Multiply Interpenetrating Polymer Networks: Preparation, Mechanical Properties, and Applications



Abstract
1. Introduction
2. Network Structures with Improved Mechanical Properties
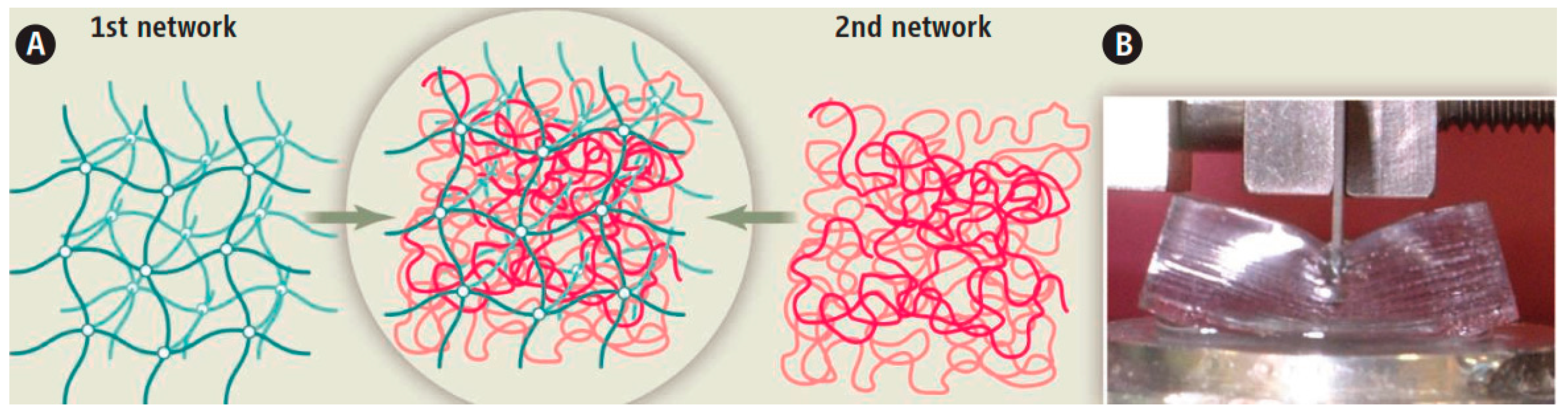
2.1. Double-Network (DN) Hydrogels
2.2. Toughening Mechanism in DN Hydrogels
2.2.1. Role of Each Network in DN Toughening
2.2.2. Interaction between the Two Network Components
2.2.3. Toughening Mechanism in DN Hydrogels
2.3. Triple-Network (TN) Hydrogels
2.3.1. Simple TN Hydrogels
2.3.2. TN Hydrogels with Microgels as the First Component
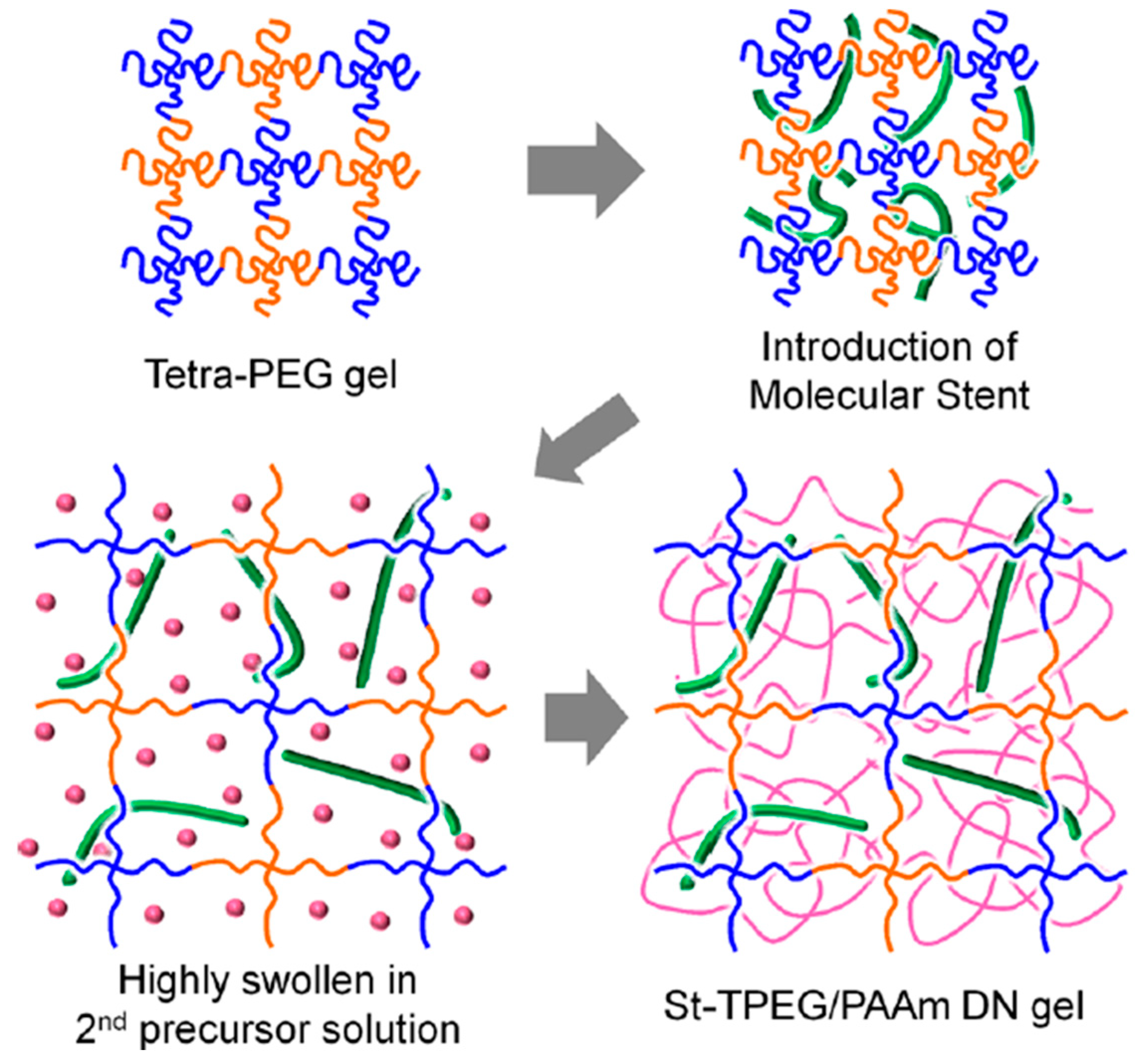
2.3.3. TN Hydrogels Containing a Linear Polyelectrolyte Stent as the Second Component
2.3.4. TN Hydrogels Prepared Using a Mold
2.3.5. TN Hydrogels Containing an Electrically Conducting Polymer as the Third Component
2.4. Quadruple-Network (QN) Hydrogels
2.5. Quintuple-Network (5×N) Hydrogels
3. Applications of the Multiply Interpenetrating Polymer Network Hydrogels
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Caccavo, D.; Cascone, S.; Lamberti, G.; Barba, A.A. Hydrogels: experimental characterization and mathematical modelling of their mechanical and diffusive behaviour. Chem. Soc. Rev. 2018, 47, 2357–2373. [Google Scholar] [CrossRef] [PubMed]
- Koetting, M.C.; Peters, J.T.; Steichen, S.D.; Peppas, N.A. Stimulus-responsive hydrogels: Theory, modern advances, and applications. Mater. Sci. Eng. R Rep. 2015, 93, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2002, 54, 3–12. [Google Scholar] [CrossRef]
- Brown, H.R. A model of the fracture of double network gels. Macromolecules 2007, 40, 3815–3818. [Google Scholar] [CrossRef]
- Okumura, Y.; Ito, K. The polyrotaxane gel: A topological gel by figure-of-eight cross-links. Adv. Mater. 2001, 7, 485–487. [Google Scholar] [CrossRef]
- Haraguchi, K.; Takehisa, T. Nanocomposite hydrogels: A unique organic‒inorganic network structure with extraordinary mechanical, optical, and swelling/de-swelling properties. Adv. Mater. 2002, 14, 1120–1124. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Gong, J.P. Why are double networks so tough? Soft Matter 2010, 6, 2583–2890. [Google Scholar] [CrossRef]
- Gong, J.P. Materials both tough and soft. Science 2014, 344, 161–162. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, D.; Tada, T.; Kurokawa, T.; Gong, J.P.; Osada, Y. Mechanically strong hydrogels with ultra-low frictional coefficients. Adv. Mater. 2005, 17, 535–538. [Google Scholar] [CrossRef]
- Chen, Y.M.; Gong, J.P.; Tanaka, M.; Yasuda, K.; Yamamoto, S.; Shinomura, M.; Osada, Y. Tuning of cell proliferation on tough gels by critical charge effect. J. Biom. Mater. Res. A 2008, 88, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qadeer, A.; Mynarcik, D.; Chen, W. Delivery of rosiglitazone from an injectable triple interpenetrating network hydrogel composed of naturally derived materials. Biomaterials 2011, 32, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Betz, A.; Qadeer, A.; Attinger, D.; Chen, W. Microfluidic formation of monodispersed spherical microgels composed of triple-network crosslinking. J. Appl. Polym. Sci. 2011, 121, 3093–3100. [Google Scholar] [CrossRef]
- Argun, A.; Can, V.; Altun, U.; Okay, O. Nonionic Double and triple network hydrogels of high mechanical strength. Macromolecules 2014, 47, 6430–6440. [Google Scholar] [CrossRef]
- Ducrot, E.; Chen, Y.; Bülters, M.; Sijbesma, R.P.; Creton, C. Toughening elastomers with sacrificial bonds and watching them break. Science 2014, 344, 186–189. [Google Scholar] [CrossRef]
- Tavsanli, B.; Can, V.; Okay, O. Mechanically strong triple network hydrogels based on hyaluronan and poly(N,N-dimethylacrylamide). Soft Matter 2015, 11, 8517–8524. [Google Scholar] [CrossRef]
- Hu, J.; Hiwatashi, K.; Kurokawa, T.; Liang, S.M.; Wu, Z.L.; Gong, J.P. Microgel-reinforced hydrogel films with high mechanical strength and their visible mesoscale fracture structure. Macromolecules 2011, 44, 7775–7781. [Google Scholar] [CrossRef]
- Hiwatashi, K.; Wu, Z.L.; Liang, S.M.; Hu, J.; Kurokawa, T.; Nakajima, T.; Gong, J.P. Structure optimization and mechanical model for microgel-reinforced hydrogels with high strength and toughness. Macromolecules 2012, 45, 5218–5228. [Google Scholar]
- Nakajima, T.; Sato, H.; Zhao, Y.; Kawahara, S.; Kurokawa, T.; Sugahara, K.; Gong, J.P. A universal molecular stent method to toughen any hydrogels based on double network concept. Adv. Funct. Mater. 2012, 22, 4426–4432. [Google Scholar] [CrossRef]
- Fukuda, Y.; Nakajima, T.; Kurokawa, T.; Sakai, T.; Chung, U.-I.; Gong, J.P. Synthesis and fracture process analysis of double network hydrogels with a well-defined first network. ACS Macro Lett. 2013, 2, 518–521. [Google Scholar]
- Nakajima, T.; Takedomi, N.; Kurokawa, T.; Furukawa, H.; Gong, J.P. A facile method for synthesizing free-shaped and tough double network hydrogels using physically crosslinked poly(vinyl alcohol) as an internal mold. Polym. Chem. 2010, 1, 693–697. [Google Scholar] [CrossRef]
- Dai, T.; Qing, X.; Lu, Y.; Xia, Y. Conducting hydrogels with enhanced mechanical strength. Polymer 2009, 50, 5236–5241. [Google Scholar] [CrossRef]
- Kishi, R.; Hiroki, K.; Tominaga, T.; Sano, K.-I.; Okuzaki, H.; Martínez, J.G.; Otero, T.F.; Osada, Y. Electro-conductive double-network hydrogels. J. Polym. Sci. B Polym. Phys. 2012, 50, 790–796. [Google Scholar] [CrossRef]
- Kishi, R.; Kubota, K.; Miura, T.; Yamaguchi, T.; Okuzaki, H.; Osada, Y. Mechanically tough double-network hydrogels with high electronic conductivity. J. Mater. Chem. C 2014, 2, 736–743. [Google Scholar] [CrossRef]
- Naficy, S.; Razal, J.M.; Spinks, G.M.; Wallace, G.G.; Whitten, P.G. Electrically conductive, tough hydrogels with pH sensitivity. Chem. Mater. 2012, 24, 3425–3433. [Google Scholar] [CrossRef]
- Es-Haghi, S.S.; Weiss, R.A. Fabrication of tough hydrogels from chemically cross-linked multiple neutral networks. Macromolecules 2016, 49, 8980–8987. [Google Scholar] [CrossRef]
- Panteli, P.A.; Patrickios, C.S. Complex Hydrogels based on multiply interpenetrated polymer networks: Enhancement of mechanical properties via network multiplicity and monomer concentration. Macromolecules 2018, 51, 7533–7545. [Google Scholar] [CrossRef]
- Panteli, P.A.; Patrickios, C.S.; Constantinou, M.; Constantinides, G. Multiple network hydrogels: A study of their nanoindentation hardness. Macromol. Symp. 2019, 385, 1800201. [Google Scholar] [CrossRef]
- Shestakova, P.; Willem, R.; Vassileva, E. Elucidation of the chemical and morphological structure of double-network (DN) hydrogels by high-resolution magic angle spinning (HRMAS) NMR spectroscopy. Chem. A Eur. J. 2011, 17, 14867–14877. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.-Y.; Zhao, X.; Illeperuma, W.R.K.; Chaudhuri, O.; Oh, K.H.; Mooney, D.J.; Vlassak, J.J.; Suo, Z. Highly stretchable and tough hydrogels. Nature 2012, 489, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Illeperuma, W.R.K.; Vlassak, J.J.; Li, J.; Suo, Z. Hybrid hydrogels with extremely high stiffness and toughness. ACS Macro Lett. 2014, 3, 520–523. [Google Scholar]
- Liu, J.; Tan, C.S.Y.; Yu, Z.; Lan, Y.; Abell, C.; Scherman, O.A. Biomimetic supramolecular polymer networks exhibiting both toughness and self-recovery. Adv. Mater. 2017, 29, 1604951. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tan, C.S.Y.; Yu, Z.; Li, N.; Abell, C.; Scherman, O.A. Tough supramolecular polymer networks with extreme stretchability and fast room-temperature self-healing. Adv. Mater. 2017, 29, 1605325. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shah, D.U.; Wang, B.; Liu, J.; Ren, X.; Ramage, M.H.; Scherman, O.A. Biomimetic supramolecular fibers exhibit water-induced supercontraction. Adv. Mater. 2018, 30, 1707169. [Google Scholar] [CrossRef] [PubMed]
- Means, A.K.; Grunlan, M.A. Modern strategies to achieve tissue-mimetic, mechanically robust hydrogels. ACS Macro Lett. 2019, 8, 705–713. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gel Components | Method | Mechanical Properties of SN | Mechanical Properties of DN | Mechanical Properties of TN | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1st Network | 2nd Network | 3rd Network | σmax (MPa) | εmax (%) | E (MPa) | σmax (MPa) | εmax (%) | E (MPa) | σmax (MPa) | εmax (%) | E (MPa) | ||
| (a) Simple Triple Networks | |||||||||||||
| AMPS-1 MBAAm-8 | AAm-2-MBAAm-0.1 | AMPS-1 MBAAm-0.1 | Compression | - | - | - | 4.60 | 65 | 0.84 | 4.80 | 57 | 2.00 | [12] |
| AMPS-1 MBAAm-0 | 9.20 | 70 | 2.10 | ||||||||||
| SAMPS-1 MBAAm-4 | DMAAm-3 MBAAm-0.1 | SAMPS-co-DMAAm (F = 0.5)-1 MBAAm-0 | Compression | 0.26 | 57 | 0.19 | - | - | - | 3.00 | 71 | 0.32 | [13] |
| SAMPS-co-DMAAm (F = 0.5)-1 MBAAm-2 | - | - | - | 2.31 | 65 | 0.65 | |||||||
| SAMPS-co-DMAAm (F = 0.5)-1 MBAAm-4 | - | - | - | 1.36 | 47 | 0.88 | |||||||
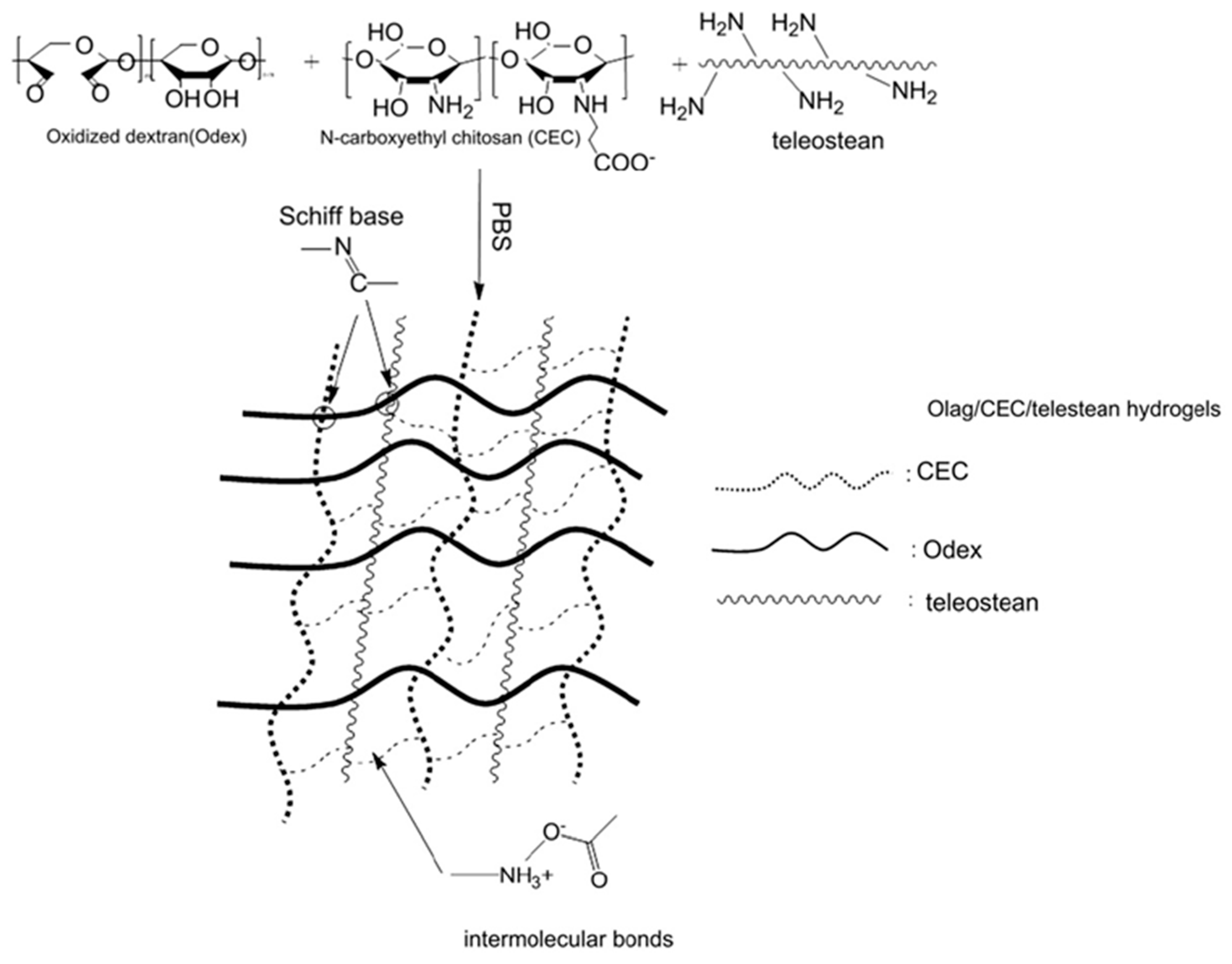
| Odex | Teleostean | CEC | - | - | - | - | - | - | - | - | - | - | [14,15] |
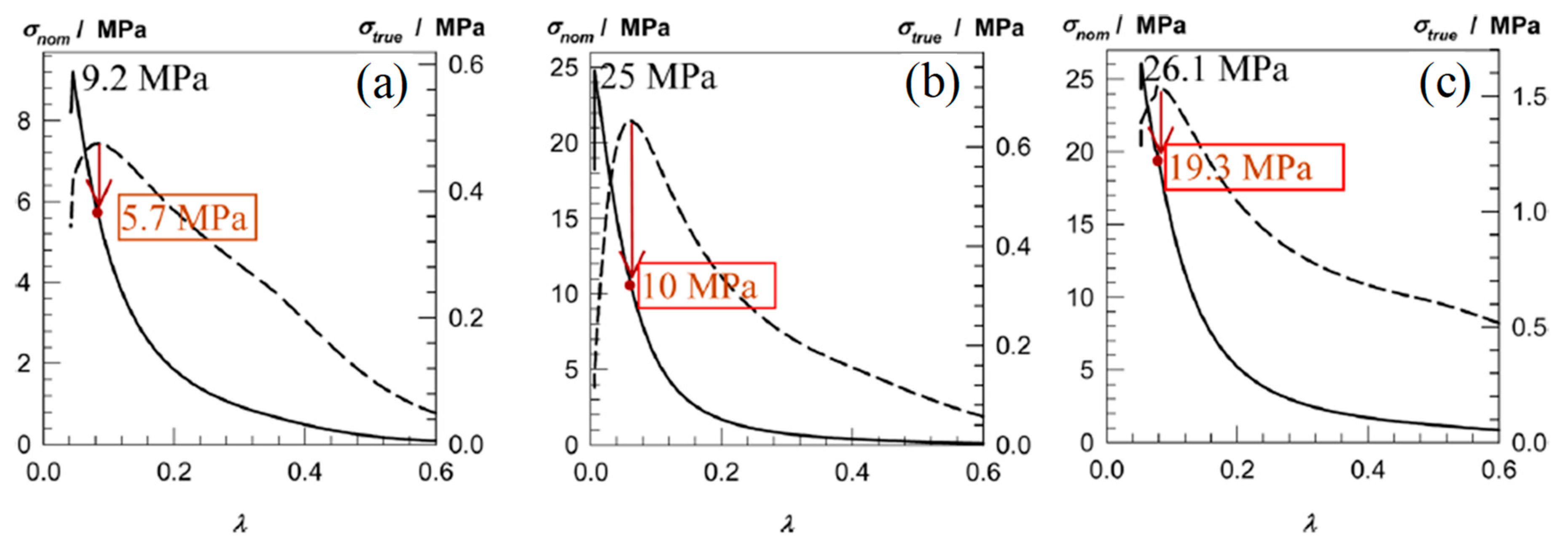
| AAm-1.40 PEGDMA-4 | AAm-0.7–7.0 MBAAm-0 | AAm-0.7–7.0 MBAAm 0 | Compression | 0.15 | 70 | - | 9.20 | 95 | - | 25.00 | 99 | 0.10 | [16] |
| DMAAm-1 PEGDMA-4–10 | DMAAm-0.5–5.0 MBAAm-0 | DMAAm-0.5–5.0 MBAAm-0 | Compression | 0.17 | 47 | - | - | - | - | 26.10 | 95 | 2.00 | |
| EA-5 BDA-1.45 | EA-9.4 BDA-0.01 | EA-9.4 BDA-0.01 | Tension | 1.20 | - | 0.60 | 10.00 | 260 | 0.80 | 16.00 | 220 | 1.50 | [17] |
| EA-5 BDA-1.45 | MA-11 BDA-0.01 | MA-11 BDA-0.01 | 0.50 | - | 0.80 | 8.00 | 240 | 1.30 | 22.00 | 260 | 2.20 | ||
| EA-5 BDA-2.81 | 0.50 | - | 1.50 | 6.50 | 190 | 2.00 | 29.00 | 190 | 4.20 | ||||
| EA-5 BDA-5.81 | 0.50 | - | 2.30 | 3.00 | 160 | 2.30 | - | - | - | ||||
| MHA (20 g·L−1) | DMAAm-3 MBAAm-0.05 | DMAAm-3 MBAAm-0.05 | Compression | 0.05 | 40 | 0.017 | 12.00 | 93 | 0.37 | 22.00 | 96 | 0.40 | [18] |
| Mi-xi, Ci-yi: Mi, xi, Ci, and yi state the abbreviation of the polymer’s name, the molar concentration of monomer, the abbreviation of the cross-linker’s name, and the cross-linker loading feed in mol% with respect to the monomer, respectively. PC: physically cross-linked. | |||||||||||||
| (b) Triple Networks with a 1st Polymer Network Based on Microgels | |||||||||||||
| SAMPS-1 MBAAm-4 | AAm-2 MBAAm-0.01 | AAm-4 MBAAm-0.01 | Compression | - | - | - | 0.15 | 130 | 0.05 | 2.46 | 1270 | 0.22 | [19,20] |
| AAm-1-MBAAm-4 | - | - | - | 0.41 | 1010 | 0.03 | 1.37 | 910 | 0.05 | [20] | |||
| DMAPAA-Q-1 MBAAm-4 | - | - | - | - | - | - | 0.94 | 970 | 0.15 | ||||
| NaSS-co-DMAEA-Q (F = 0.5)-1-MBAAm-4 | - | - | - | - | - | - | 0.75 | 530 | 0.07 | ||||
| SAMPS+DMAPAA-Q (F = 0.5)-1 MBAAm-4 | - | - | - | - | - | - | 0.51 | 410 | 0.07 | ||||
| (c) Triple Networks with a Linear Polyelectrolyte Stent as the 2nd Polymeric Component | |||||||||||||
| AAm-1.2 MBAAm-4 | AMPS-1 MBAAm-0 | AAm-2 MBAAm-0.02 | Tension | - | - | - | - | - | - | 0.83 | 1000 | - | [21] |
| DMAAm-0.7 MBAAm-3 | - | - | - | - | - | - | 1.95 | - | - | ||||
| DMAAm-1 MBAAm-2 | - | - | - | - | - | - | 1.57 | - | - | ||||
| NIPAAm-0.7 MBAAm-2 | - | - | - | - | - | - | 1.02 | - | - | ||||
| AAc-1 MBAAm-4 | AMPS-0.7 MBAAm-0 | AAm-2 MBAAm-0.02 | Tension | - | - | - | - | - | 0.067 | 0.70 | - | - | |
| AAm-1 MBAAm-4 | 0.031 | - | - | - | - | 0.15 | 0.69 | - | - | ||||
| HEA-1 MBAAm-4 | 0.037 | - | 0.054 | - | - | - | 0.82 | - | - | ||||
| NaSS-1 MBAAm-0 | - | - | - | 0.34 | - | - | |||||||
| DMAPAA-Q-1 MBAAm-0 | - | - | - | 0.47 | - | - | |||||||
| DMAEA-1 MBAAm-0 | - | - | - | 0.47 | - | - | |||||||
| TPEG-2 × 10−5 | AMPS-0.6 MBAAm-0 | Tension | - | - | - | 0.20 | 600 | - | 2 | 2200 | - | [22] | |
| AMPS-1.0 MBAAm-0 | - | - | - | ||||||||||
| (d) Triple Networks with a Mold Used in the 1st Polymeric Component | |||||||||||||
| PVA-PC | AMPS-1 MBAAm-4 | AAm-2-MBAAm-0 | Tension | - | - | - | 0.30 | 300 | - | 0.80 | 900 | - | [23] |
| (e) Triple Networks Containing an Electrically Conducting Polymer as the 3rd Component | |||||||||||||
| AAc-1.5 MBAAm-6 | AAc-6 MBAAm-0.1 | EDOT (0.48 M)-PNaSS (0.10 M) PC | Tension | 0.10 | 37 | - | 0.60 | 61 | - | 1.00 | 68 | - | [24] |
| EDOT-PNaSS (No. 2) PC | 1.10 | 70 | - | ||||||||||
| EDOT-PNaSS (No. 3) PC | 1.30 | 72 | - | ||||||||||
| EDOT-PNaSS (No. 4) PC | 0.60 | 61 | - | 1.60 | 73 | - | |||||||
| EDOT-NaSS (No. 5) PC | 1.80 | 78 | - | ||||||||||
| AMPS-1 MBAAm-4 | AAm-2 MBAAm-0.1 | EDOT (No. 1) | Tension | - | - | - | 1.19 | 134 | 0.37 | 1.38 | 154 | 0.33 | [25] |
| EDOT (No. 2) | 2.07 | 235 | 0.56 | ||||||||||
| NaSS-1 MBAAm-10 | DMAAm-1.5 MBAAm-0 | PEDOT | Compression | - | - | - | 0.39 | 45 | 0.71 | 1.27 | 45 | 3.48 | [26] |
| DMAAm-2.0 MBAAm-0 | 1.08 | 81 | 0.95 | 1.98 | 76 | 2.97 | |||||||
| PEGMA-0.18 MBAAm-4 | AAc-2.78 MBAAm-0.1 | EDOT (0.46 M) + NaPSS (0.48 M) PC | Tension | - | - | - | 0.48 | 340 | 0.40 | 0.60 | 240 | 0.11 | [27] |
| Compression | 8.50 | 81 | - | 11.60 | 78 | - | |||||||
| Chemical Structure | Name |
|---|---|
 | AAc, Acrylic acid |
 | AAm, Acrylamide |
 | AMPS, 2-Acrylamido-2-methylpropane sulfonic acid |
 | CEC, Ν-Carboxyethyl chitosan |
 | DMAAm, Ν,Ν-Dimethylacrylamide |
 | DMAEA-Q, 2-(Trimethylamino)ethyl acrylate, chloride quaternary salt |
 | DMAPAA-Q, 3-(Acrylamidopropyl)trimethylammonium chloride |
 | EDOT, 3,4-Ethylenedioxythiophene |
 | EΑ, Ethyl acrylate |
 | MHA, Methacrylated hyaluronan |
 | HEA, 2-Hydroxyethyl acrylate |
 | MΑ, Methyl acrylate |
 | NaSS, Sodium 4-styrenesulfonate |
 | NIPAAm, N-Isopropylacrylamide |
 | SΑMPS, 2-Acrylamido-2-methyl-propane sulfonic acid sodium salt |
 | ODEX, Partially oxidized dextran |
 | PEGMA, Poly(ethylene glycol) methyl ether methacrylate |
 | PVA, Poly(vinyl alcohol) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panteli, P.A.; Patrickios, C.S. Multiply Interpenetrating Polymer Networks: Preparation, Mechanical Properties, and Applications. Gels 2019, 5, 36. https://doi.org/10.3390/gels5030036
Panteli PA, Patrickios CS. Multiply Interpenetrating Polymer Networks: Preparation, Mechanical Properties, and Applications. Gels. 2019; 5(3):36. https://doi.org/10.3390/gels5030036
Chicago/Turabian StylePanteli, Panayiota A., and Costas S. Patrickios. 2019. "Multiply Interpenetrating Polymer Networks: Preparation, Mechanical Properties, and Applications" Gels 5, no. 3: 36. https://doi.org/10.3390/gels5030036
APA StylePanteli, P. A., & Patrickios, C. S. (2019). Multiply Interpenetrating Polymer Networks: Preparation, Mechanical Properties, and Applications. Gels, 5(3), 36. https://doi.org/10.3390/gels5030036