
Evidence of Many-Body Interactions in the Virial Coefficients of Polyelectrolyte Gels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Virial Coefficients (A2 and A3) of Polyelectrolyte and Neutral Polymer Gels
= (RT/V1) (A2 φ2 + A3 φ3 + … )
2.2. Estimation of Πelast
2.3. Estimation of Πion
2.4. Scattering from Polymer Gels and the Estimation of the Osmotic Compressibility
2.5. Osmotic Pressure Estimates of Polyelectrolyte Gels and Virial Coefficients
2.6. Scattering from Polymer Gels—An Alternative Approach to Estimating Osmotic Properties of Hydrogels
= φ2 (RT/V1) (2 A2 φ + 3 A3 φ2 + …) + (4/3) Gs
2.7. Future Directions
3. Conclusions
4. Materials and Methods
4.1. Preparation Poly(acrylic Acid) and Polystyrene Sulfonate Gels
4.2. Preparation of DNA and HA Gels
4.3. Preparation of Poly(vinyl Acetate) Gels
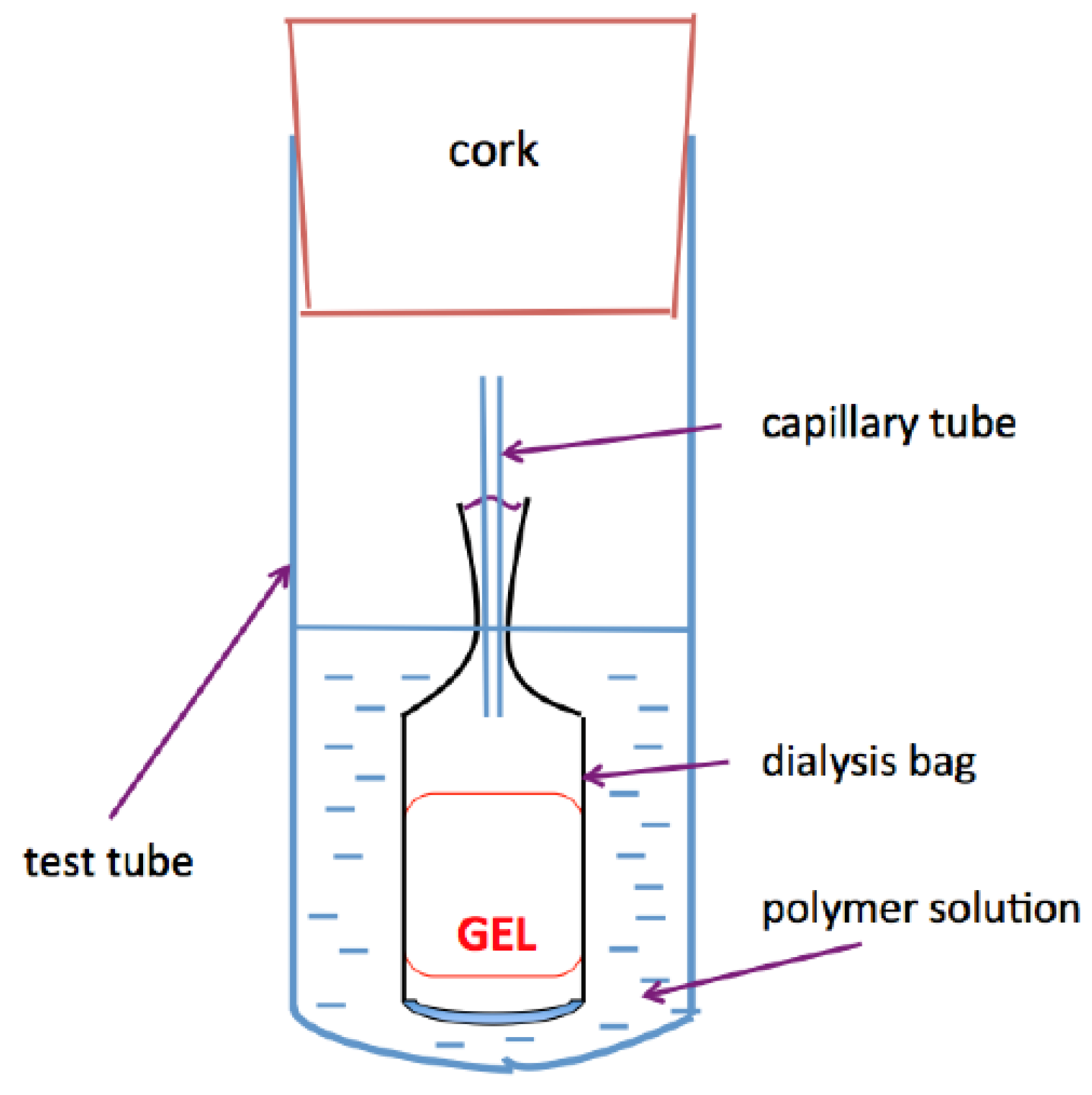
4.4. Osmotic Swelling Pressure Measurements
4.5. Shear Modulus Measurements
4.6. Small-Angle Neutron Scattering
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Viliegenthart, G.A.; Lekkerkerker, H.N.W. Predicting the gas–liquid critical point from the second virial coefficient. J. Chem. Phys. 2000, 112, 5364–5369. [Google Scholar] [CrossRef]
- Noro, M.G.; Frenkel, D. Extended corresponding-states behavior for particles with variable range attractions. J. Chem. Phys. 2000, 113, 2941–2944. [Google Scholar] [CrossRef]
- MacDowell, L.J.; Menduińa, C.; Vega, C.; de Miguel, E. Critical properties of molecular fluids from the virial series. J. Chem. Phys. 2003, 119, 11367–11373. [Google Scholar] [CrossRef]
- Janećek, J.R.; Boublik, T. Critical properties of non-spherical molecule fluids from the virial expansion. Mol. Phys. 2000, 98, 93–99. [Google Scholar] [CrossRef]
- Nezbeda, I.; Smith, W.R. On the calculation of the critical temperature from the second virial coefficient. Fluid Phase Equilib. 2004, 216, 183–186. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Douglas, J.F.; Dudowicz, J.; Freed, K.F. Lattice model of equilibrium polymerization. VI. Measures of fluid “complexity” and search for generalized corresponding states. J. Chem. Phys. 2007, 127, 224901. [Google Scholar] [CrossRef] [PubMed]
- Audus, D.J.; de Pablo, J.J. Polymer informatics: Opportunities and challenges. ACS Macro Lett. 2017, 6, 1078–1082. [Google Scholar] [CrossRef]
- Wood, R.H.; Thompson, P.T. Differences between pair and bulk hydrophobic interactions. Proc. Natl. Acad. Sci. USA 1999, 87, 946–949. [Google Scholar] [CrossRef]
- Raschke, T.M.; Tsai, J.; Levitt, M. Quantification of the hydrophobic interaction by simulations of the aggregation of small hydrophobic solutes in water. Proc. Natl. Acad. Sci. USA 2001, 98, 5965–5969. [Google Scholar] [CrossRef]
- Rankin, B.L.; Ben-Amotz, D.; van der Post, S.T.; Bakker, H.J.J. Contacts between alcohols in water are random rather than hydrophobic. Phys. Chem. Lett. 2015, 6, 688–692. [Google Scholar] [CrossRef]
- Watanabe, K.; Anderson, H.C. Molecular dynamics study of the hydrophobic interaction in an aqueous solution of krypton. J. Phys. Chem. 1986, 90, 795–802. [Google Scholar] [CrossRef]
- Koga, K. Osmotic second virial coefficient of methane in water. J. Phys. Chem. B 2013, 117, 12619–12624. [Google Scholar] [CrossRef]
- De Gennes, P.G. Special features of water soluble polymers. Pure Appl. Chem. 1992, 64, 1585–1588. [Google Scholar] [CrossRef][Green Version]
- Bekiranov, B.; Bruinsma, R.; Pincus, P. Solution behavior of polyethylene oxide in water as a function of temperature and pressure. Phys. Rev. E 1977, 55, 577–585. [Google Scholar] [CrossRef]
- Benjamin, K.M.; Schultz, A.J.; Kofke, D.A. Virial coefficients of polarizable water: Applications to thermodynamic properties and molecular clustering. J. Phys. Chem. C 2007, 111, 16021–16027. [Google Scholar] [CrossRef]
- Benjamin, K.M.; Singh, J.K.; Schultz, A.J.; Kofke, D.A. Higher-order virial coefficients of water models. J. Phys. Chem. B 2007, 111, 11463–11473. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.J.; Kofke, D.A.; Harvey, A.H. Molecular-based virial coefficients of CO2-H2O mixtures. AIChE J. 2015, 61, 3029–3037. [Google Scholar] [CrossRef]
- Ghosh, T.; García, A.E.; Garde, S. Water-mediated three-particle interactions between hydrophobic solutes: Size, pressure, and salt effects. J. Phys. Chem. B 2003, 107, 612–617. [Google Scholar] [CrossRef]
- Ben-Amotz, D. Water-mediated hydrophobic interactions. Ann. Rev. Phys. Chem. 2016, 67, 617–638. [Google Scholar] [CrossRef]
- Garberoglio, G.; Jankowoski, P.; Szalewicz, K.; Harvey, A.H. Fully quantum calculations of the second and third virial coefficients of water and its isotopologues from ab initio potentials. Faraday Discuss. 2018, 212, 467–497. [Google Scholar] [CrossRef]
- Eldrod, M.J.; Saykally, R.J. Many-body effects in intermolecular forces. Chem. Rev. 1994, 94, 1975–1997. [Google Scholar] [CrossRef]
- Cisneros, G.A.; Wikfeldt, K.T.; Ojamae, L.; Lu, J.; Xu, Y.; Torabifard, H.; Bartok, A.P.; Csányi, G.; Molinero, V.; Paesani, F. Modeling molecular interactions in water: From pairwise to many-body potential energy functions. Chem. Rev. 2016, 116, 7501–7528. [Google Scholar] [CrossRef] [PubMed]
- Dudowicz, J.; Freed, K.F.; Douglas, J.F. Lattice model of equilibrium polymerization IV. Influence of activation, chemical initiation, chain scission and fusion, and chain stiffness on polymerization and phase separation. J. Chem. Phys. 2003, 119, 12645–12666. [Google Scholar] [CrossRef]
- Pitzer, K.S. The volumetric and thermodynamic properties of fluids. I. Theoretical basis and virial coefficients. J. Am. Chem. Soc. 1955, 77, 3427–3433. [Google Scholar] [CrossRef]
- Pitzer, K.S.; Lippmann, D.Z.; Curl, R.F.; Huggins, C.M.; Petersen, D.E. The volumetric and thermodynamic properties of fluids. II. Compressibility factor, vapor pressure and entropy of vaporization. J. Am. Chem. Soc. 1955, 77, 3433–3440. [Google Scholar] [CrossRef]
- De Bruyn, J.R.; Goldstein, R.E. Comment on: Rectilinear diameters and extended corresponding states. J. Chem. Phys. 1991, 95, 9424–9425. [Google Scholar] [CrossRef][Green Version]
- Dudowicz, J.; Freed, K.F.; Douglas, J.F. New patterns of polymer blend miscibility associated with monomer shape and size asymmetry. J. Chem. Phys. 2002, 116, 9983–9996. [Google Scholar] [CrossRef]
- Dudowicz, J.; Freed, K.F.; Douglas, J.F. Beyond Flory-Huggins theory: New classes of blend miscibility associated with monomer structural asymmetry. Phys. Rev. Lett. 2002, 88, 095503. [Google Scholar] [CrossRef]
- Zhang, Y.; Douglas, J.F.; Ermi, B.D.; Amis, E.J. Influence of counterion valency on the scattering properties of highly charged polyelectrolyte solutions. J. Chem. Phys. 2001, 114, 3299–3313. [Google Scholar] [CrossRef]
- Chremos, A.; Douglas, J.F. Communication: Counter-ion solvation and anomalous low-angle scattering in salt-free polyelectrolyte solutions. J. Chem. Phys. 2017, 147, 241103. [Google Scholar] [CrossRef] [PubMed]
- Chremos, A.; Douglas, J.F. Polyelectrolyte association and solvation. J. Chem. Phys. 2018, 149, 163305. [Google Scholar] [CrossRef]
- Horkay, F.; Tasaki, I.; Basser, P.J. Osmotic swelling of polyacrylate hydrogels in physiological salt solutions. Biomacromolecules 2000, 1, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Horkay, F.; Tasaki, I.; Basser, P.J. Effect of monovalent−divalent cation exchange on the swelling of polyacrylate hydrogels in physiological salt solutions. Biomacromolecules 2001, 2, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Horkay, F.; Hecht, A.; Basser, P.J.; Geissler, E. Comparison between neutral gels and neutralized polyelectrolyte gels in the presence of divalent cations. Macromolecules 2001, 34, 4285–4287. [Google Scholar] [CrossRef]
- Horkay, F. Effect of the ionic environment on the supramolecular structure and thermodynamics of DNA gels. Macromol. Symp. 2019, 385, 1800199. [Google Scholar] [CrossRef]
- Horkay, F.; Basser, P.J.; Londono, D.J.; Hecht, A.; Geissler, E. Ions in hyaluronic acid solutions. J. Chem. Phys. 2009, 131, 184902. [Google Scholar] [CrossRef]
- Treloar, L.R.G. The Physics of Rubber Elasticity, 3rd ed.; Clarendon: Oxford, UK, 1975. [Google Scholar]
- Lin, D.C.; Douglas, J.F.; Horkay, F. Development of minimal models of the elastic properties of flexible and stiff polymer networks with permanent and thermoreversible cross-links. Soft Matter 2010, 6, 3548–3561. [Google Scholar] [CrossRef] [PubMed]
- Horkay, F.; Basser, P.J.; Hecht, A.; Geissler, E. Calcium induced volume transition in polyacrylate hydrogels swollen in physiological salt solutions. Macromol. Biosci. 2002, 2, 207–213. [Google Scholar] [CrossRef]
- Mussel, M.; Basser, P.J.; Horkay, F. Effects of mono-and divalent cations on the structure and thermodynamic properties of polyelectrolyte gels. Soft Matter 2019, 15, 4153–4161. [Google Scholar] [CrossRef]
- Yin, D.W.; Horkay, F.; Douglas, J.F.; de Pablo, J. Molecular simulation of the swelling of polyelectrolyte gels by monovalent and divalent counterions. J. Chem. Phys. 2008, 129, 154902. [Google Scholar] [CrossRef]
- Baumann, C.G.; Smith, S.B.; Bloomfield, V.A.; Bustamante, C. Ionic effects on the elasticity of single DNA molecules. Proc. Natl. Acad. Sci. USA 1997, 94, 6185–6190. [Google Scholar] [CrossRef]
- Guilbaud, S.; Salomé, L.; Destainville, N.; Manghi, M. Dependence of DNA persistence length on ionic strength and ion type. Phys. Rev. Lett. 2019, 122, 028102. [Google Scholar] [CrossRef] [PubMed]
- Chremos, A.; Douglas, J.F. Influence of higher valent ions on flexible polyelectrolyte stiffness and counter-ion distribution. J. Chem. Phys. 2016, 144, 164904. [Google Scholar] [CrossRef] [PubMed]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1971. [Google Scholar]
- Ornstein, L.S.; Zernike, F. Accidental deviations of density and opalescence at the critical point of a single substance. K. Ned. Akad. Wet. Amst. Proc. Sec. Sci. 1914, 17, 793–806. [Google Scholar]
- Sears, V.F. Neutron scattering lengths and cross sections. Neutron News 1992, 3, 26–37. [Google Scholar] [CrossRef]
- Horkay, F.; Basser, P.J.; Hecht, A.M.; Geissler, E. Chondroitin sulfate in solution: Effects of mono-and divalent salts. Macromolecules 2012, 45, 2882–2890. [Google Scholar] [CrossRef]
- Glatter, O.; Kratky, O. Small-Angle X-ray Scattering; Academic Press: London, UK, 1982. [Google Scholar]
- Forster, S.; Schmidt, M. Polyelectrolytes in solution. Adv. Polym. Sci. 1995, 120, 51–133. [Google Scholar]
- Horkay, F.; Zrinyi, M.; Geissler, E.; Hecht, A.M. Elasticity of polymer gels: Effect of solvent quality on the shear modulus of Poly (Vinyl Acetate) networks. Makromol. Chem. Macromol. Symp. 1990, 40, 195–202. [Google Scholar] [CrossRef]
- McQuarrie, D.A. Statistical Mechanics; Harper and Row: New York, NY, USA, 1976. [Google Scholar]
- Ricka, J.; Tanaka, T. Swelling of ionic gels: Quantitative performance of the Donnan theory. Macromolecules 1984, 17, 2916–2921. [Google Scholar] [CrossRef]
- Ricka, J.; Tanaka, T. Phase transition in ionic gels induced by copper complexation. Macromolecules 1985, 18, 83–85. [Google Scholar] [CrossRef]
- Suzuki, A.; Suzuki, H. Hysteretic behavior and irreversibility of polymer gels by pH change. J. Chem. Phys. 1995, 103, 4706–4710. [Google Scholar] [CrossRef]
- Hirotsu, S.; Hirokawa, Y.; Tanaka, T. Volume-phase transitions of ionized N-isopropylacrylamide gels. J. Chem. Phys. 1987, 87, 1392–1395. [Google Scholar] [CrossRef]
- Horkay, F.; Chremos, A.; Douglas, J.F.; Jones, R.; Lou, J.; Xia, Y. Comparative experimental and computational study of synthetic and natural bottlebrush polyelectrolyte solutions. J. Chem. Phys. 2021, 155, 074901. [Google Scholar] [CrossRef]
- Tokita, M.; Tanaka, T. Reversible decrease of gel-solvent friction. Science 1991, 253, 1121–1123. [Google Scholar] [CrossRef]
- Tokita, M.; Tanaka, T. Friction coefficient of polymer networks of gels. J. Chem. Phys. 1991, 95, 4613–4619. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Ishii, R.; Matsui, J.; Suzuki, A.; Tokita, M. A simple technique to measure the friction coefficient between polymer network of hydrogel and water. Jpn. J. Appl. Phys. 2005, 44, 8196–8200. [Google Scholar] [CrossRef]
- Tanaka, T. Dynamics of critical concentration fluctuations in gels. Phys. Rev. A 1978, 17, 763–766. [Google Scholar] [CrossRef]
- Horkay, F.; Basser, P.J. Composite hydrogel model of cartilage predicts its load-bearing ability. Sci. Rep. 2020, 10, 8103. [Google Scholar] [CrossRef]
- Bosse, A.W.; Douglas, J.F. The osmotic virial formulation of the free energy of polymer mixing. J. Chem. Phys. 2015, 143, 104903. [Google Scholar] [CrossRef]
- Ghosh, S.; Mukherjee, A.; Arroyave, R.; Douglas, J.F. Impact of particle arrays on phase separation composition patterns. J. Chem. Phys. 2020, 152, 224902. [Google Scholar] [CrossRef]
- Shin, Y. Brangwynne Liquid phase condensation in cell physiology and disease. Science 2017, 357, 1253. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Altmeyer, M. Phase separation: Linking cellular compartmentalization to disease. Trends Cell Biol. 2016, 26, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Zbinden, A.; Pérez-Berlanga, M.M.; De Rossi, P.; Polymenidou, M. Phase separation and neuro-degenerative diseases: A disturbance in the force. Dev. Cell 2020, 55, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Sugitani, M.; Kobayashi, T.; Tanaka, T. Synthesis of poly (acrylic acid) gel beads. Polym. Prepr. 1987, 36, 2876. [Google Scholar]
- Horkay, F.; Chremos, A.; Douglas, J.F.; Jones, R.L.; Lou, L.; Xia, Y. Systematic investigation of synthetic polyelectrolyte bottlebrush solutions by neutron and dynamic light scattering, osmometry, and molecular dynamics simulation. J. Chem. Phys. 2020, 152, 194904. [Google Scholar] [CrossRef]
- Teo, Y.C.; Xia, Y. Importance of macromonomer quality in the ring-opening metathesis polymerization of macromonomers. Macromolecules 2015, 48, 5656–5662. [Google Scholar] [CrossRef]
- Teo, Y.C.; Xia, Y. Facile synthesis of macromonomers via ATRP–nitroxide radical coupling and well-controlled brush block copolymers. Macromolecules 2019, 52, 81–87. [Google Scholar] [CrossRef]
- Horkay, F.; Basser, P.J. Osmotic observations on chemically cross-linked DNA gels in physiological salt solutions. Biomacromolecules 2004, 5, 232–237. [Google Scholar] [CrossRef]
- Horkay, F.; Burchard, W.; Geissler, E.; Hecht, A.M. Thermodynamic properties of poly (vinyl alcohol) and poly (vinyl alcohol-vinyl acetate) hydrogeLS. Macromolecules 1993, 26, 1296–1303. [Google Scholar] [CrossRef]
- Horkay, F.; Zrinyi, M. Studies on the mechanical and swelling behavior of polymer networks based on the scaling concept 4. Extension of the scaling approach to gels swollen to equilibrium in a diluent of arbitrary activity. Macromolecules 1982, 15, 1306–1310. [Google Scholar] [CrossRef]
- Vink, H. Precision measurements of osmotic pressure in concentrated polymer solutions. Eur. Polym. J. 1971, 7, 1411–1419. [Google Scholar] [CrossRef]
- NIST Cold Neutron Research Facility, NG3 and NG7 30 m SANS Instruments Data Acquisition Manual. 2002. Available online: https://www.nist.gov/ncnr/ng7-sans-small-angle-neutron-scattering (accessed on 15 December 2021).








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horkay, F.; Douglas, J.F. Evidence of Many-Body Interactions in the Virial Coefficients of Polyelectrolyte Gels. Gels 2022, 8, 96. https://doi.org/10.3390/gels8020096
Horkay F, Douglas JF. Evidence of Many-Body Interactions in the Virial Coefficients of Polyelectrolyte Gels. Gels. 2022; 8(2):96. https://doi.org/10.3390/gels8020096
Chicago/Turabian StyleHorkay, Ferenc, and Jack F. Douglas. 2022. "Evidence of Many-Body Interactions in the Virial Coefficients of Polyelectrolyte Gels" Gels 8, no. 2: 96. https://doi.org/10.3390/gels8020096
APA StyleHorkay, F., & Douglas, J. F. (2022). Evidence of Many-Body Interactions in the Virial Coefficients of Polyelectrolyte Gels. Gels, 8(2), 96. https://doi.org/10.3390/gels8020096