devi, R.; Yadav, S.; Pundir, C. Amperometric determination of xanthine in fish meat by zinc oxide nanoparticle/chitosan/multiwalled carbon nanotube/polyaniline composite film bound xanthine oxidase. Analyst 2012, 137, 754–759. [Google Scholar] [CrossRef]
- Devi, R.; Yadav, S.; Pundir, C. Au-colloids–polypyrrole nanocomposite film based xanthine biosensor. Colloids Surfaces A: Physicochem. Eng. Asp. 2012, 394, 38–45. [Google Scholar] [CrossRef]
- Koh, A.; Lu, Y.; Schoenfisch, M.H. Fabrication of Nitric Oxide-Releasing Porous Polyurethane Membranes-Coated Needle-type Implantable Glucose Biosensors. Anal. Chem. 2013, 85, 10488–10494. [Google Scholar] [CrossRef]
- Devi, R.; Narang, J.; Yadav, S.K.; Pundir, C.S. Amperometric determination of xanthine in tea, coffee, and fish meat with graphite rod bound xanthine oxidase. J. Anal. Chem. 2012, 67, 273–277. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LbL Assembly Scheme at Pt Electrode | n | Sensitivity a (nA/μM) | Correction Coefficient b (R2) | Response Time, c tr-95% (s) |
|---|---|---|---|---|
| HMTES | ||||
| HMTES (XOx)/PU | 5 | 1.02 | 0.9926 | 39.3 (±2.8) |
| HMTES (XOx) + C3-MPCs/PU | 5 | 0.95 | 0.9663 | 38.7 (±1.3) |
| HMTES (XOx) + C4-MPCs/PU | 5 | 1.86 | 0.9997 | 31.3 (±0.8) |
| HMTES (XOx) + C6-MPCs/PU * | 10 | 2.34 | 0.9995 | 19.1 (±1.5) |
| HMTES (XOx)/HMTES/PU | 6 | 0.40 | 0.9995 | 42.0 (±2.4) |
| HMTES (XOx) + C3-MPCs/HMTES/PU | 4 | 0.47 | 0.9761 | 26.8 (± 2.7) |
| HMTES (XOx) + C4-MPCs/HMTES/PU | 4 | 0.76 | 0.9960 | 23.0 (±1.1) |
| HMTES (XOx) + C6-MPCs/HMTES/PU | 3 | 0.99 | 0.9997 | 22.4 (±1.9) |
| TEES | ||||
| TEES (XOx)/PU | 5 | 2.06 | 0.9895 | 59.2 (±4.1) |
| TEES (XOx) + C3-MPCs/PU | 4 | 2.70 | 0.9856 | 27.2 (±9.3) |
| TEES (XOx) + C4-MPCs/PU * | 4 | 3.08 | 0.9915 | 19.4 (±2.9) |
| TEES (XOx) + C6-MPCs/PU | 3 | 2.67 | 0.9894 | 55.5 (±2.1) |
| TEES (XOx)/TEES/PU | 6 | No signal | --- | --- |
| TEES (XOx) + C3-MPCs/TEES/PU | - | --- | --- | --- |
| TEES (XOx) + C4-MPCs/TEES/PU | - | --- | --- | --- |
| TEES (XOx) + C6-MPCs/TEES/PU | - | --- | --- | --- |
| LbL Assembly Scheme at Pt Electrode | Eapp (V) | Method | n | (XAN) Spike (μM) | (XAN) Found (μM) | % Recovery |
|---|---|---|---|---|---|---|
| Synthetic Urine | ||||||
| HMTES (XOx) + C6-MPCs/PU | +0.65 | CC | 8 | 80.5 | 78.3(±1.7) | 97.2(±2.1) |
| HMTES (XOx) + C6-MPCs/PU | +0.4 | CC | 7 | 80.5 | 79.3(±2.1) | 98.5(±2.6) |
| TEES (XOx) + C6-MPCs/PU | +0.65 | CC | 4 | 300.1 | 233.6(±26.7) | 77.8(±8.9) |
| TEES (XOx) + C6-MPCs/PU | +0.4 | CC | 4 | 300.1 | 213.9(±28.7) | 71.3(±9.6) |
| TEES (XOx) + C4-MPCs/PU | +0.65 | CC | 4 | 80.5 | 57.8(±9.6) | 71.8(±7.9) |
| TEES (XOx) + C4-MPCs/PU | +0.4 | CC | 3 | 80.5 | 49.2(±3.8) | 61.1(±4.7) |
| Sea Bass | ||||||
| HMTES (XOx) + C6-MPCs/PU | +0.65 | CC | 7 | 42.0 | 23.6(±2.1) | 59.5(±5.0) |
| HMTES (XOx) + C6-MPCs/PU | +0.65 | SA | 6 | 40.3 | 42.2(±1.1) | 104.8(±2.6) |
| TEES (XOx) + C6-MPCs/PU | +0.65 | CC | 4 | 42.0 | 19.2(±3.6) | 45.7(±8.7) |
| TEES (XOx) + C6-MPCs/PU | +0.65 | SA | Fouled | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, Q.M.; Wemple, A.H.; Leopold, M.C. Nanomaterial-Doped Xerogels for Biosensing Measurements of Xanthine in Clinical and Industrial Applications. Gels 2023, 9, 437. https://doi.org/10.3390/gels9060437
Dang QM, Wemple AH, Leopold MC. Nanomaterial-Doped Xerogels for Biosensing Measurements of Xanthine in Clinical and Industrial Applications. Gels. 2023; 9(6):437. https://doi.org/10.3390/gels9060437
Chicago/Turabian StyleDang, Quang Minh, Ann H. Wemple, and Michael C. Leopold. 2023. "Nanomaterial-Doped Xerogels for Biosensing Measurements of Xanthine in Clinical and Industrial Applications" Gels 9, no. 6: 437. https://doi.org/10.3390/gels9060437
APA StyleDang, Q. M., Wemple, A. H., & Leopold, M. C. (2023). Nanomaterial-Doped Xerogels for Biosensing Measurements of Xanthine in Clinical and Industrial Applications. Gels, 9(6), 437. https://doi.org/10.3390/gels9060437