Exploration of Dynamic Elastic Modulus Changes on Glioblastoma Cell Populations with Aberrant EGFR Expression as a Potential Therapeutic Intervention Using a Tunable Hyaluronic Acid Hydrogel Platform



Abstract
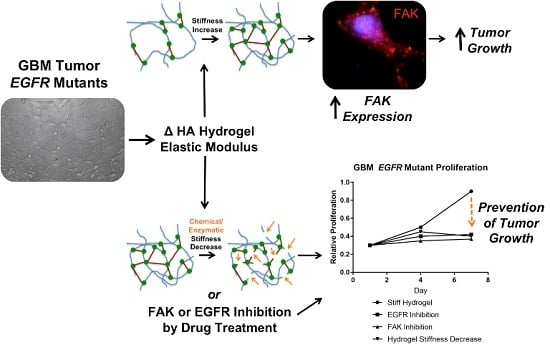
:
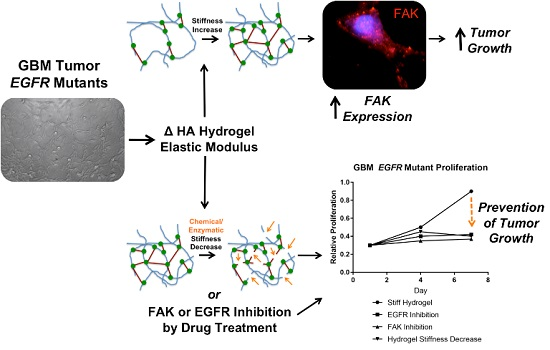
1. Introduction
2. Results and Discussion
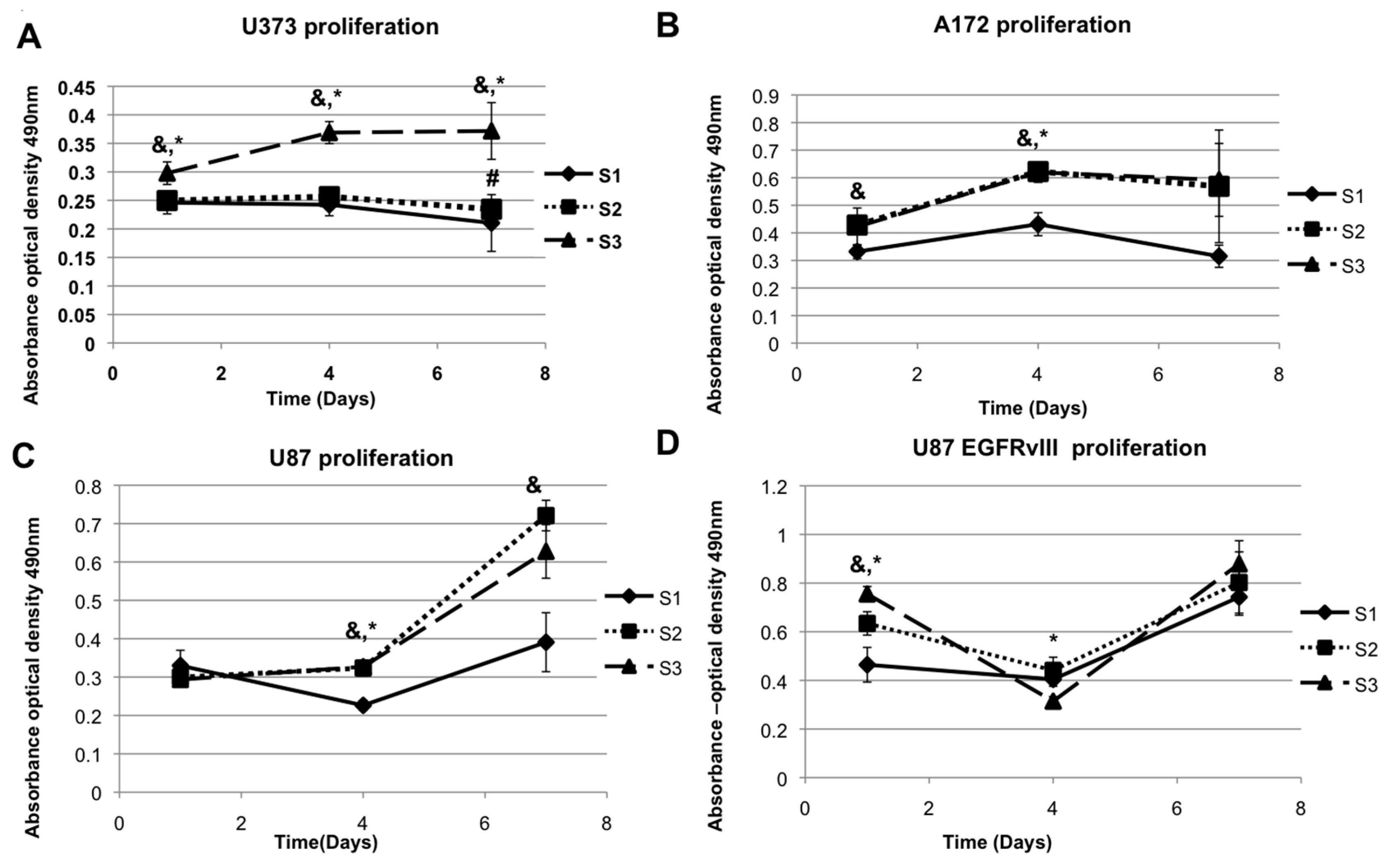
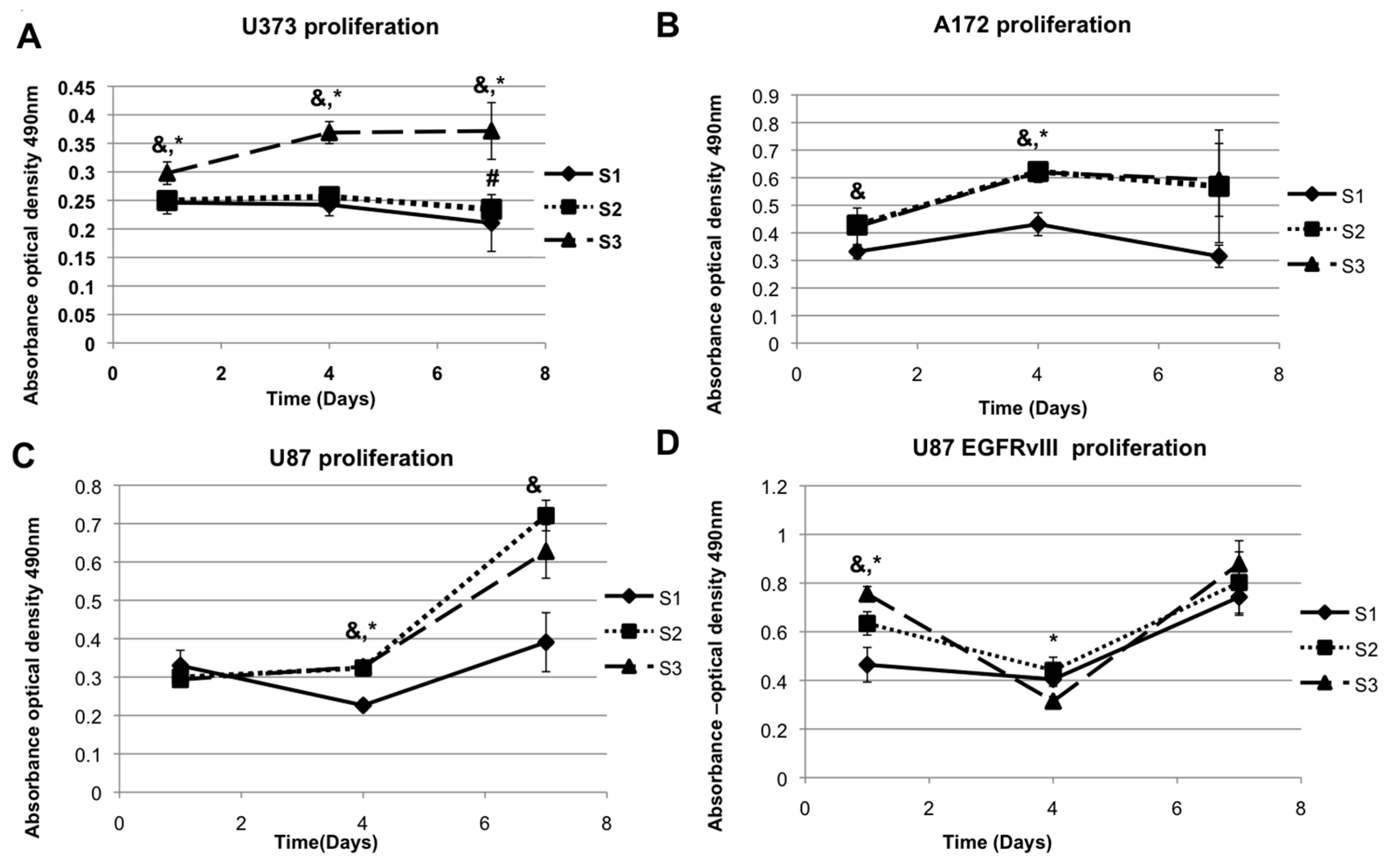
2.1. Elastic Modulus-Dependent Proliferation Assays
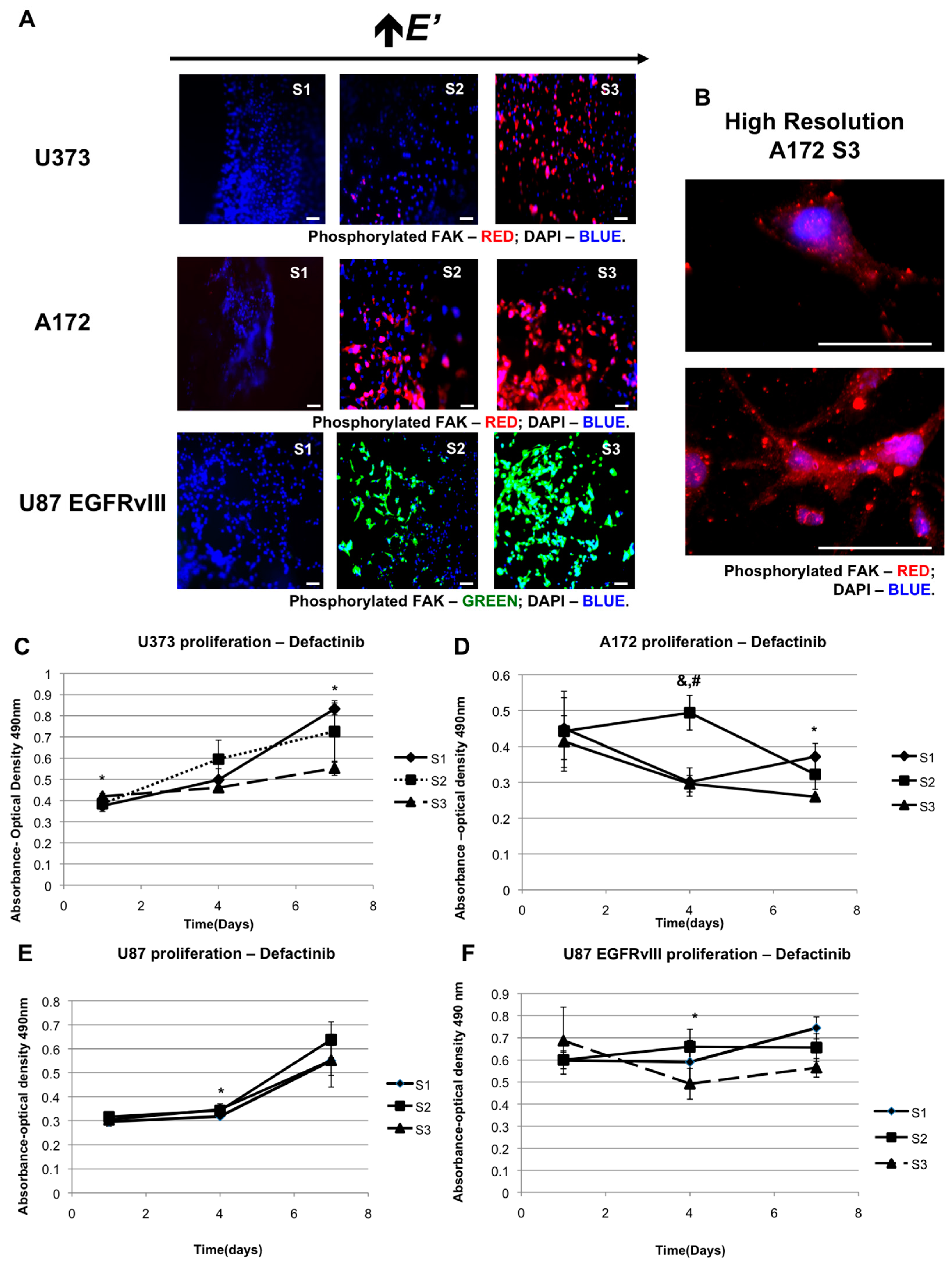
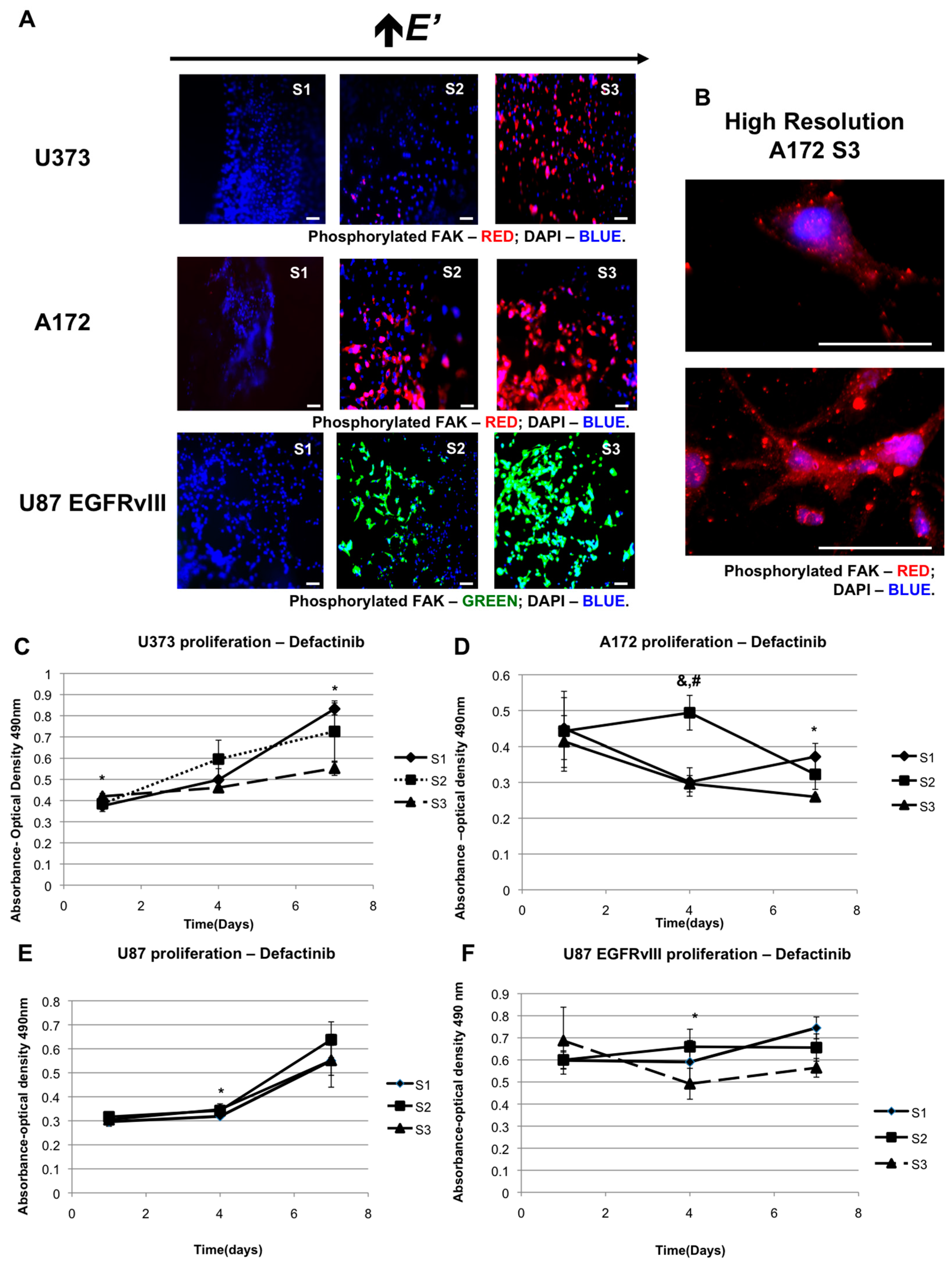
2.2. The Role of FAK in Elastic Modulus-Dependent Cell Proliferation
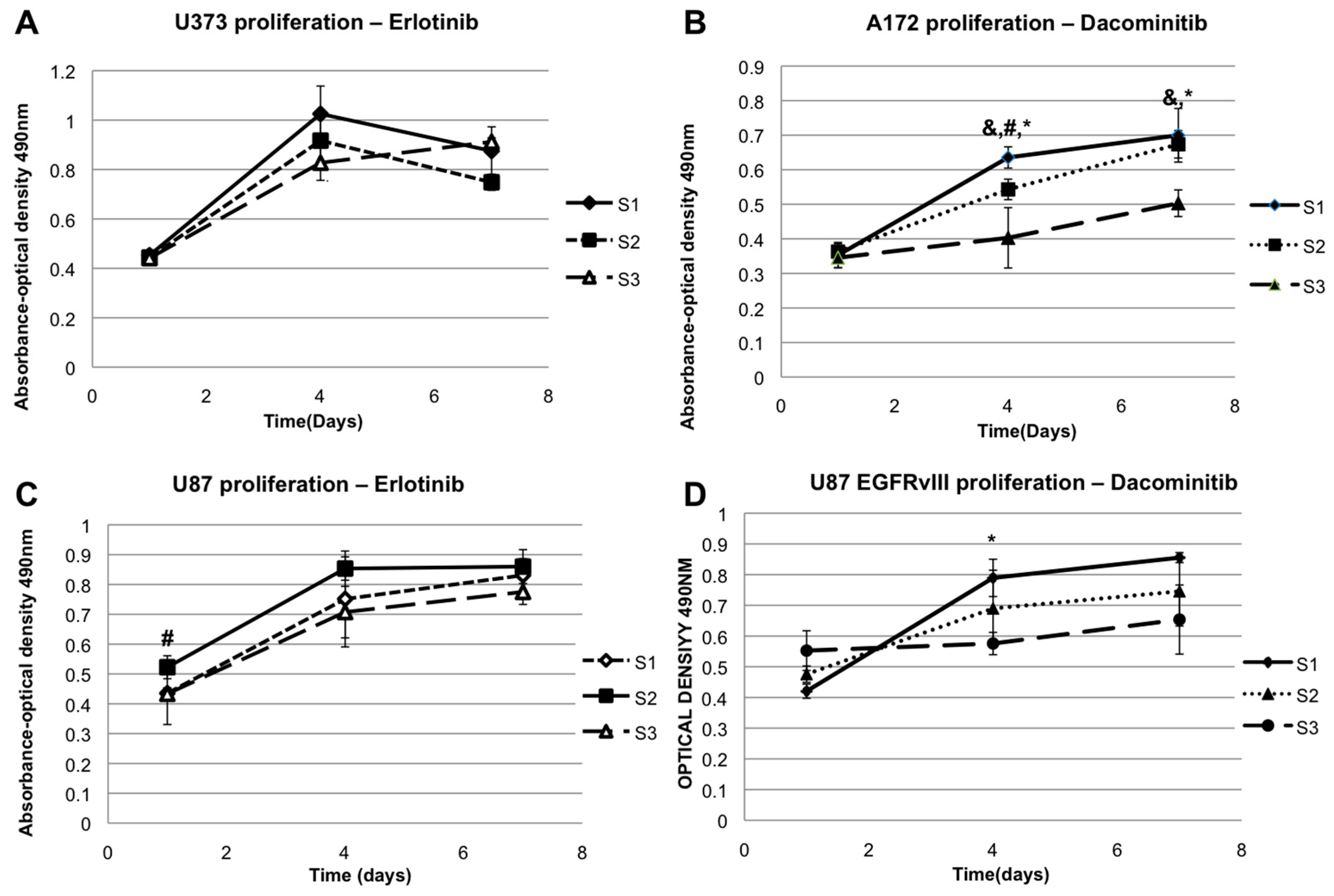
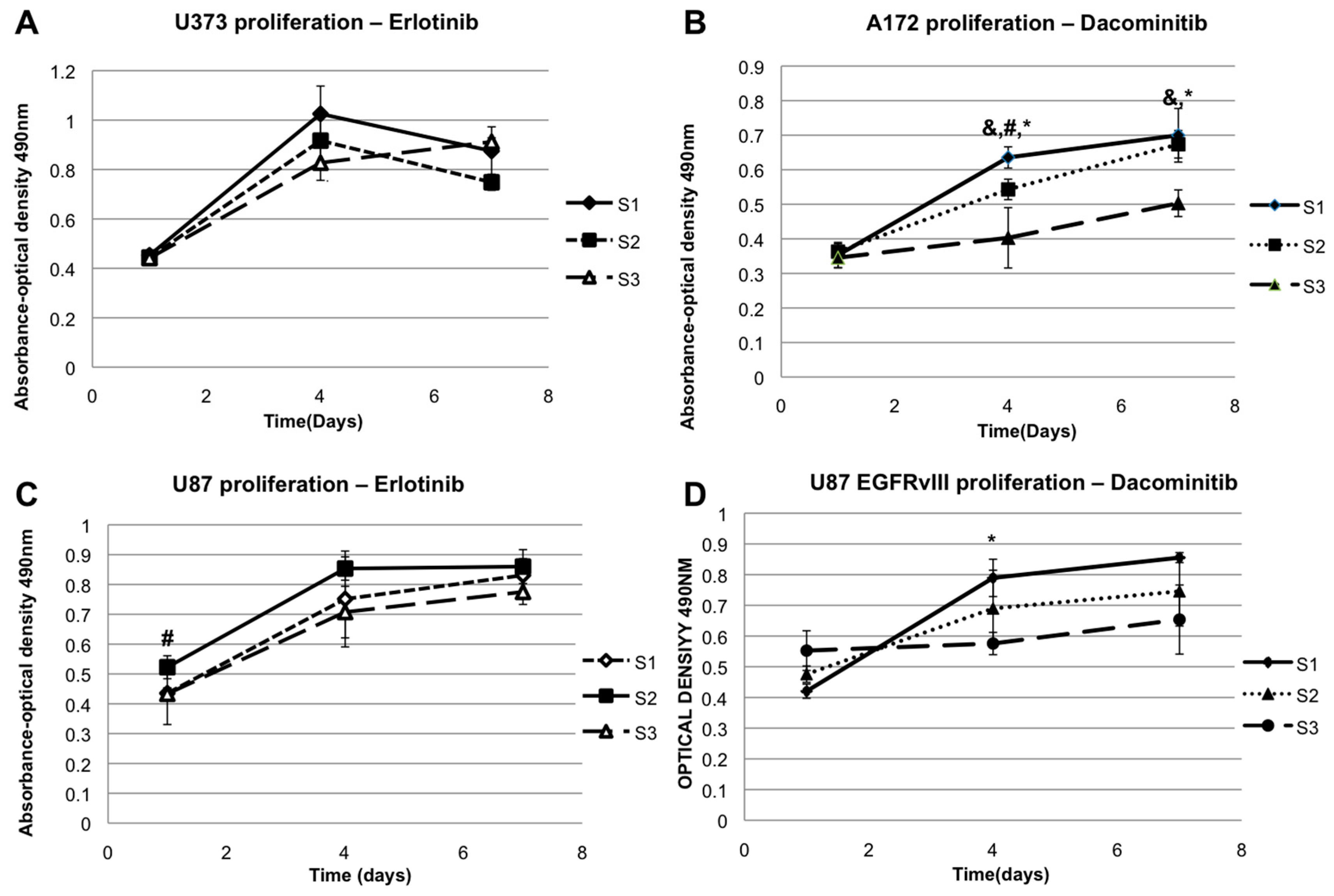
2.3. The Effect of EGFR Inhibition on Elastic Modulus-Dependent Cell Proliferation
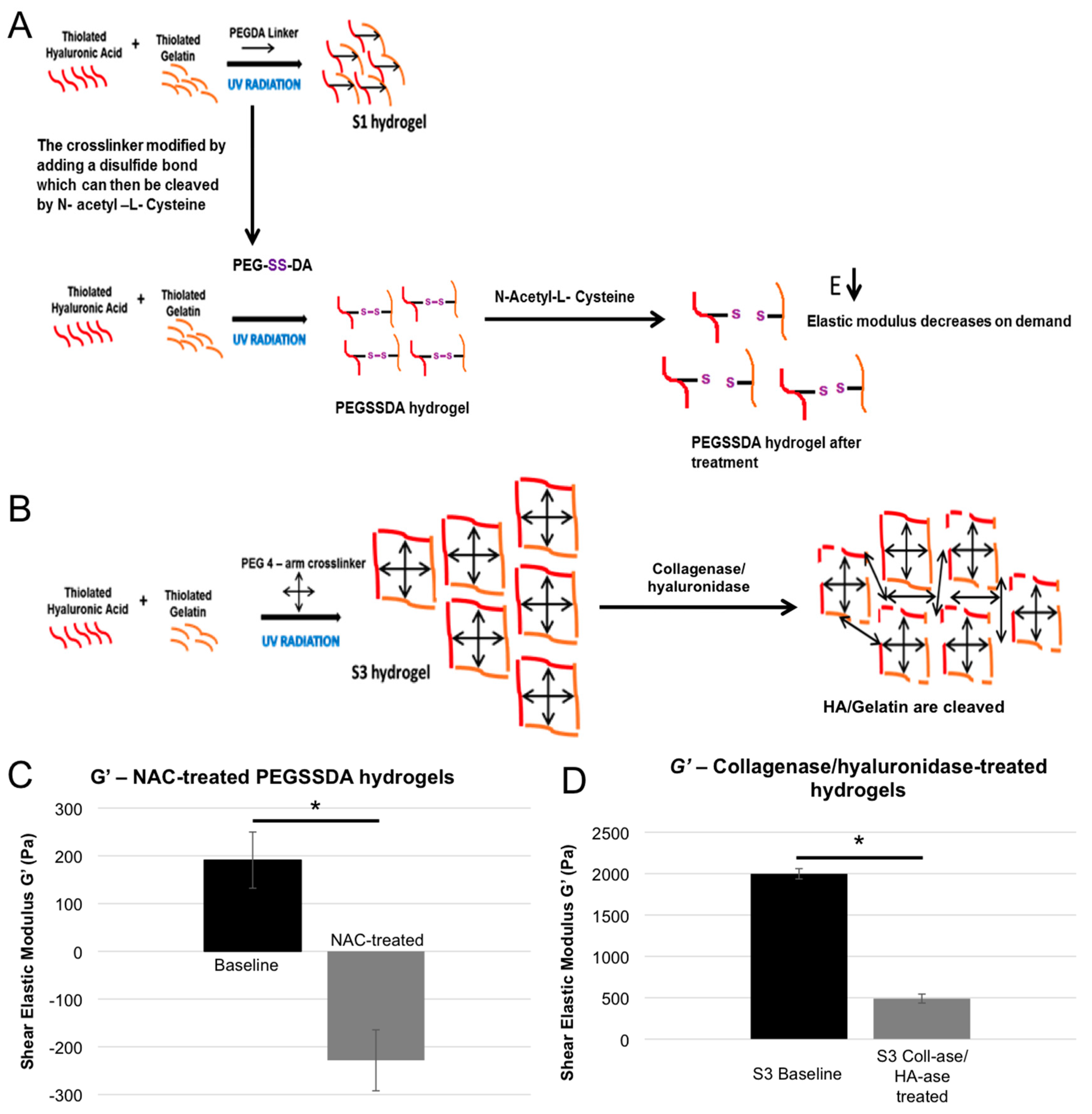
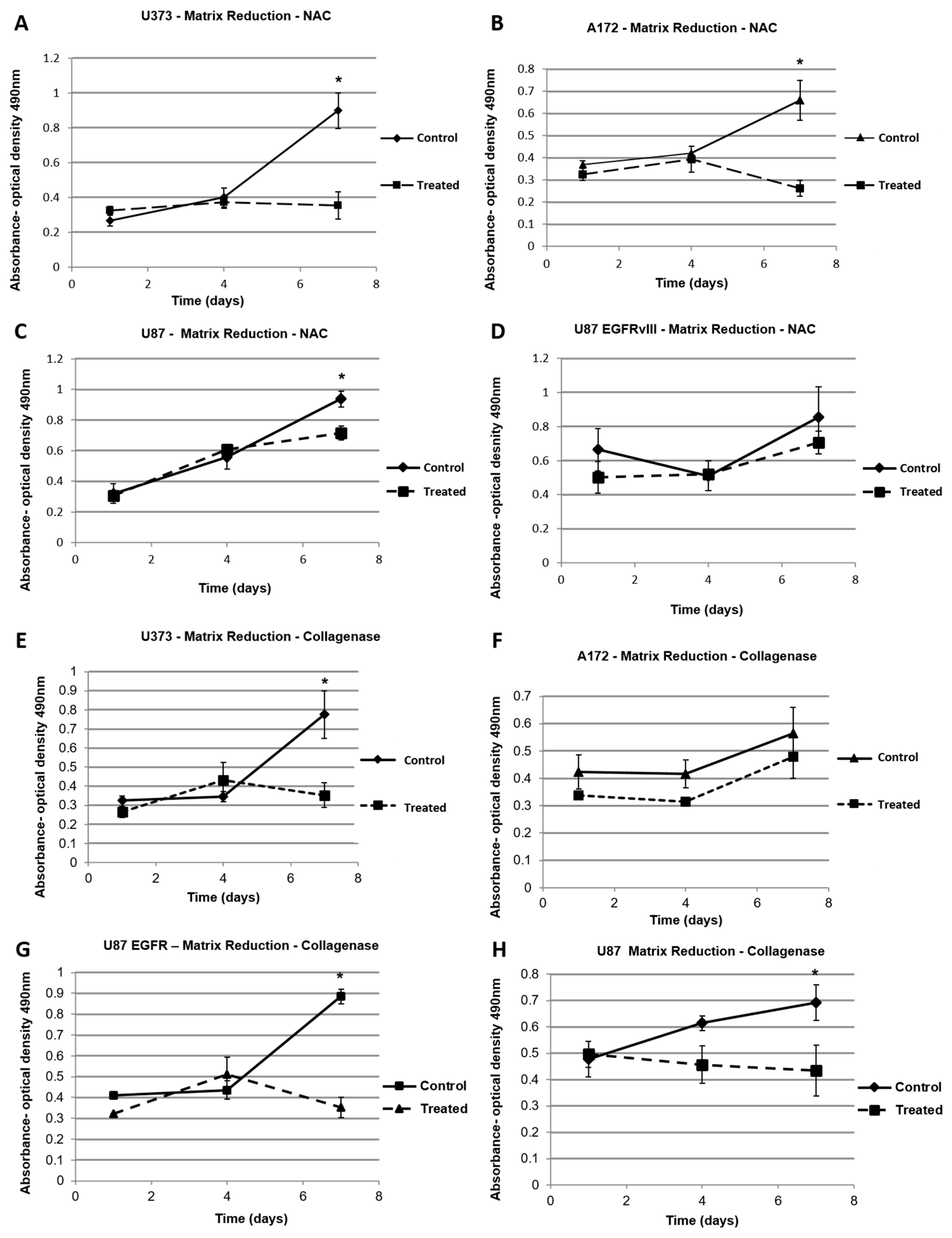
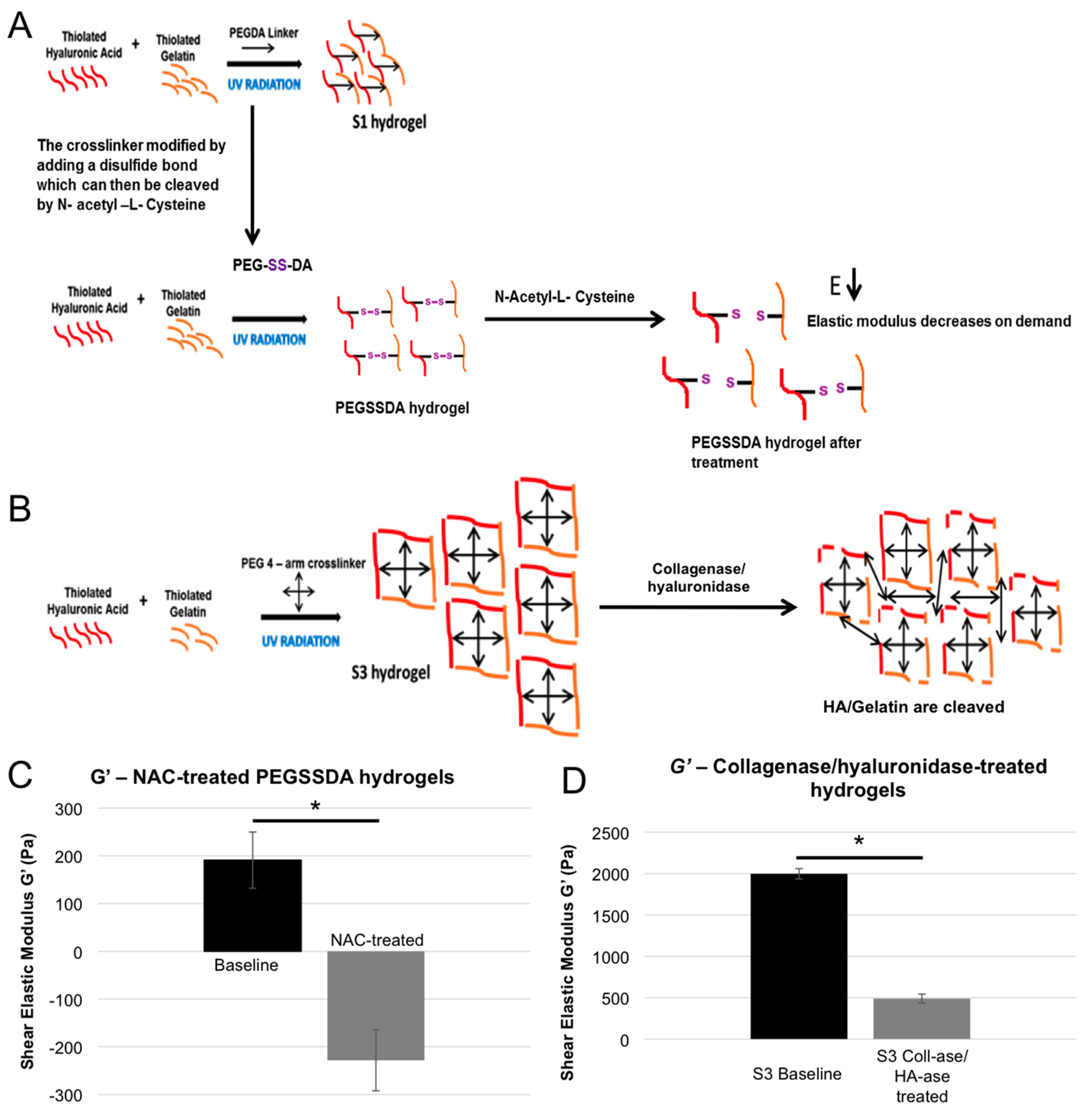
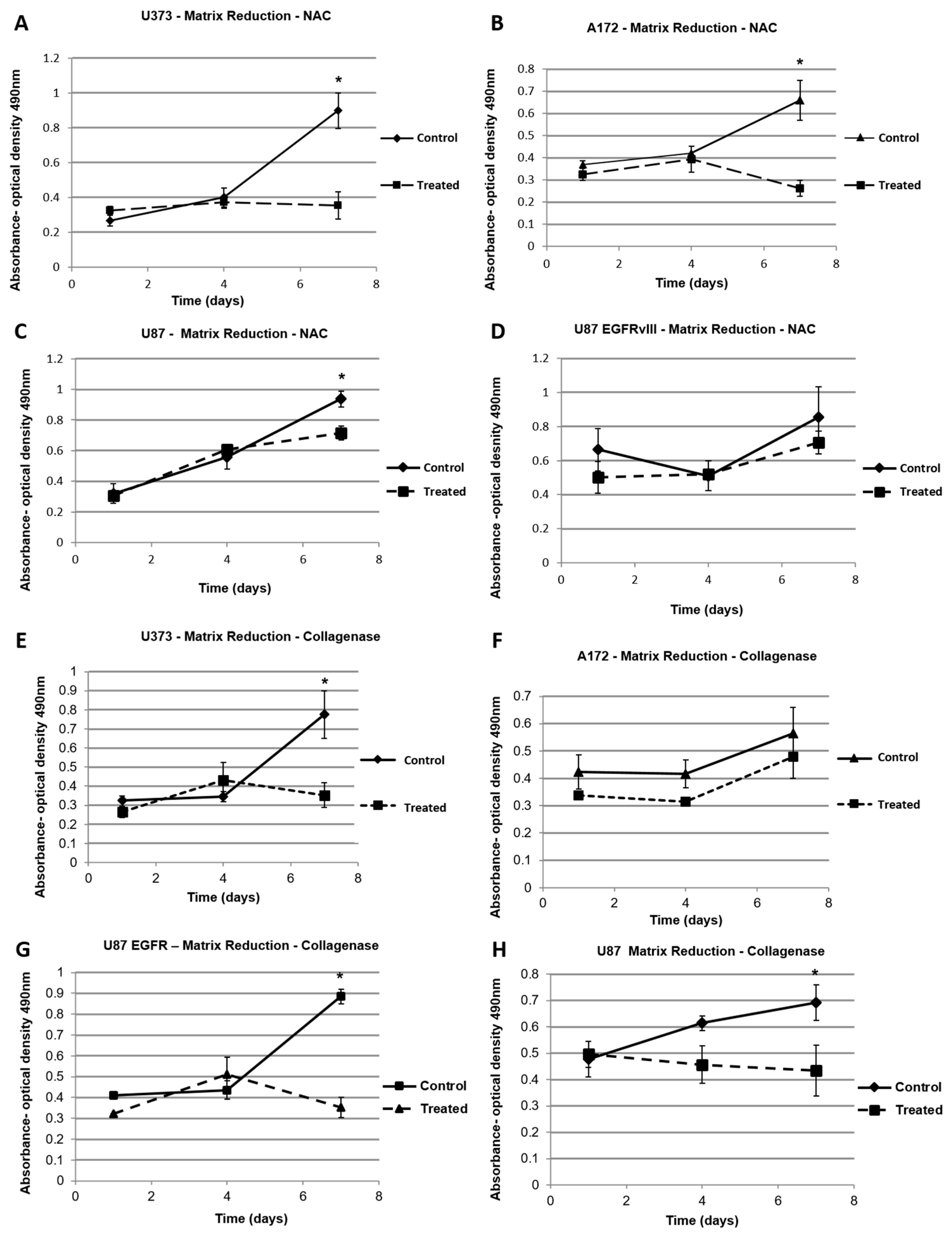
2.4. Dynamic Elastic Modulus Reduction as a Therapeutic Application
3. Conclusions
4. Materials and Methods
4.1. Cells Employed in Elastic Modulus Experiments
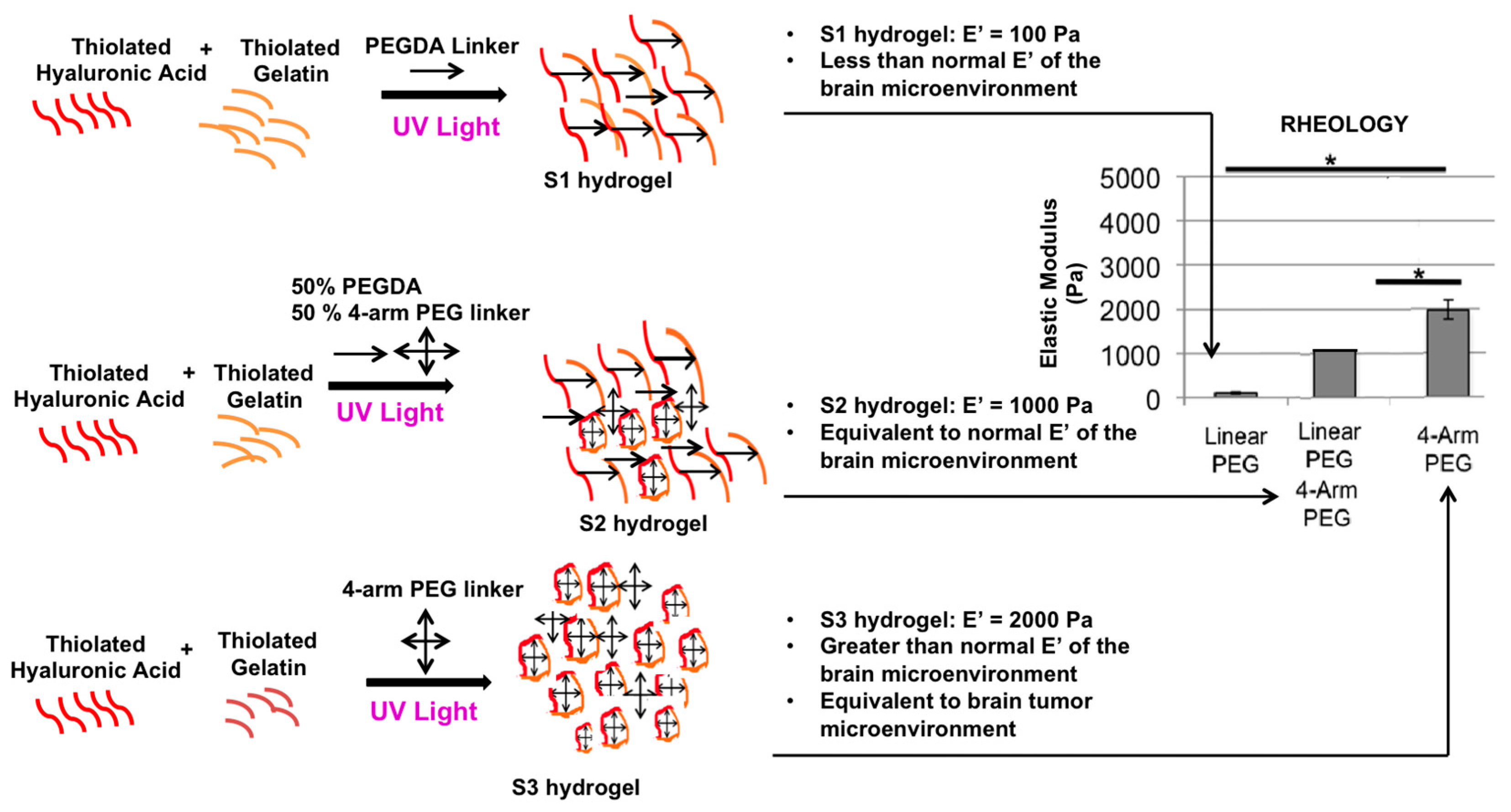
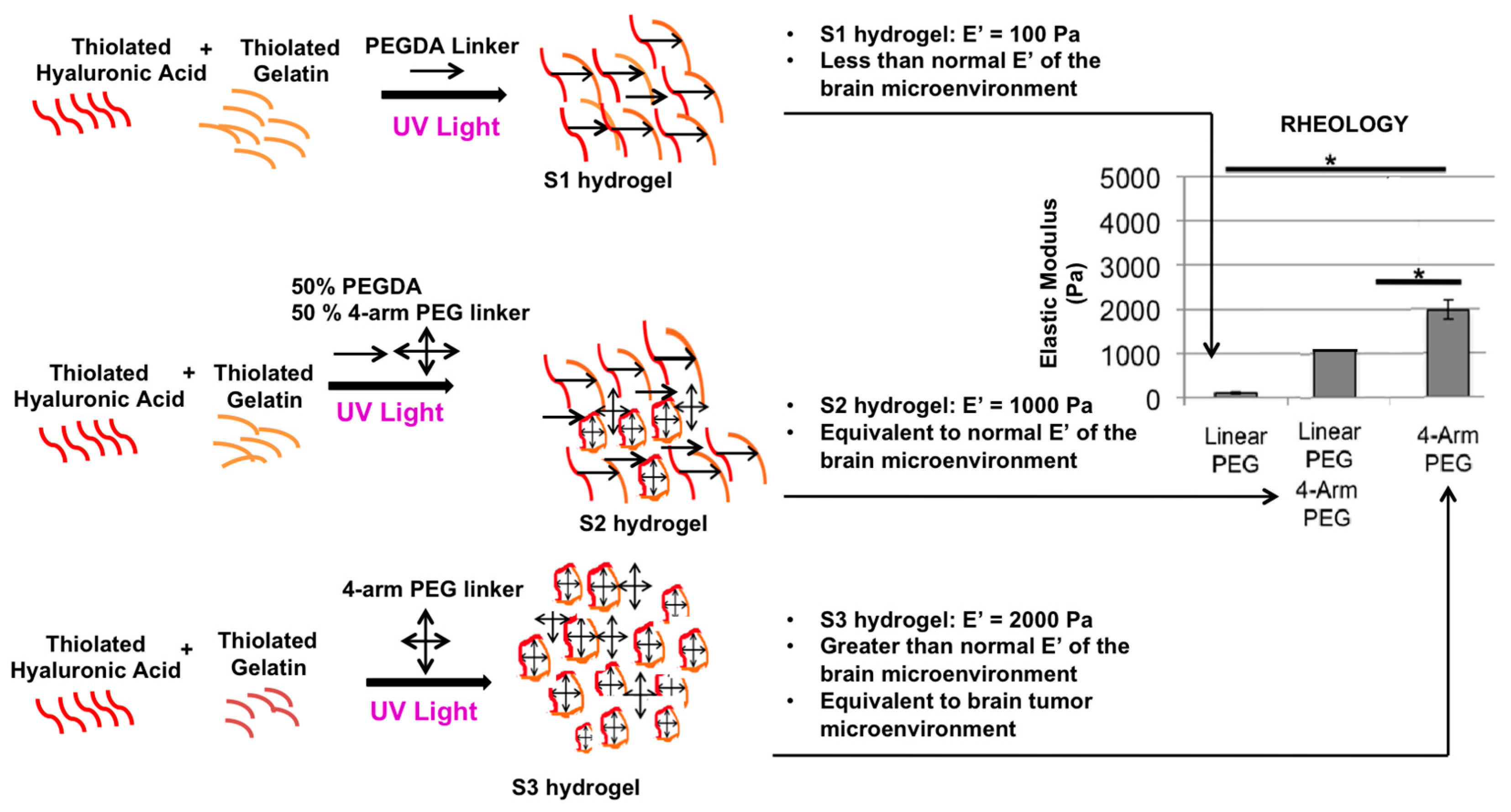
4.2. Hydrogels
4.3. Proliferation Assays
4.4. Immunofluorescence Staining for FAK Phosphorylation
4.5. FAK and EGFR Inhibition
4.7. Hydrogel Reduction Through a Breakable Cross-linker
4.8. Hydrogel Reduction through Enzymatic Degradation
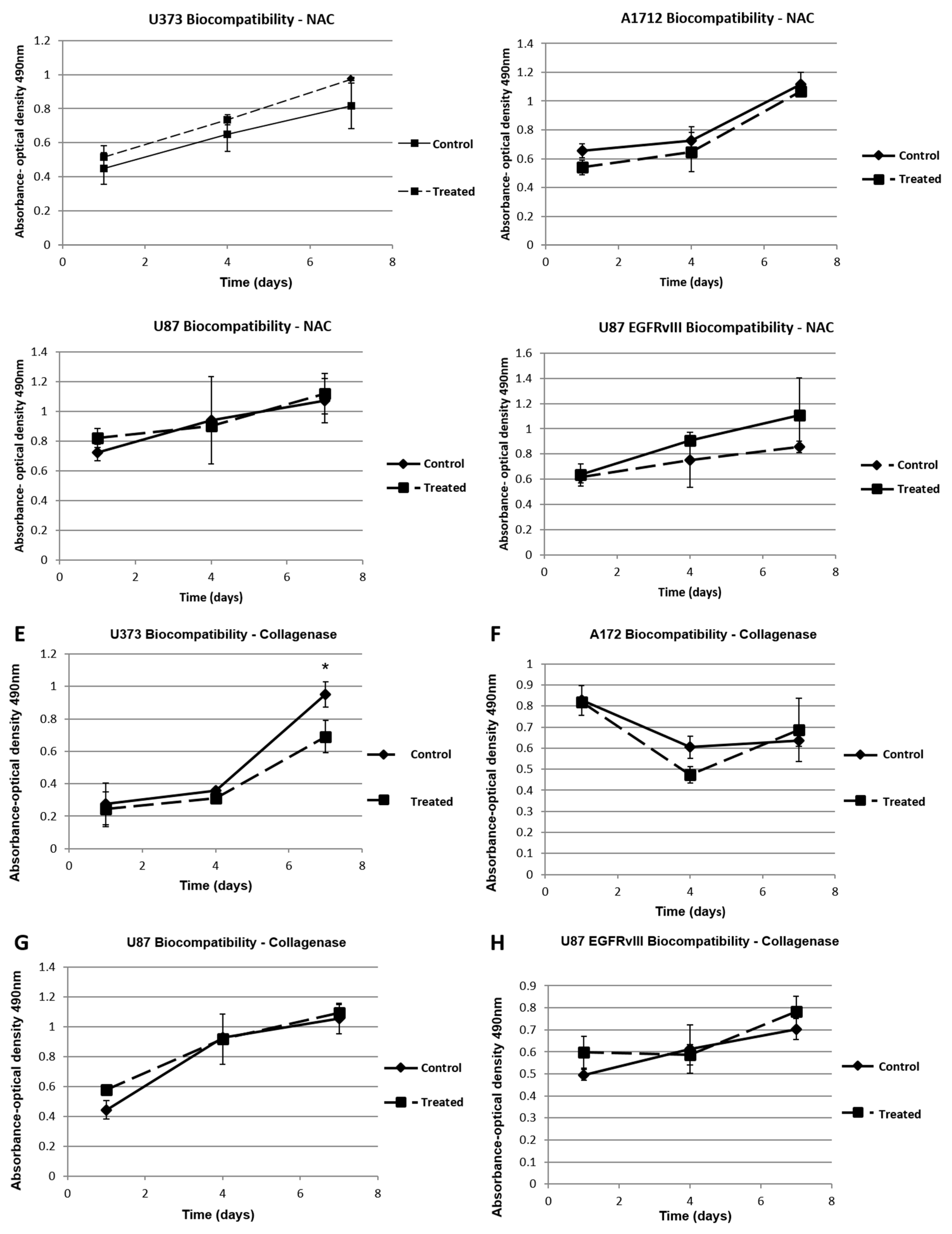
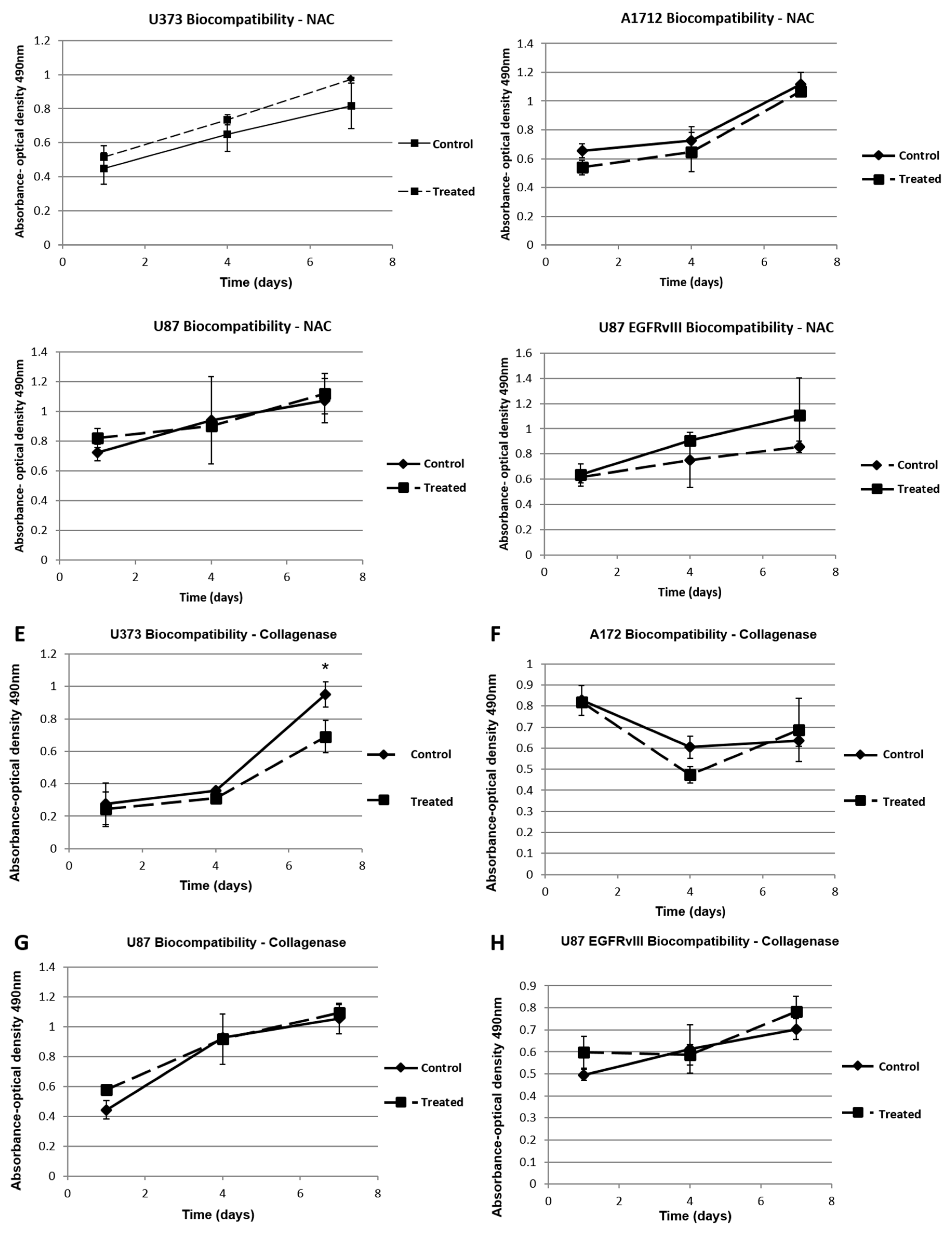
4.9. NAC and Collagenase/Hyaluronidase Biocompatibility Verification
4.10. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Optn: Organ Procurement and Transplantation Network Data Report. Avaliable online: http://optn.transplant.hrsa.gov/ (accessed on 31 March 2017).
- Dolecek, T.A.; Propp, J.M.; Stroup, N.E.; Kruchko, C. Cbtrus statistical report: Primary brain and central nervous system tumors diagnosed in the united states in 2005–2009. Neuro-Oncology 2012, 14, v1–v49. [Google Scholar] [CrossRef] [PubMed]
- Behin, A.; Hoang-Xuan, K.; Carpentier, A.F.; Delattre, J.Y. Primary brain tumours in adults. Lancet 2003, 361, 323–331. [Google Scholar] [CrossRef]
- Ruoslahti, E. Brain extracellular matrix. Glycobiology 1996, 6, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Bonneh-Barkay, D.; Wiley, C.A. Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009, 19, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Bellail, A.C.; Hunter, S.B.; Brat, D.J.; Tan, C.; van Meir, E.G. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 2004, 36, 1046–1069. [Google Scholar] [CrossRef] [PubMed]
- Laurent, T.C.; Fraser, J.R. Hyaluronan. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1992, 6, 2397–2404. [Google Scholar]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Delpech, B.; Maingonnat, C.; Girard, N.; Chauzy, C.; Maunoury, R.; Olivier, A.; Tayot, J.; Creissard, P. Hyaluronan and hyaluronectin in the extracellular matrix of human brain tumour stroma. Eur. J. Cancer 1993, 29, 1012–1017. [Google Scholar] [CrossRef]
- Duck, F.A. Mechanics of Biological Systems and Materials. In Physical Properties of Tissues—A Comprehensive Reference; Academic Press: Cambridge, MA, USA, 1990; Volume 2. [Google Scholar]
- Murphy, M.C.; Huston, J., 3rd; Jack, C.R., Jr.; Glaser, K.J.; Manduca, A.; Felmlee, J.P.; Ehman, R.L. Decreased brain stiffness in Alzheimer’s disease determined by magnetic resonance elastography. J. Magn. Reson. Imaging JMRI 2011, 34, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Mahesparan, R.; Tysnes, B.B.; Read, T.A.; Enger, P.O.; Bjerkvig, R.; Lund-Johansen, M. Extracellular matrix-induced cell migration from glioblastoma biopsy specimens in vitro. Acta Neuropathol. 1999, 97, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Elias, D.; Sideris, L.; Pocard, M.; de Baere, T.; Dromain, C.; Lassau, N.; Lasser, P. Incidence of unsuspected and treatable metastatic disease associated with operable colorectal liver metastases discovered only at laparotomy (and not treated when performing percutaneous radiofrequency ablation). Ann. Surg. Oncol. 2005, 12, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.B.; Harris, R.; Fletcher, S.W. The rational clinical examination. Does this patient have breast cancer? The screening clinical breast examination: Should it be done? How? JAMA 1999, 282, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Hauck, C.R.; Sieg, D.J. Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 1999, 71, 435–478. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Huang, G.; Ho, B.; Yemma, M.; Morrison, C.D.; Lee, J.; Eliceiri, B.P.; Cance, W.G. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol. Cancer Ther. 2013, 12, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, M.; Hecker, T.P.; Gladson, C.L. FAK signaling in anaplastic astrocytoma and glioblastoma tumors. Cancer J. 2003, 9, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Kalman, B.; Szep, E.; Garzuly, F.; Post, D.E. Epidermal growth factor receptor as a therapeutic target in glioblastoma. Neuromol. Med. 2013, 15, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The egfrviii variant in glioblastoma multiforme. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, R.; Ji, X.D.; Harmon, R.C.; Lazar, C.S.; Gill, G.N.; Cavenee, W.K.; Huang, H.J. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA 1994, 91, 7727–7731. [Google Scholar] [CrossRef] [PubMed]
- Fenstermaker, R.A.; Ciesielski, M.J. EGFR intron recombination in human gliomas: Inappropriate diversion of V(D)J recombination? Curr. Genom. 2007, 8, 163–170. [Google Scholar] [CrossRef]
- Tomar, A.; Schlaepfer, D.D. A PAK-activated linker for EGFR and FAK. Dev. Cell 2010, 18, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Malkki, H. Trial watch: Glioblastoma vaccine therapy disappointment in phase iii trial. Nat. Rev. Neurol. 2016, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, T.A.; de Juan Pardo, E.M.; Kumar, S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef] [PubMed]
- Hegedüs, B.; Marga, F.; Jakab, K.; Sharpe-Timms, K.L.; Forgacs, G. The interplay of cell–cell and cell–matrix interactions in the invasive properties of brain tumors. Biophys. J. 2006, 91, 2708–2716. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Mack, D.; Kapetanovic, E.; Atala, A.; Jackson, J.D.; Yoo, J.; Soker, S. Bioprinted amniotic fluid-derived stem cells accelerate healing of large skin wounds. Stem Cells Transl. Med. 2012, 1, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Alivon, M.; Beaussier, H.; Boutouyrie, P. Aortic stiffness as a tissue biomarker for predicting future cardiovascular events in asymptomatic hypertensive subjects. Ann. Med. 2012, 44 (Suppl. 1), S93–S97. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, K.; Gastaneda, B.; Zhang, M.; Nigwekar, P.; di Sant’Agnese, P.A.; Joseph, J.V.; Strang, J.; Rubens, D.J.; Parker, K.J. Tissue elasticity properties as biomarkers for prostate cancer. Cancer Biomark. 2008, 4, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Sebens, S.; Seidl, D.; Lehnert, H.; Hass, R. Interaction of tumor cells with the microenvironment. Cell Commun. Signal. CCS 2011, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Weaver, V.M. Mechanics, malignancy, and metastasis: The force journey of a tumor cell. Cancer Metast. Rev. 2009, 28, 113–127. [Google Scholar] [CrossRef]
- Grundy, T.J.; de Leon, E.; Griffin, K.R.; Stringer, B.W.; Day, B.W.; Fabry, B.; Cooper-White, J.; O’Neill, G.M. Differential response of patient-derived primary glioblastoma cells to environmental stiffness. Sci. Rep. 2016, 6, 23353. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, G.D. Simplifying the extracellular matrix for 3-D cell culture and tissue engineering: A pragmatic approach. J. Cell Biochem. 2007, 101, 1370–1383. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, G.D. Engineering a clinically-useful matrix for cell therapy. Organogenesis 2008, 4, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Kang, H.W.; Mead, I.; Bishop, C.; Shupe, T.; Lee, S.J.; Jackson, J.; Yoo, J.; Soker, S.; et al. A hydrogel bioink toolkit for mimicking native tissue biochemical and mechanical properties in bioprinted tissue constructs. Acta Biomater. 2015, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Kang, H.W.; Seol, Y.J.; Forsythe, S.D.; Bishop, C.; Shupe, T.; Soker, S.; Atala, A. Bioprinting cellularized constructs using a tissue-specific hydrogel bioink. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Murphy, S.V.; Crowell, K.; Mack, D.; Atala, A.; Soker, S. A tunable hydrogel system for long-term release of cell-secreted cytokines and bioprinted in situ wound cell delivery. J. Biomed. Mater. Res. B Appl. Biomater. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Skardal, A.; Prestwich, G.D. Engineered extracellular matrices with cleavable crosslinkers for cell expansion and easy cell recovery. Biomaterials 2008, 29, 4521–4531. [Google Scholar] [CrossRef] [PubMed]
- Umesh, V.; Rape, A.D.; Ulrich, T.A.; Kumar, S. Microenvironmental stiffness enhances glioma cell proliferation by stimulating epidermal growth factor receptor signaling. PLoS ONE 2014, 9, e101771. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Forsythe, S.; Atala, A.; Soker, S. A reductionist metastasis-on-a-chip platform for in vitro tumor progression modeling and drug screening. Biotechnol. Bioeng. 2016, 113, 2020–2032. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Rodman, C.; Atala, A.; Soker, S. Liver-tumor hybrid organoids for modeling tumor growth and drug response in vitro. Ann. Biomed. Eng. 2015, 43, 2361–2373. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Soker, S.; Hall, A.R. In situ patterned micro 3d liver constructs for parallel toxicology testing in a fluidic device. Biofabrication 2015, 7, 031001. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Smith, L.; Bharadwaj, S.; Atala, A.; Soker, S.; Zhang, Y. Tissue specific synthetic ecm hydrogels for 3-d in vitro maintenance of hepatocyte function. Biomaterials 2012, 33, 4565–4575. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Zhang, J.; Prestwich, G.D. Bioprinting vessel-like constructs using hyaluronan hydrogels crosslinked with tetrahedral polyethylene glycol tetracrylates. Biomaterials 2010, 31, 6173–6181. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.J.; Fisher, C.; Scheffler, K.; Wan, R.; Maleki, H.; Liu, H.; Sun, Y.; Simmons, A.C.; Birngruber, R.; Lilge, L. Polyacrylamide gel substrates that simulate the mechanical stiffness of normal and malignant neuronal tissues increase protoporphyin ix synthesis in glioma cells. J. Biomed. Opt. 2015, 20, 098002. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Chin, L.; Georges, P.C.; Byfield, F.J.; Bucki, R.; Kim, R.; Weaver, M.; Wells, R.G.; Marcinkiewicz, C.; Janmey, P.A. Compression stiffening of brain and its effect on mechanosensing by glioma cells. New J. Phys. 2014, 16, 075002. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Mack, D.; Atala, A.; Soker, S. Substrate elasticity controls cell proliferation, surface marker expression and motile phenotype in amniotic fluid-derived stem cells. J. Mech. Behav. Biomed. Mater. 2013, 17, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Spatz, J.P.; Bershadsky, A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009, 10, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, Y.; Wang, C.; Sun, L.; Mei, C.; Yao, W.; Liu, Y.; Shi, Y.; Qiu, S.; Fan, J.; et al. The effect of epidermal growth factor receptor variant III on glioma cell migration by stimulating ERK phosphorylation through the focal adhesion kinase signaling pathway. Arch. Biochem. Biophys. 2010, 502, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.E.; Furnari, F.B.; Cavenee, W.K. Targeting EGFR for treatment of glioblastoma: Molecular basis to overcome resistance. Curr. Cancer Drug Targets 2012, 12, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Schulte, A.; Liffers, K.; Kathagen, A.; Riethdorf, S.; Zapf, S.; Merlo, A.; Kolbe, K.; Westphal, M.; Lamszus, K. Erlotinib resistance in EGFR-amplified glioblastoma cells is associated with upregulation of egfrviii and pi3kp110delta. Neuro-Oncology 2013, 15, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shah, K. Multiple lesions in receptor tyrosine kinase pathway determine glioblastoma response to pan-ERBB inhibitor PF-00299804 and PI3K/mTOR dual inhibitor PF-05212384. Cancer Biol. Ther. 2014, 15, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, M.; Das, S.; Berns, E.J.; Kim, J.; Stupp, S.I.; Kessler, J.A. Nanofiber-mediated inhibition of focal adhesion kinase sensitizes glioma stemlike cells to epidermal growth factor receptor inhibition. Neuro-Oncology 2013, 15, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Ananthanarayanan, B.; Kim, Y.; Kumar, S. Elucidating the mechanobiology of malignant brain tumors using a brain matrix-mimetic hyaluronic acid hydrogel platform. Biomaterials 2011, 32, 7913–7923. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, L.J.; Brangwynne, C.P.; Kasza, K.E.; Filippidi, E.; Gordon, V.D.; Deisboeck, T.S.; Weitz, D.A. Glioma expansion in collagen i matrices: Analyzing collagen concentration-dependent growth and motility patterns. Biophys. J. 2005, 89, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Gordon, V.D.; Valentine, M.T.; Gardel, M.L.; Andor-Ardo, D.; Dennison, S.; Bogdanov, A.A.; Weitz, D.A.; Deisboeck, T.S. Measuring the mechanical stress induced by an expanding multicellular tumor system: A case study. Exp. Cell Res. 2003, 289, 58–66. [Google Scholar] [CrossRef]
- Lu, Z.; Jiang, G.; Blume-Jensen, P.; Hunter, T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol. Cell. Biol. 2001, 21, 4016–4031. [Google Scholar] [CrossRef] [PubMed]
- Muthupillai, R.; Lomas, D.J.; Rossman, P.J.; Greenleaf, J.F.; Manduca, A.; Ehman, R.L. Magnetic resonance elastography by direct visualization of propagating acoustic strain waves. Science 1995, 269, 1854–1857. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lin, Y.; Han, J.C.; Xi, Z.N.; Shen, H.; Gao, P.Y. Magnetic resonance elastography of brain tumors: Preliminary results. Acta Radiol. 2007, 48, 327–330. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stiffness Sensitivity Summary | |||
|---|---|---|---|
| Cell Line | Proliferation Rate Highest in Normal Condition | Proliferation Rate Highest in Defactinib | Proliferation Rate Highest in EGFR Inhibition |
| U373-MG | S3 *,& | S1 * | No distinct preference |
| A172 | S2 & and S3 * (DAY 4) | S1 * | S1 * and S2 & |
| U87-MG | S2 & | S2 | No distinct preference |
| U87-EGFRvIII | S3 | S1 | S1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivakumar, H.; Strowd, R.; Skardal, A. Exploration of Dynamic Elastic Modulus Changes on Glioblastoma Cell Populations with Aberrant EGFR Expression as a Potential Therapeutic Intervention Using a Tunable Hyaluronic Acid Hydrogel Platform. Gels 2017, 3, 28. https://doi.org/10.3390/gels3030028
Sivakumar H, Strowd R, Skardal A. Exploration of Dynamic Elastic Modulus Changes on Glioblastoma Cell Populations with Aberrant EGFR Expression as a Potential Therapeutic Intervention Using a Tunable Hyaluronic Acid Hydrogel Platform. Gels. 2017; 3(3):28. https://doi.org/10.3390/gels3030028
Chicago/Turabian StyleSivakumar, Hemamylammal, Roy Strowd, and Aleksander Skardal. 2017. "Exploration of Dynamic Elastic Modulus Changes on Glioblastoma Cell Populations with Aberrant EGFR Expression as a Potential Therapeutic Intervention Using a Tunable Hyaluronic Acid Hydrogel Platform" Gels 3, no. 3: 28. https://doi.org/10.3390/gels3030028
APA StyleSivakumar, H., Strowd, R., & Skardal, A. (2017). Exploration of Dynamic Elastic Modulus Changes on Glioblastoma Cell Populations with Aberrant EGFR Expression as a Potential Therapeutic Intervention Using a Tunable Hyaluronic Acid Hydrogel Platform. Gels, 3(3), 28. https://doi.org/10.3390/gels3030028