



Recent Progress in Gels for Neuropathic Pain


Abstract

1. Introduction
2. Pharmacological Treatment of Neuropathic Pain
3. Development of Analgesic-Containing Gels Useful for NeP
3.1. Capsaicin
3.2. Tramadol
3.2.1. Gels for Transdermal Use
3.2.2. Gels for Parenteral Use
3.3. Gabapentin
3.4. Pregabalin
3.5. Amitriptyline
3.6. Other Substances
3.7. Associations of Analgesic Substances
4. Conclusions and Perspectives
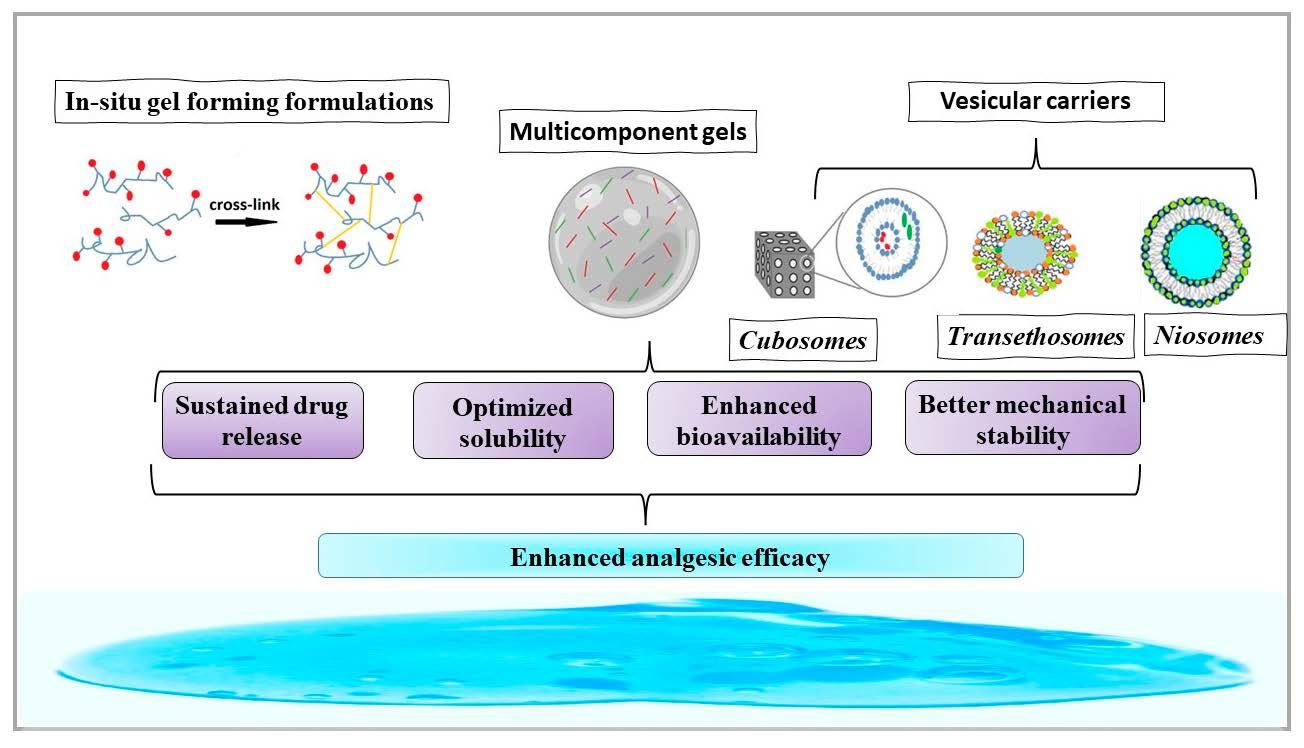
- incorporation of vesicular carriers, such as niosomes, transethosomes, and cubosomes, with enhanced skin penetration and drug delivery compared to conventional liposomes [118]. These carriers protected the encapsulated drug from degradation and acted as local reservoirs of the drug. All the included studies revealed that the utilization of vesicular carriers as drug delivery systems enables a profound interaction with the skin layers, leading to sustained and efficient drug release for a wide range of drugs. However, a better understanding of this interaction would allow further optimization of the formulas.
- use of in situ gel-forming formulations which also ensured a prolonged and constant drug release. Furthermore, thermoresponsive polymers that can undergo a reversible sol–gel transition in response to temperature changes have been used in the preparation of injectable gels for drug delivery. In situ cross-linking was used as a trigger to stimulate this transformation.
- use of multicomponent gels that contain more than one type of polymer. They offer better mechanical stability and tunable properties.
- co-delivery of multiple drugs in a single gel formulation can improve treatment efficacy and reduce side effects.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Terminology|International Association for the Study of Pain. Available online: https://www.iasp-pain.org/resources/terminology/ (accessed on 17 October 2022).
- RTreede, R.-D.; Jensen, T.S.; Campbell, J.N.; Cruccu, G.; Dostrovsky, J.O.; Griffin, J.W.; Hansson, P.; Hughes, R.; Nurmikko, T.; Serra, J. Neuropathic pain: Redefinition and a grading system for clinical and research purposes. Neurology 2007, 70, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.; et al. Neuropathic pain. Nat. Rev. Dis. Prim. 2017, 16, 17002. [Google Scholar] [CrossRef] [PubMed]
- Nickel, F.T.; Seifert, F.; Lanz, S.; Maihöfner, C. Mechanisms of neuropathic pain. Eur. Neuropsychopharmacol. 2012, 22, 81–91. [Google Scholar] [CrossRef] [PubMed]
- DiBonaventura, M.D.; Sadosky, A.; Concialdi, K.; Hopps, M.; Kudel, I.; Parsons, B.; Cappelleri, J.C.; Hlavacek, P.; Alexander, A.H.; Stacey, B.R.; et al. The prevalence of probable neuropathic pain in the US: Results from a multimodal general-population health survey. J. Pain Res. 2017, 10, 2525–2538. [Google Scholar] [CrossRef] [PubMed]
- Mbrah, A.K.; Nunes, A.P.; Hume, A.L.; Zhao, D.; Jesdale, B.M.; Bova, C.; Lapane, K.L. Prevalence and treatment of neuropathic pain diagnoses among U.S. nursing home residents. Pain 2021, 163, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Taams, E.N.; Drenthen, J.; Hanewinckel, R.; Ikram, M.A.; van Doorn, A.P. Prevalence and Risk Factor Profiles for Chronic Axonal Polyneuropathy in the General Population. Neurology 2022, 20, e2234–e2240. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, R.; Mozzillo, E.; Di Candia, F.; Rosanio, F.M.; Leonardi, L.; Liguori, A.; Micheli, F.; Cauvin, V.; Franzese, A.; Piona, C.A.; et al. A systematic review of the prevalence, risk factors and screening tools for autonomic and diabetic peripheral neuropathy in children, adolescents and young adults with type 1 diabetes. Acta Diabetol. 2022, 59, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Langley, P.C.; Van Litsenburg, C.; Cappelleri, J.C.; Carroll, D. The burden associated with neuropathic pain in Western Europe. J. Med. Econ. 2012, 16, 85–95. [Google Scholar] [CrossRef]
- Freynhagen, R.; Baron, R.; Gockel, U.; Tölle, T.R. painDETECT: A new screening questionnaire to identify neuropathic components in patients with back pain. Curr. Med. Res. Opin. 2006, 22, 1911–1920. [Google Scholar] [CrossRef]
- Freynhagen, R.; Bennett, I.M. Diagnosis and management of neuropathic pain. BMJ 2009, 339, b3002. [Google Scholar] [CrossRef]
- O’connor, A.B. Neuropathic Pain. Pharmacoeconomics 2009, 27, 95–112. [Google Scholar] [CrossRef]
- Hans, G.; Masquelier, E.; De Cock, P. The diagnosis and management of neuropathic pain in daily practice in Belgium: An observational study. BMC Public Health 2007, 7, 170. [Google Scholar] [CrossRef] [PubMed]
- Torrance, N.; Smith, B.H.; Watson, M.C.; Bennett, I.M. Medication and treatment use in primary care patients with chronic pain of predominantly neuropathic origin. Fam. Pract. 2007, 24, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Schultheis, B.C.; Hanes, M.C.; Jolly, S.M.; Chakravarthy, K.V.; Deer, T.R.; Levy, R.M.; Hunter, C.W. A Comprehensive Algorithm for Management of Neuropathic Pain. Pain Med. 2019, 20, S2–S12. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Fang, P.; Xiang, D.; Yang, Y. Topical treatments for diabetic neuropathic pain (Review). Exp. Ther. Med. 2019, 17, 1963–1976. [Google Scholar] [CrossRef] [PubMed]
- Jorge, L.L.; Feres, C.C.; Telles-Dias, P. Topical preparations for pain relief: Efficacy and patient adherence. J. Pain Res. 2010, 4, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.; Nair, A.B.; Shah, H.; Jacob, S.; Shehata, T.; Morsy, M.A. Enhancement in antinociceptive and anti-inflammatory effects of tramadol by transdermal proniosome gel. Asian J. Pharm. Sci. 2019, 15, 786–796. [Google Scholar] [CrossRef]
- Kopsky, D.J.; Hesselink, J.M.K. High Doses of Topical Amitriptyline in Neuropathic Pain: Two Cases and Literature Review. Pain Pract. 2011, 12, 148–153. [Google Scholar] [CrossRef]
- Cruccu, G.; Sommer, C.; Anand, P.; Attal, N.; Baron, R.; Garcia-Larrea, L.; Haanpaa, M.; Jensen, T.S.; Serra, J.; Treede, R.-D. EFNS guidelines on neuropathic pain assessment: Revised 2009. Eur. J. Neurol. 2010, 17, 1010–1018. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef]
- Price, R.; Smith, D.; Franklin, G.; Gronseth, G.; Pignone, M.; David, W.S.; Armon, C.; Perkins, B.A.; Bril, V.; Rae-Grant, A.; et al. Oral and Topical Treatment of Painful Diabetic Polyneuropathy: Practice Guideline Update Summary. Neurology 2022, 98, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Shinu, P.; Morsy, M.A.; Nair, A.B.; Al Mouslem, A.K.; Venugopala, K.N.; Goyal, M.; Bansal, M.; Jacob, S.; Deb, P.K. Novel Therapies for the Treatment of Neuropathic Pain: Potential and Pitfalls. J. Clin. Med. 2022, 11, 3002. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.A.; Derry, S.; Aldington, D.; Cole, P.; Wiffen, P.J. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst. Rev. 2012, 12, 8242. [Google Scholar] [CrossRef]
- Macone, A.; Otis, J.A.D. Neuropathic Pain. Semin. Neurol. 2018, 38, 644–653. [Google Scholar] [CrossRef]
- Maloney, J.; Pew, S.; Wie, C.; Gupta, R.; Freeman, J.; Strand, N. Comprehensive Review of Topical Analgesics for Chronic Pain. Curr. Pain Headache Rep. 2021, 25, 7. [Google Scholar] [CrossRef]
- Cavalli, E.; Mammana, S.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The neuropathic pain: An overview of the current treatment and future therapeutic approaches. Int. J. Immunopathol. Pharmacol. 2019, 33, 1–10. [Google Scholar] [CrossRef]
- Attal, N.; de Andrade, D.C.; Adam, F.; Ranoux, D.; Teixeira, M.J.; Galhardoni, R.; Raicher, I.; Üçeyler, N.; Sommer, C.; Bouhassira, D. Safety and efficacy of repeated injections of botulinum toxin A in peripheral neuropathic pain (BOTNEP): A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2016, 15, 555–565. [Google Scholar] [CrossRef]
- Gabriel, M.; Sharma, V. Antidepressant discontinuation syndrome. Can. Med. Assoc. J. 2017, 189, E747. [Google Scholar] [CrossRef]
- Attal, N. Pharmacological treatments of neuropathic pain: The latest recommendations. Rev. Neurol. 2018, 175, 46–50. [Google Scholar] [CrossRef]
- Nolano, M.; Simone, D.A.; Wendelschafer-Crabb, G.; Johnson, T.; Hazen, E.; Kennedy, W.R. Topical capsaicin in humans: Parallel loss of epidermal nerve fibers and pain sensation. Pain 1999, 81, 135–145. [Google Scholar] [CrossRef]
- Aoki, K.R.; Guyer, B. Botulinum toxin type A and other botulinum toxin serotypes: A comparative review of biochemical and pharmacological actions. Eur. J. Neurol. 2001, 8, 21–29. [Google Scholar] [CrossRef]
- Mitchell, K.; Bates, B.D.; Keller, J.M.; Lopez, M.; Scholl, L.; Navarro, J.; Madian, N.; Haspel, G.; Nemenov, I.M.; Iadarola, M.J. Ablation of Rat TRPV1-Expressing Adelta/C-Fibers with Resiniferatoxin: Analysis of Withdrawal Behaviors, Recovery of Function and Molecular Correlates. Mol. Pain 2010, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Trompetto, C.; Abbruzzese, G.; Berardelli, A. Central effects of botulinum toxin type A: Evidence and supposition. Mov. Disord. 2004, 19, S60–S64. [Google Scholar] [CrossRef]
- O’Neill, J.; Brock, C.; Olesen, A.E.; Andresen, T.; Nilsson, M.; Dickenson, A.H. Unravelling the Mystery of Capsaicin: A Tool to Understand and Treat Pain. Pharmacol. Rev. 2012, 64, 939–971. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, C.; Shi, F.; Firempong, C.K.; Yu, J.; Xu, X.; Zhang, W. Preparation, characterization, and pharmacokinetics study of capsaicin via hydroxypropyl-beta-cyclodextrin encapsulation. Pharm. Biol. 2015, 54, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Capsaicin: Uses, Interactions, Mechanism of Action|DrugBank Online. Available online: https://go.drugbank.com/drugs/DB06774 (accessed on 21 April 2023).
- Zhuang, H.; Zheng, J.P.; Gao, H.; De Yao, K. In vitro biodegradation and biocompatibility of gelatin/montmorillonite-chitosan intercalated nanocomposite. J. Mater. Sci. Mater. Med. 2007, 18, 951–957. [Google Scholar] [CrossRef]
- Wang, X.-H.; Su, T.; Zhao, J.; Wu, Z.; Wang, D.; Zhang, W.-N.; Wu, Q.-X.; Chen, Y. Fabrication of polysaccharides-based hydrogel films for transdermal sustained delivery of Ibuprofen. Cellulose 2020, 27, 10277–10292. [Google Scholar] [CrossRef]
- MAbu Ghalia, M.; Dahman, Y. Radiation crosslinking polymerization of poly (vinyl alcohol) and poly (ethylene glycol) with controlled drug release. J. Polym. Res. 2015, 22, 3319–3326. [Google Scholar] [CrossRef]
- Grond, S.; Sablotzki, A. Clinical pharmacology of tramadol. Clin. Pharmacokinet. 2004, 43, 879–923. [Google Scholar] [CrossRef]
- Smyj, R.; Wang, X.-P.; Han, F. Tramadol Hydrochloride. Profiles Drug Subst. Excip. Relat. Methodol. 2013, 38, 463–494. [Google Scholar] [CrossRef]
- FDA. MedWatch: The FDA Safety Information and Adverse Event Reporting Program. Available online: www.fda.gov/medwatch (accessed on 20 April 2023).
- Bockbrader, H.N.; Wesche, D.; Miller, R.; Chapel, S.; Janiczek, N.; Burger, P. A Comparison of the Pharmacokinetics and Pharmacodynamics of Pregabalin and Gabapentin. Clin. Pharmacokinet. 2010, 49, 661–669. [Google Scholar] [CrossRef]
- Ben-Menachem, E. Pregabalin Pharmacology and Its Relevance to Clinical Practice. Epilepsia 2004, 45, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Singh, N.; Jaggi, A. Pregabalin in Neuropathic Pain: Evidences and Possible Mechanisms. Curr. Neuropharmacol. 2014, 12, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.-W.; Lee, H.J.; Kim, H.-J.; Chang, S.-H.; Park, D.J. Two cases of pregabalin neurotoxicity in chronic kidney disease patients. Clin. Kidney J. 2011, 4, 138. [Google Scholar] [CrossRef] [PubMed]
- FDA; CDER. High Blood Pressure. Available online: https://www.fda.gov/ (accessed on 20 April 2023).
- Amitriptyline Active Not Recruiting Phase 4 Trials for Depressive Disorder Treatment|DrugBank Online. Available online: https://go.drugbank.com/drugs/DB00321/clinical_trials?conditions=DBCOND0024243&phase=4&purpose=treatment&status=active_not_recruiting (accessed on 20 April 2023).
- Graff-Radford, S.B.; Shaw, L.R.; Naliboff, B.N. Amitriptyline and Fluphenazine in the Treatment of Postherpetic Neuralgia. Clin. J. Pain 2000, 16, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.; Nimmo, W.; Grant, I. Bioavailability, Pharmacokinetics, and Analgesic Activity of Ketamine in Humans. J. Pharm. Sci. 1982, 71, 539–542. [Google Scholar] [CrossRef]
- Wang, R.; Gan, J.; Li, R.; Duan, J.; Zhou, J.; Lv, M.; Qi, R. Controlled delivery of ketamine from reduced graphene oxide hydrogel for neuropathic pain: In vitro and in vivo studies. J. Drug Deliv. Sci. Technol. 2020, 60, 101964. [Google Scholar] [CrossRef]
- Mion, G.; Villevieille, T. Ketamine Pharmacology: An Update (Pharmacodynamics and Molecular Aspects, Recent Findings). CNS Neurosci. Ther. 2013, 19, 370–380. [Google Scholar] [CrossRef]
- Baclofen|DrugBank Online. Available online: https://go.drugbank.com/articles/A245323 (accessed on 20 April 2023).
- Romito, J.W.; Turner, E.R.; Rosener, A.J.; Coldiron, L.; Udipi, A.; Nohrn, L.; Tausiani, J.; Romito, B.T. Baclofen therapeutics, toxicity, and withdrawal: A narrative review. SAGE Open Med. 2021, 9, 2197. [Google Scholar] [CrossRef]
- Ertzgaard, P.; Campo, C.; Calabrese, A. Efficacy and safety of oral baclofen in the management of spasticity: A rationale for intrathecal baclofen. J. Rehabil. Med. 2017, 49, 193–203. [Google Scholar] [CrossRef]
- Davies, N.M.; Anderson, K.E. Clinical Pharmacokinetics of Naproxen. Clin. Pharmacokinet. 1997, 32, 268–293. [Google Scholar] [CrossRef]
- Falany, C.N.; Ström, P.; Swedmark, S. Sulphation of o-desmethylnaproxen and related compounds by human cytosolic sulfotransferases. Br. J. Clin. Pharmacol. 2005, 60, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.; Bolinske, T.; Sinatra, E.; Akhtar, S.; Albert, G.; Allison, S.; Anwar, M.; Arita, H.; Barker, A.; Bekhit, M.H.; et al. Naproxen. In The Essence of Analgesia and Analgesics; Cambridge University Press: Cambridge, UK, 2011; pp. 221–225. [Google Scholar] [CrossRef]
- Naproxen: Uses, Interactions, Mechanism of Action|DrugBank Online. Available online: https://go.drugbank.com/drugs/DB00788 (accessed on 21 April 2023).
- Bushra, R.; Aslam, N. An Overview of Clinical Pharmacology of Ibuprofen. Oman Med. J. 2010, 25, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Anderson, K.E. Clinical Pharmacokinetics of Diclofenac. Clin. Pharmacokinet. 1997, 33, 184–213. [Google Scholar] [CrossRef]
- Todd, P.A.; Sorkin, E.M. Diclofenac Sodium. A reappraisal of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs 1988, 35, 244–285. [Google Scholar] [CrossRef]
- Campbell, J.N.; Stevens, R.; Hanson, P.; Connolly, J.; Meske, D.S.; Chung, M.-K.; Lascelles, B.D.X. Injectable Capsaicin for the Management of Pain Due to Osteoarthritis. Molecules 2021, 26, 778. [Google Scholar] [CrossRef]
- CHMP. Qutenza; INN-Capsaicin. Available online: www.ema.europa.eu/contact (accessed on 21 March 2023).
- The 6 Best Capsaicin Creams: Uses, Options, and Risks. Available online: https://www.medicalnewstoday.com/articles/best-capsaicin-creams (accessed on 21 March 2023).
- Groninger, H.; Schisler, R.E. Topical Capsaicin for Neuropathic Pain #255. J. Palliat. Med. 2012, 15, 946–947. [Google Scholar] [CrossRef]
- Contri, R.V.; Katzer, T.; Pohlmann, A.R.; Guterres, S.S. Chitosan Hydrogel Containing Capsaicinoids-Loaded Nanocapsules: An Innovative Formulation for Topical Delivery. Soft Mater. 2010, 8, 370–385. [Google Scholar] [CrossRef]
- Choi, A.-J.; Kim, C.-J.; Cho, Y.-J.; Hwang, J.-K.; Kim, C.-T. Characterization of Capsaicin-Loaded Nanoemulsions Stabilized with Alginate and Chitosan by Self-assembly. Food Bioprocess Technol. 2011, 4, 1119–1126. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Xu, Z.C. Synthesis and Properties Research on the Nanocapsulated Capsaicin by Simple Coacervation Method. J. Dispers. Sci. Technol. 2008, 29, 687–695. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Hong, C.-T.; Chiu, W.-T.; Fang, J.-Y. In vitro and in vivo evaluations of topically applied capsaicin and nonivamide from hydrogels. Int. J. Pharm. 2001, 224, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wen, X.; Pan, X.; Wang, R.; Chen, B.; Wu, C. Design and In Vitro Evaluation of Capsaicin Transdermal Controlled Release Cubic Phase Gels. AAPS PharmSciTech 2010, 11, 1405–1410. [Google Scholar] [CrossRef]
- Wang, X.-R.; Gao, S.-Q.; Niu, X.-Q.; Li, L.-J.; Ying, X.-Y.; Hu, Z.-J.; Gao, J.-Q. Capsaicin-loaded nanolipoidal carriers for topical application: Design, characterization, and in vitro/in vivo evaluation. Int. J. Nanomed. 2017, 12, 3881–3898. [Google Scholar] [CrossRef] [PubMed]
- Aylanc, V.; Ertosun, S.; Akyuz, L.; Bilican, B.K.; Gokdag, S.; Bilican, I.; Cakmak, Y.S.; Yilmaz, B.A.; Kaya, M. Natural β-chitin-protein complex film obtained from waste razor shells for transdermal capsaicin carrier. Int. J. Biol. Macromol. 2020, 155, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhou, Y.; Han, K.; Qin, L.; Dian, L.; Li, G.; Pan, X.; Wu, C. Characterization of cubosomes as a targeted and sustained transdermal delivery system for capsaicin. Drug Des. Dev. Ther. 2015, 9, 4209–4218. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.Z.; Mohammed, A.A.; Ibrahim, M.M. Technology overview and drug delivery application of proniosome. Pharm. Dev. Technol. 2016, 22, 302–311. [Google Scholar] [CrossRef]
- Jacob, S.; Nair, A.B.; Aldhubiab, B.E. Preparation and evaluation of niosome gel containing acyclovir for enhanced dermal deposition. J. Liposome Res. 2017, 27, 283–292. [Google Scholar] [CrossRef]
- Natori, N.; Shibano, Y.; Hiroki, A.; Taguchi, M.; Miyajima, A.; Yoshizawa, K.; Kawano, Y.; Hanawa, T. Preparation and Evaluation of Hydrogel Film Containing Tramadol for Reduction of Peripheral Neuropathic Pain. J. Pharm. Sci. 2022, 112, 132–137. [Google Scholar] [CrossRef]
- Sundar, V.D.; Divya, P.; Dhanaraju, M.D. Design Development and Characterisation of Tramadol Hydrochloride Loaded Transethosomal Gel Formulation for Effective Pain Management. Indian J. Pharm. Educ. Res. 2020, 54, s88–s97. [Google Scholar] [CrossRef]
- Tavakoli, J.; Wang, J.; Chuah, C.; Tang, Y. Natural-based Hydrogels: A Journey from Simple to Smart Networks for Medical Examination. Curr. Med. Chem. 2020, 27, 2704–2733. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Nair, A.B.; Shah, J.; Sreeharsha, N.; Gupta, S.; Shinu, P. Emerging Role of Hydrogels in Drug Delivery Systems, Tissue Engineering and Wound Management. Pharmaceutics 2021, 13, 357. [Google Scholar] [CrossRef]
- De Araújo, D.; dos Santos, A.C.M.; Akkari, A.C.S.; Ferreira, I.R.S.; Páscoli, M.; Guilherme, V.A.; de Paula, E.; Fraceto, L.; de Lima, R.; Melo, P.D.S.; et al. Poloxamer-based binary hydrogels for delivering tramadol hydrochloride: Sol-gel transition studies, dissolution-release kinetics, in vitro toxicity, and pharmacological evaluation. Int. J. Nanomed. 2015, 10, 2391–2401. [Google Scholar] [CrossRef] [PubMed]
- Barati, M.; Samani, S.M.; Jahromi, L.P.; Ashrafi, H.; Azadi, A. Controlled-release in-situ gel forming formulation of tramadol containing chitosan-based pro-nanogels. Int. J. Biol. Macromol. 2018, 118, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Cline, A.E.; Turrentine, J.E. Compounded Topical Analgesics for Chronic Pain. Dermatitis 2016, 27, 263–271. [Google Scholar] [CrossRef]
- Boardman, L.A.; Cooper, A.S.; Blais, L.R.; Raker, C.A. Topical Gabapentin in the Treatment of Localized and Generalized Vulvodynia. Obstet. Gynecol. 2008, 112, 579–585. [Google Scholar] [CrossRef]
- Shahid, M.; Subhan, F.; Ahmad, N.; Ali, G.; Akbar, S.; Fawad, K.; Sewell, R. Topical gabapentin gel alleviates allodynia and hyperalgesia in the chronic sciatic nerve constriction injury neuropathic pain model. Eur. J. Pain 2016, 21, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.; Subhan, F.; Ahmad, N.; Sewell, R.D.E. Efficacy of a topical gabapentin gel in a cisplatin paradigm of chemotherapy-induced peripheral neuropathy. BMC Pharmacol. Toxicol. 2019, 20, 51. [Google Scholar] [CrossRef]
- Martin, C.J.; Alcock, N.; Hiom, S.; Birchall, J.C. Development and Evaluation of Topical Gabapentin Formulations. Pharmaceutics 2017, 9, 31. [Google Scholar] [CrossRef]
- Khosravi, N.; Youseftabar-Miri, L.; Divsar, F.; Hallajian, S.; Hafezi, K. Development and evaluation of chitosan-g-poly(acrylic acid-co-acrylamide) hydrogel composite containing gabapentin for in vitro controlled release. J. Mol. Struct. 2022, 1270, 133934. [Google Scholar] [CrossRef]
- Shakshuki, A.; Yeung, P.; Agu, R.U. Compounded gabapentin for neuropathic pain: Stability and beyond-use date (BUD) in some commonly used bases. J. Am. Pharm. Assoc. 2019, 59, 514–520. [Google Scholar] [CrossRef]
- Le, U.M.; Baltzley, S.; AlGhananeem, A. Gabapentin in Elastic Liposomes: Preparation, Characterization, Drug Release, and Penetration Through Porcine Skin. Int. J. Pharm. Compd. 2018, 22, 498–503. [Google Scholar] [PubMed]
- Arafa, M.G.; Ayoub, B.M. DOE Optimization of Nano-based Carrier of Pregabalin as Hydrogel: New Therapeutic & Chemometric Approaches for Controlled Drug Delivery Systems. Sci. Rep. 2017, 7, 41503. [Google Scholar] [CrossRef]
- Haddad, N.; Hasian, J. Effect of combination of some Polymers with Carbopol 940 on Pregabalin Release Rate from Emulgels. Res. J. Pharm. Technol. 2022, 15, 2003–2009. [Google Scholar] [CrossRef]
- Nagao, M.; Tajima, M.; Sugiyama, E.; Shinouchi, R.; Shibata, K.; Yoshikawa, M.; Yamamoto, T.; Sato, V.H.; Nobe, K.; Sato, H. Evaluation of in vitro transdermal permeation, mass spectrometric imaging, and in vivo analgesic effects of pregabalin using a pluronic lecithin organogel formulation in mice. Pharmacol. Res. Perspect. 2022, 10, e00919. [Google Scholar] [CrossRef] [PubMed]
- Cevik, O.; Gidon, D.; Kizilel, S. Visible-light-induced synthesis of pH-responsive composite hydrogels for controlled delivery of the anticonvulsant drug pregabalin. Acta Biomater. 2015, 11, 151–161. [Google Scholar] [CrossRef]
- Barton, D.L.; Wos, E.J.; Qin, R.; Mattar, B.I.; Green, N.B.; Lanier, K.S.; Bearden, J.D.; Kugler, J.W.; Hoff, K.L.; Reddy, P.S.; et al. A double-blind, placebo-controlled trial of a topical treatment for chemotherapy-induced peripheral neuropathy: NCCTG trial N06CA. Support. Care Cancer 2010, 19, 833–841. [Google Scholar] [CrossRef]
- Ho, K.-Y.; Huh, B.K.; White, W.D.; Yeh, C.-C.; Miller, E.J. Topical Amitriptyline Versus Lidocaine in the Treatment of Neuropathic Pain. Clin. J. Pain 2008, 24, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Shakshuki, A.; Yeung, P.; Agu, R.U. Compounded Topical Amitriptyline for Neuropathic Pain: In Vitro Release from Compounding Bases and Potential Correlation with Clinical Efficacy. Can. J. Hosp. Pharm. 2020, 73, 133. [Google Scholar] [CrossRef]
- Feturi, F.G.; Weinstock, M.; Zhao, W.; Zhang, W.; Schnider, J.T.; Erbas, V.E.; Oksuz, S.; Plock, J.A.; Rohan, L.; Spiess, A.M.; et al. Mycophenolic Acid for Topical Immunosuppression in Vascularized Composite Allotransplantation: Optimizing Formulation and Preliminary Evaluation of Bioavailability and Pharmacokinetics. Front. Surg. 2018, 5, 20. [Google Scholar] [CrossRef]
- Nasr-Alla, S.M.; El-Nabarawi, M.; Bendas, E.; El-Ridy, M.; Abdel-Jaleel, G.; Nasr-Alla, S.M. Formulation and evaluation of topical niosomal gel of baclofen. J. Chem. Pharm. Res. 2015, 7, 277–288. [Google Scholar]
- Syed, M.; Rao, D.; Rao, V.; Nagarani, G. Formu Lation and E Va Lu Ation of Contr O Lle D Re Le Ase of Top Ical ge l bac lofe N. EMCare Cover. J. Pharmanest Int. J. Adv. Pharm. Sci. 2023, 6, 5. [Google Scholar]
- Yussef, A.; Fayez, S.; Sakran, W. Formulation and Evaluation of Baclofen Polymeric Nanoparticles for Transdermal Delivery In-vitro and Ex-vivo Optimization. J. Adv. Pharm. Res. 2021, 5, 285–296. [Google Scholar] [CrossRef]
- Kocot-Kępska, M.; Zajączkowska, R.; Mika, J.; Kopsky, D.J.; Wordliczek, J.; Dobrogowski, J.; Przeklasa-Muszyńska, A. Topical Treatments and Their Molecular/Cellular Mechanisms in Patients with Peripheral Neuropathic Pain—Narrative Review. Pharmaceutics 2021, 13, 450. [Google Scholar] [CrossRef]
- Barakat, N.S. Evaluation of Glycofurol-Based Gel as a New Vehicle for Topical Application of Naproxen. AAPS PharmSciTech 2010, 11, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kuang, Y.; Gao, Y.; Du, X.; Shi, J.; Xu, B. d-Amino Acids Boost the Selectivity and Confer Supramolecular Hydrogels of a Nonsteroidal Anti-Inflammatory Drug (NSAID). J. Am. Chem. Soc. 2012, 135, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Vilaça, H.; Hortelão, A.C.; Castanheira, E.M.; Queiroz, M.J.R.; Hilliou, L.; Hamley, I.W.; Martins, J.A.; Ferreira, P.M. Dehydrodipeptide Hydrogelators Containing Naproxen N-Capped Tryptophan: Self-Assembly, Hydrogel Characterization, and Evaluation as Potential Drug Nanocarriers. Biomacromolecules 2015, 16, 3562–3573. [Google Scholar] [CrossRef]
- Chen, H.; Chang, X.; Du, D.; Li, J.; Xu, H.; Yang, X. Microemulsion-based hydrogel formulation of ibuprofen for topical delivery. Int. J. Pharm. 2006, 315, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Mauri, E.; Rossetti, A.; Mozetic, P.; Schiavon, C.; Sacchetti, A.; Rainer, A.; Rossi, F. Ester coupling of ibuprofen in hydrogel matrix: A facile one-step strategy for controlled anti-inflammatory drug release. Eur. J. Pharm. Biopharm. 2019, 146, 143–149. [Google Scholar] [CrossRef]
- Mahmood, S.; Almurisi, S.H.; Al-Japairai, K.; Hilles, A.R.; Alelwani, W.; Bannunah, A.M.; Alshammari, F.; Alheibshy, F. Ibuprofen-Loaded Chitosan–Lipid Nanoconjugate Hydrogel with Gum Arabic: Green Synthesis, Characterisation, In Vitro Kinetics Mechanistic Release Study and PGE2 Production Test. Gels 2021, 7, 254. [Google Scholar] [CrossRef]
- Andrade-Vivero, P.; Fernandez-Gabriel, E.; Alvarez-Lorenzo, C.; Concheiro, A. Improving the Loading and Release of NSAIDs from pHEMA Hydrogels by Copolymerization with Functionalized Monomers. J. Pharm. Sci. 2007, 96, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Uzaraga, I.; Gerbis, B.; Holwerda, E.; Gillis, D.; Wai, E. Topical amitriptyline, ketamine, and lidocaine in neuropathic pain caused by radiation skin reaction: A pilot study. Support. Care Cancer 2011, 20, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Portugal, F.; Araújo, A.; Silva, C.; Campos, M.; Valentim, A. Combination gel of 2% amitriptyline and 0.5% ketamine to treat refractory erythromelalgia pain—A case report of pain control success. Rev. Esp. Anestesiol. Reanim. 2021, 68, 293–296. [Google Scholar] [CrossRef]
- Kaku, M.; Vinik, A.; Simpson, D.M. Pathways in the Diagnosis and Management of Diabetic Polyneuropathy. Curr. Diabetes Rep. 2015, 15, 35–609. [Google Scholar] [CrossRef]
- Sawynok, J.; Zinger, C. Topical amitriptyline and ketamine for post-herpetic neuralgia and other forms of neuropathic pain. Expert Opin. Pharmacother. 2015, 17, 601–609. [Google Scholar] [CrossRef]
- Silva, C.; Topgaard, D.; Kocherbitov, V.; Sousa, J.; Pais, A.; Sparr, E. Stratum corneum hydration: Phase transformations and mobility in stratum corneum, extracted lipids and isolated corneocytes. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2647–2659. [Google Scholar] [CrossRef]
- Bhowmik, D. Topical Drug Delivery System. Available online: https://www.researchgate.net/publication/304716203 (accessed on 20 March 2023).
- Van Nooten, F.; Treur, M.; Pantiri, K.; Stoker, M.; Charokopou, M. Capsaicin 8% Patch Versus Oral Neuropathic Pain Medications for the Treatment of Painful Diabetic Peripheral Neuropathy: A Systematic Literature Review and Network Meta-analysis. Clin. Ther. 2017, 39, 787–803.e18. [Google Scholar] [CrossRef]
- Das, B.; Nayak, A.K.; Mallick, S. Nanovesicles for delivery of antifungal drugs. Appl. Nanovesicular Drug Deliv. 2022, 2022, 383–397. [Google Scholar] [CrossRef]

{kind=link}
| Pharmacological Treatment of NeP | ||||||
|---|---|---|---|---|---|---|
| First-Line Therapy | Second-Line Therapy | Third-Line Therapy | ||||
| TCA Amitriptyline | SNRI Duloxetine Venlafaxine | Gabapentinoids Pregabalin Gabapentin | Topical treatment Capsaicine Lidocaine | Tramadol | Strong opioids Morphine Oxycodone | Botulinum toxin type A |
| Painful neuropathy Nerve injury pain Postherpetic neuralgia Central postpartum pain Spinal cord injury | Diabetic neuropathy Fibromyalgia Back pain Diabetic pain | Spinal cord injury Herpetic neuralgia Phantom limb syndrome | Localized NeP: Postherpetic neuralgia Diabetic pain Regional pain syndrome Diabetic pain Central Pain | Chemotherapy-induced peripheral neuropathy Phantom limb syndrome | Diabetic pain Postherpetic neuralgia Central pain | Focal peripheral neuropathy and allodynia |
| Substance | Absorbtion | Metabolism | Elimination | Half-life | Physicochemical Characteristics | References |
|---|---|---|---|---|---|---|
| Capsaicin |
Oral administration 50 to 90% Intravenous/subcutaneous administration in the brain and spinal cord about 5 times higher than in blood; in the liver about 3 times higher than in the blood. Topical administration: rapidly and well absorbed through the skin. | Significant first-pass effect of liver after oral administration | Renal excretion |
Following oral ingestion approximately 24.9 ± 5.0 min. Following topical application approximately 24 h | Low water solubility. pKa (Strongest Acidic): 9.93 pKa (Strongest Basic): −1.4 Melting point: 65 °C | [35,36,37] |
| Tramadol | Oral administration: rapidly and almost completely absorbed, with a bioavailability of 75% | Extensive first-pass metabolism in the liver | Primarily through metabolism by the liver; metabolites are excreted primarily by the kidneys. | 5–6 h | Tramadol hydrochloride salt is highly water-soluble. The pH range of tramadol hydrochloride salt: 4.0–5.5. The melting point of tramadol: 180–184 °C. The solubility of tramadol in ethanol and chloroform makes it possible to incorporate it into hydro-alcoholic or lipophilic gel formulations. | [41,42] |
| Gabapentin | The oral bioavailability is inversely proportional to the administered dose. | Not appreciably metabolized | In the urine as unchanged drug. | 5–7 h | pKa: 3.7 Octanol/water partition coefficient: 1.24, indicating that it is highly hydrophilic. | [43,44] |
| Pregabalin | Oral administration bioavailability ≥90% regardless of the dose | Less than 2% is metabolized | Almost exclusively eliminated in the urine. | 6.3 h | Good water solubility. pKa: 4.2. Octanol/water partition coefficient: −1.35 Low molecular weight −159.23 g/mol | [45,46,47,48] |
| Amitriptyline | Rapidly absorbed following oral administration | Suffers from first-pass metabolism. The main active metabolite is nortriptyline. | Amitriptyline and its metabolites are mainly excreted in the urine. | About 25 h | Highly lipophilic, small-molecular-weight compound. Low water solubility: pKa: 9.4 Octanol/water partition coefficient: 3.3. High molecular weight: 313.87 g/mol. | [49,50] |
| Ketamine | Absorption is very rapid and the bioavailability is around 93%. | Mainly hepatic metabolism. | Manly urine excretion mainly in the form of metabolites | 186 min | pKa: 7.5 Molecular mass: 238 g/mol Ketamine is typically administered intravenously, and its use is associated with several side effects. | [51,52,53] |
| Baclofen | Oral bioavailability: 70% to 85% | About 15% of the oral dose is metabolized in the liver. | 70–80% is eliminated in an unchanged form by renal excretion. | 2–6 h after oral administration. | Water solubility: 4 mg/mL pKa: 9.62 + 0.1 (amino group) 3.87 + 0.1 (carboxyl group) Adverse effects following oral administration affect between 25% and 75% of patients. | [54,55,56] |
| Naproxen | Peak plasma concentration after 1 h, peak plasma concentration for naproxen (free acid) is observed after 2 h | Heavily metabolized in the liver. | Approximately 95% of naproxen and its metabolites can be identified in the urine | 12–17 h | Water solubility: 15.9 mg/L pKa: 4.15 Melting point: 152 °C | [57,58,59,60] |
| Ibuprofen | Very well absorbed orally. | Rapidly metabolized and biotransformed in the liver to the formation of major metabolites. | Eliminated in the urine. | 1.2–2 h | Water solubility: 21 mg/L pKa: 5.3 Melting point: 75–77 °C | [61] |
| Diclofenac | Totally absorbed from the gastrointestinal tract. | Substantial first-pass metabolism | 60–70% urinary elimination and 30% elimination through feces | 2 h | Water solubility: 2.37 mg/mL pKa: 4.15 Melting point: 283–285 °C | [62,63] |
| Substance | Gel Type | Method of Preparation | Formula with Optimal In Vitro Parameters | Role of Agents | Parameter | Advantages of the New Formulation in the Treatments of Neuropathic Pain | References |
|---|---|---|---|---|---|---|---|
| Capsaicin | Hydrogel | Dissolution The certain quantity of polymer and drug was added into pH 4 buffer with continuous stirring for 1 h. | 0.075% capsaicin, 6% CMC-Na | CMC, synthetic water-soluble cellulose added as matrix for drug delivery systems. | Rate of release 12.69 ± 0.58 g/cm2 per ½ h | Advantages The cumulative quantity of capsaicin 4 h after the application is bigger than that of marketed creams. CMC-Na showed better bio-adhesion to the skin, resulting in a prolonged time of location at the site of application and enhanced permeation efficacy. CMC-Na hydrogels can produce a fast onset and sustained duration of capsaicin release, in contrast to cream bases. Disadvantages All individuals revealed more powerful pungent sensation of CMC-Na hydrogels than cream bases. | [71] |
| Capsaicin | Biogel | Dissolution Capsaicin was weighed into the monooleate and heated to melt at 45 °C. After the mixture was homogeneously vortexed, propylene glycol and water were added, then homogeneously vortexed. | 2.5 mg/g capsaicin, 63% glycerol monooleate, 7% propylene glycol 30% | Monooleate—solvent Propylene glycol, organic solvent decreases the cubic phase gel viscosity and it is a skin penetration enhancer. | Rate of release 33% | Cubic phase gels based on the ternary phase diagram of the monooleate–propylene glycol–water system offer transdermal controlled release of capsaicin. After 108 h, around 30% of capsaicin was released from the gels. | [72] |
| Capsaicin | Emulsion gel | Hot melt homogenization technique Capsaicin and melted lipids were thoroughly mixed using a magnetic stirrer, forming a clear lipid phase. A separate aqueous phase with Tween 80 was prepared. The hot lipid phase was then gradually added to the hot aqueous phase and homogenized. | Capsaicin, carbopol, ethanol, PEG400 | Ethanol—solvent PEG400—solvent Carbopol, a rheology modifier and extended-release polymers that enhance the viscosity of gel and skin permeation and offers sustained release | Rate of release 59.2% Permeation rate (skin of Sprague–Dawley rats) 7.2% (after 24 h) | The capsaicin-loaded nanoparticles containing solution showed a higher analgesic efficacy than the capsaicin-loaded nanoparticles containing gel in the Hot-plate test. The capsaicin-based nanoparticles formulations presented increased analgesic efficacy than capsaicin cream at the same concentration in the Hot-plate test due to the higher permeation ability and improved skin retention. The gel induced slightly less irritation than the solutions. | [73] |
| Capsaicin | Biogel | Passive loading technique Capsaicin was dissolved in methanol using a sonicator. After adding natural β-chitin–protein complex film, the mix was vortexed at 120 rpm and incubated in a shaker at room temperature for 48 h. | 100 mg capsaicin, 8 mL methanol, 200 mg β-chitin–protein complex | Methanol—solvent β-chitin–protein complex—natural polymer β-chitin–protein complex film has a finer structure in contrast to synthetically made chitosan films. | Rate of release pH 4.0: 50.49% (48 h) pH 5.5: 59.81% (72 h) pH 7.4: 59.02% (96 h) | β-chitin–protein complex film offers a prolonged release rate of capsaicin The maximum capsaicin release was at pH 7.4, which is slightly higher than that of the skin, after 96 h. | [74] |
| Capsaicin | Hydrogel | Interfacial deposition of preformed polymer method An organic phase containing Eudragit RS100®, capsaicinoids mixture, acetone, and capric/caprylic triglyceride was injected in an aqueous phase containing polysorbate 80. After mixing, the solvents were removed with a reduced-pressure evaporator. | 5 mg capsaicinoids mixture, 3.5% chitosan, 1.5% lactic acid, 100 mg Eudragit RS100® | Lactic acid-pH modifier Eudragit RS100®-polymer Chitosan, a polysaccharide, that presents enhanced skin bioadhesion, film-forming capability, and wound-healing promotion | Rate of release 81 ± 1% (96 h) | The best formula had the lowest quantity of lactic acid, which provides a tolerable pH value (4.34 ± 0.11) and good biocompatibility. | [68] |
| Capsaicin | Emulsion gels | High shear mixing Capsaicin was added to a melted mixture of phytantriol and poloxamer 407 at 60 °C. This mixture was stirred with water, and after equilibrating for 48 h at room temperature, a cubic phase gel formed. | 10.88 mg capsaicin, 0.3047 g PL F127, 3.0096 g phytantriol. | Phytantriol—solvent | Rate of release F1: 41% F2: 33% Permeation rate (skin of Sprague–Dawley rats) F1: 0.32 ± 0.05 μg·cm−1·h−1 F2: 0.18 ± 0.02 μg·cm−2·h−1 | Phytantriol forms cubosomes determining a sustained release of capsaicine (longer than that of on-market products). Cubosomes formulations induced no obvious irritation to the skin. | [75] |
| Tramadol | Organogel | Coacervation phase separation method Proniosome components and tramadol were mixed with absolute ethyl alcohol and heated in a thermostatic water bath. Further, phosphate buffer (pH 7.4) was added on the water bath. After cooling down to room temperature, the solutions was mixed with HPMC. | 100 mg tramadol, 1800 mg Span 80, 1800 mg lecithin, 100 mg cholesterol | Lecithin—nonionic surfactant Cholesterol—nonionic surfactant Span 80—hydrophobic surfactant | Rate of release 60% (6 h) Permeation rate (skin of New Zealand Wistar albino rats) 2300 µg/cm−1 (in 24 h) | The tramadol gel demonstrated higher analgesic efficacy than oral tablets in rat tests and showed significant anti-inflammatory effects. This formula had improved entrapment efficiency and transdermal flux compared to tramadol cream. The low transition temperature of Span80 contributed to a fluid state, facilitating drug transport into the skin. | [18] |
| Tramadol | Hydrogel | Irradiation with electron beams Tramadol solution was created by diluting tramadol injection in purified water. HPMC hydrogel films were purified and dried before being turned into xerogels. The xerogels were then submerged in tramadol solution for 24 h to obtain hydrogel films containing tramadol (30 or 50 kGy dose). | 20% tramadol, 20% hydroxypropyl methylcellulose | HPMC—cellulosic polymer HPMC interaction with the drug is minimum and do not inhibit the sustained release of the drug HPMC produced a transparent film | Rate of release 100% (after 240 min) Permeation rate (skin of mice) 1.36 mg/cm2 (in 240 min) | The tramadol-containing gel showed a higher analgesic efficacy than oral tramadol tablets in acetic-acid-induced abdominal writhing test in rats. The new formula presented an enhanced release and skin permeation compared to standard formulations. The amount of tramadol released from the hydrogel film and the amount of skin permeation increased by changing the electron dose. | [78] |
| Tramadol | Organogel | Cold method The aqueous phase was heated at 30 °C and then delivered to the organic phase dropwise with constant mixing. The stir was continued for 45 min to bring the transethosomal dispersions that were supplemented for size reduction by probe sonication. | 100 mg tramadol, 3% α phosphatidylcholine from egg yolk, 0.5% cremophor EL-35 | α phosphatidylcholine—lipid carrier; showed low viscosity Cremophor EL-35—edge activator | Rate of release 79.98% (8 h) | The new formula exhibited a controlled rate of release compared to standard formulations. The use of edge activator in formulation increased the skin permeability. | [79] |
| Tramadol | Hydrogel | Dispersion Chitosan was dispersed in an acetic acid solution, and tramadol and poloxamer F-127 were dissolved in it. The mixture was placed in an ice bath for 30 min, then glycerophosphate disodium salt hydrate and pentasodium triphosphate were added. Finally, the mixture was moved to a 37 °C water bath, where gel formation occurred after about 1–1.5 min. | 20% tramadol, 1% chitosan, 20% PL F-127, 14.5% glycerophosphate disodium salt hydrate, 0.5% pentasodium triphosphate | Chitosan, a natural polysaccharide that can form gels by interacting with other polymers or through physical and chemical cross-linking. It can form hydrogels by swelling in water. | Rate of release 80% (8 h) | Chitosan extends the release of drug compared to tramadol-containing gel. | [83] |
| Tramadol | Hydrogel | Cold method Tramadol was dispersed in various solutions containing PL 407 alone or in binary systems with PL 188 and were retained at 4 °C under magnetic stirring. The PL concentrations were selected in order to obtain a thermoreversible gel at minimum possible final concentration with a maximum final PL concentration of 35% (weight per weight [w/w]). | 20 mg × mL −1 tramadol, 25% PL 407, 10% PL 188 | PL 407 and PL 188—nonionic surfactants | Rate of release 100% (after 4 h) | PL 407 and PL 188-based binary hydrogels showed controlled release of tramadol. Subsequently, they had extended duration of analgesia (72 h) compared to tramadol solution in tail-flick test. This leads to the possibility of reapplying every 48–72 h at lower doses. The gel showed an enhanced release rate at pH 7.4 and 37 °C. | [82] |
| Gabapentin | Hydrogel | Dissolution Gabapentin was dissolved into de-ionized water, and methyl and propyl hydroxybenzoate were dissolved in a permeation enhancer solvent. The enhancer mixture was added to the aqueous mixture and mixed for 5 min. | 6% gabapentin, 0.75% carbopol | Carbopol—synthetic polymer carbomer | Mean flux value 2661.62 ± 50.39 Permeation rate (“nonhy-drated” human epidermal membrane) 7.56 ± 5.50 mcg/cm2/h | Carbopol can incorporate low-molecular-weight compounds, such as gabapentin. Prehydrated and non-prehydrated membranes had similar gabapentin permeability, but prehydrated ones showed less variability. | [88] |
| Gabapentin | Emulsion gel | - | 10% gabapentin, xanthan gum hydrocolloid, polyacrylamide | - | - | A 10% w/w topical gabapentin gel applied thrice daily demonstrated strong antiallodynic and antihyperalgic effects in Hot-plate and von Frey test. Topical use of the gel potentially avoids dose titration in neuropathic pain patients, reduces pain similarly to systemic gabapentin, and avoids related side effects. | [86] |
| Gabapentin | Emulsion gel | - | 10% gabapentin, xanthan gum hydrocolloid, polyacrylamide | - | - | Topical application of gabapentin highly reduced cisplatin-associated neuropathic allodynia and heat-hypoalgesia. It might offer an alternative for neuropathic pain relief in patients treated with chemotherapy or those intolerant to systemic medications’ side effects. | [87] |
| Gabapentin | Hydrogel | Copolymerization The chitosan-g-poly(acrylic acid-co-acrylamide) hydrogel was obtained following the reaction between a radical initiator (ammonium persulfate) and a cross-linking agent (N,N′-methylene bisacrylamide). | Gabapentin, acrylic acid, acrylamide monomers, chitosan | Chitosan—natural polymer Acrylamide monomers—cross-linkers | Rate of release 90% in the first 2 h | Acrylic acid and acrylamide monomers overcome the disadvantages of chitosan by cross-linking. In vitro simulation of gabapentin release from hydrogel in stomach-like (pH 1.2) and physiological buffer (pH 7.4) conditions showed direct drug diffusion from loaded gel samples in both pHs, with faster diffusion in pH 1.2 due to swelled samples. | [89] |
| Pregabalin | Organogel | Mixing A 1% aqueous solution of pregabalin was mixed with propylene glycol. This solution was then mixed with the oil phase solution of a PLO gel kit. | 0.4% pregabalin, lecithin, isopropyl palmitate, PL 407 | PL 407—block copolymer Lecithin—absorption enhancer Isopropyl palmitate—solvent for lecithin | Permeation rate (skin of mice) 0.7 µg/mL (after 120 h) | A significant analgesic effect was observed 1.5 h after application using von Frey test in mice streptozotocin-induced diabetic neuropathy. | [94] |
| Pregabalin | Hydrogel | High shear mixing A transparent gel was obtained by spraying with continuous stirring on the water surface 2% w/w gel bases (HPMC and carbopol 934). Then a uniform and clear solution was formed under continuous stirring by dissolving the pregabalin in the polymer dispersion. | 5 g niosomes (1.5 g pregabalin F1 Span 60: cholesterol, 4:1 F2 Span 60: cholesterol, 4:4 F3 Span 60: cholesterol, 4:7) in HPMC or carbopol | Cholesterol enhanced the entrapment efficiency. Span 60, nonionic surfactant, gave the highest entrapment efficiency and stability of niosomes. | Release rate 32.2 ± 0.02% Permeation rate (skin of rats) 28.34% (in a period of 8 h) | - | [92] |
| Pregabalin | Hydrogel | Photopolymerization under both UV lights The pH-responsive layer was prepared using poly(methacrylic acid-g-ethylene glycol) as the macromer, eosin Y as the photoinitiator, and triethanolamine as the co-initiator. Hydrophobic domains were added by incorporating cross-linked styrene-butadiene-styrene (SBS) 30 copolymer in the pH-sensitive prepolymer. | 3.6 g pregabalin, 2.0 g Poly(ethylene glycol) monomethyl ether monomethacrylate | - | Rate of release 86.4% at neutral pH Permeation rate (human fibroblast cell line): 28.34% over a period of 8 h | The gel was swollen and transparent at pH 7.0, while opaque at pH 2.2. Hydrogels formed with UV and visible light showed reversibility of swelling, responding to repeated pH variations in both low (pH 2.2) and high (pH 7.0) buffers. | [95] |
| Pregabalin | Emulsion gel | High shear mixing Span 80 was dissolved in castor oil (oil phase). Pregabalin and tween 80 were dissolved in distilled water, mixed with methyl and propyl parabens dissolved in propylene glycol (aqueous phase). Both phases were heated separately to 70–80 °C, before combining the oil phase with the aqueous phase. | 2 g pregabalin, 10 g propylene glycol, 4 g Tween 80, 1 g Span 80, 10 g castor oil, 0.4 g carbopol 940. | Carbopol—polymer | Rate of release 30% in 20 min and 93% in 360 min | Carbopol in the specified amount resulted in the best drug release rate. | [93] |
| Amitriptyline | Organogel | High shear mixing Amitriptyline was mixed with Poloxamer 30% after being dissolved in sterile water. Isopropyl lecithin was mixed with the poloxamer component. | 5% amitryptiline, 30% PL, lecithin-isopropyl myristate | - | - | No significant change in pain intensity vs. topical lidocaine in patients with postherpetic neuralgia, chronic postsurgical pain, and painful peripheral neuropathy | [97] |
| Amitriptyline | Organogel | High shear mixing Amitriptyline was finely powdered and mixed with ethoxy diglycol to obtain a smooth paste. Mediflo PLO gel was added. | 10% amitryptiline, ethoxy diglycol, Mediflo PLO gel | - | Rate of release 53.2% Permeation rate (Strat-M membrane) 9.3% (over 24 h) | Mediflo PLO gel resulted in the highest release rate at 32 ± 0.5 °C and pH 7.4. However, Lipoderm base and Emollient Cream resulted in a lower cumulative permeation relative to Mediflo PLO gel. | [98] |
| Ketamine | Hydrogel | High shear mixing Ketamine was added in the Pluronic® F127 stabilized reduced graphene oxide dispersion and kept under mild stirring for 24 h. The viscosity of the ketamine-loaded Pluronic® F127 stabilized reduced graphene oxide dispersion was increased by adding 2% Carbopol 94 for topical application. | 5% ketamine, 0.1 μg/mL pluronic F127 stabilized reduced graphene oxide, 2% Carbopol 940 | Pluronic F127 stabilized reduced graphene oxide offered a prolonged release of ketamine due to the unique π-π stacking interaction between ketamine and reduced graphene oxide. | Permeation rate 120.0 μg/cm2/h | Ketamine-loaded reduced graphene oxide demonstrated sustained analgesic effect (24 h) compared to control hydrogel (4 h) in the tail-flick study. This alternative for neuropathic pain treatment using Pluronic® F127 graphene oxide hydrogel avoids side effects and skin irritation associated with other administration routes. | [52] |
| Baclofen | Organogel | Thin-film hydration method | 5% baclofen niosomes (baclofen/Span 60/40/cholesterol) in 1% Carbopol 934 | Span60/40—nonionic surfactants Cholesterol, enhancer of niosomal membrane rigidity. | Rate of release 62.75% (in 24 h)–for niosomes Permeation rate (cellulose membrane) 99.51% (in 24 h) at a maintained temperature of 37 ± 0.5 °C and pH 5.5 | Reduced carrageenan-induced paw edema similar to oral marketed tablets. Topically applied niosomes improve the residence time of drugs in the stratum corneum and epidermis, while decreasing the systemic absorption of drug Carbopol prints faster permeation to the skin. Niosomes offer a prolonged and controlled release of the drug. | [100] |
| Baclofen | Hydrogel | High shear mixing | F13: baclofen in Carbopol 934 (1:4) F18: baclofen in xanthan (1:6) | Carbopol/Xanthan-thickening agent, stabilizer, controlled release | Rate of release F13: 98.98% (after 24 h) Permeation rate (skin of Albino rats) F13: 4.27 µg/cm2 /h | Carbopol and xanthan as gelling agents showed enhanced permeation through the skin and release rate. | [101] |
| Naproxen | Inorganic gel | Dissolution Gantrez AN 119 and glycofurol were homogenized to form a clear dispersion, which was degassed under vacuum and stored at room temperature for 1 day before use. Naproxen was dissolved before the addition of the polymer. | 5% naproxen, 2.5% Gantrez, glycofurol | Gantrez—copolymer, gelling and bioadhesive agent. Glycofurol—good adhesiveness and spreadability. | Rate of release 584.78 ± 32.8 μg/cm2 Permeation rate (skin of Sprague–Dawley rats) 161.168 ± 29.2 μg/cm2/h | No skin irritation Glycofurol use results in high drug permeation. | [104] |
| Naproxen | Hydrogel | Synthetic procedures | Naproxen, diphenylalanine, N-hydroxysuccinimide | Diphenylalanine-D-amino acid | Rate of release 35.8% after 24 h | Minimal adverse effects | [105] |
| Ibuprofen | Hydrogel | High shear mixing The clear microemulsion-based hydrogel was prepared by completely dissolving the xanthan gum in the microemulsion under stirring. | 3% ibuprofen, 3% ethyl oleate, 20% Tween 80, 10% propylene glycol, 1.5% xanthan gum | Xanthan gum increases viscosity. Ethyl oleate enhances the solubilizing capacity of microemulsion systems. Tween 80 acts as a surfactant, and propylene glycol as a cosurfactant. | Rate of release 38.06 ± 1.04 (µg × cm−2 h−1) Permeation rate (porcine ear skin) 7.61 ± 0.21 × 10−3 cm × h−1 | Ethyl oleate determines excellent skin permeation rate of ibuprofen. | [107] |
| Ibuprofen | Hydrogel | Dissolution Carbomer 974P was dissolved in a phosphate-buffered saline solution and pH was adjusted to 7.8 with 1 M NaOH. Agarose was added and the mixture was microwave-irradiated (500 W) for 30 s at 80 °C to initiate the condensation reaction. After cooling to 55 °C, the mixture was poured into steel cylindrical molds for gelation. | 2.8% ibuprofen in carbomer 974P | - | Rate of release 80% after 48 h | Rate of release was pH-sensitive: at acidic or basic pH levels, increased hydrolytic rate was reported, with the alkaline conditions showing the fastest release (+10% cumulative release in acidic medium and +20% cumulative release in alkaline medium at 24 h, compared to neutral pH)- | [108] |
| Ibuprofen | Hydrogel | Freeze–thaw cycle Ibuprofen was dissolved in sodium hydroxide and polyethylene glycol. Chitosan was dissolved in 2% acetic acid and brought to 20 mL with distilled water to give a 2% w/v concentration. The drug–polymer suspension was obtained by adding the Ibuprofen solution to the chitosan solution. A nanoconjugate with a gum arabic gel matrix was formed by dispersing gum arabic propylene glycol solution and polyvinylpyrrolidone under continuous stirring and then incorporating the ibuprofen nanoconjugate into the gum arabic matrix solution. | 50 mg ibuprofen, 4% phospholipon 90G, 2% chitosan, 10% polyvinyl alcohol, 2.5% gum arabic | Gum arabic—reduced the crystallinity of ibuprofen | Rate of release 90% (after approximately 13 h) | Nanoconjugate hydrogel ibuprofen-loaded chitosan–PC90G showed sustained and controlled release, surpassing the disadvantages associated with the oral dosage form. | [109] |
| Ibuprofen/Diclofenac | Inorganic gel | Dissolution Polymerization Ethyleneglycol dimethacrylate, N-(3-aminopropyl) methacrylamide, and 4-vinyl-pyridine were dissolved in 2-Hydroxyethyl methacrylate. The initiator 2, 20-azobis(isobutyronitrile) was added, and the monomer solution was injected into a mold. Polymerization occurred for 12 h at 50 °C followed by 24 h at 70 °C. The gels were then submerged in boiling water for 15 min to remove unreacted monomers. The resulting 10.5 mm diameter discs were washed in water, 0.9% NaCl, and 0.1 M HCl, and finally dried at 40 °C. | 5 mg/g diclofenac, 18 mg/g ibuprofen, 4-vinyl-pyridine, N-(3-aminopropyl) methacrylamide, ethyleneglycol dimethacrylate | 4-vinyl-pyridine and N-(3-aminopropyl) methacrylamide remarkably increased the amount of ibuprofen and diclofenac loaded. | Rate of release 60% for both ibuprofen and diclofenac Sustained release for 24 h for ibuprofen Sustained release for 1 week for diclofenac. | No difference was observed in the release rate at both pH 5.8 and 8.0. | [110] |
| Ibuprofen | Biogel | Layer-by-layer self-assembly method The chitosan hydrochloride and sodium cellulose sulfate were dissolved in distilled water and ultrasonized. This mixture was used as the intermediate layer solution. Aqueous solutions of 2% (w/v) sodium cellulose sulfate and 2% (w/v) sodium tripolyphosphate were used as the upper and lower layer solutions, respectively. | 5% ibuprofen 2% sodium tripolyphosphate in chitosan hydrochloride/sodium cellulose sulfate (4:1) | Sodium cellulose sulfate—polyanionic polymer chitosan—natural polymer | Rate of release 27.2 ± 1.0% (in the first 60 min) 35.3 ± 1.88% (after 1440 min) Permeation rate (skin of mice) 3140.44 ± 159.89 mg/cm (24 h) | Sodium cellulose sulfate, chitosan hydrochloride, and sodium tripolyphosphate determine a favorable sustained release profile, no cytotoxicity, and good biocompatibility. | [39] |
| Baclofen, amitriptyline, ketamine | Organogel | - | 10 mg baclofen, 40 mg amitriptyline, 20 mg ketamine in PL lecithin | - | - | Double-blind, placebo-controlled trial (chemotherapy-induced peripheral neuropathy) Slight improvement in sensory pain and motor scale vs. placebo Topical gel was well tolerated, without evident systemic toxicity | [96] |
| Baclofen, amitriptyline, ketamine | Organogel | - | 10 mg baclofen, 40 mg amitriptyline, and 20 mg ketamine in PL lecithin | - | - | Prospective, single-arm, cohort pilot study (neuropathic pain caused by radiation skin reaction): reduction in pain intensity, sharpness, burning, sensitivity, itchiness, and unpleasantness, at 30 min posttreatment and at 2 weeks posttreatment. The gel may prove effective in relieving pain in subjects who do not respond to standard treatment, such as opioids. The efficacy of gel in reducing burning pain may be explained by blocking the release of chemical mediators or stimulation of ion channels by drugs, such as amitriptyline, ketamine | [111] |
| Amitriptyline, ketamine | - | - | 2% amitriptyline and 0.5% ketamine in methylcellulose | - | - | Case report (patient with erythromelalgia): topical gel decreased the intensity of pain, and enhanced the functional status and quality of life of a patient (which had not responded satisfactory to oral paracetamol, NSAIDs, gabapentin, and amitriptyline). The gel made it possible to stop all other pain medications of patient. Due to biochemical effects of amitriptyline and ketamine, the gel could avert vasodilation and decrease the redness and high skin temperature characteristic of this disorder. | [112] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pușcașu, C.; Zanfirescu, A.; Negreș, S. Recent Progress in Gels for Neuropathic Pain. Gels 2023, 9, 417. https://doi.org/10.3390/gels9050417
Pușcașu C, Zanfirescu A, Negreș S. Recent Progress in Gels for Neuropathic Pain. Gels. 2023; 9(5):417. https://doi.org/10.3390/gels9050417
Chicago/Turabian StylePușcașu, Ciprian, Anca Zanfirescu, and Simona Negreș. 2023. "Recent Progress in Gels for Neuropathic Pain" Gels 9, no. 5: 417. https://doi.org/10.3390/gels9050417
APA StylePușcașu, C., Zanfirescu, A., & Negreș, S. (2023). Recent Progress in Gels for Neuropathic Pain. Gels, 9(5), 417. https://doi.org/10.3390/gels9050417