
Grafting Techniques towards Production of Peptide-Tethered Hydrogels, a Novel Class of Materials with Biomedical Interest



Abstract
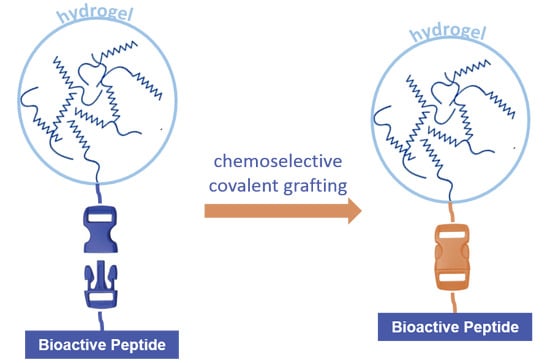
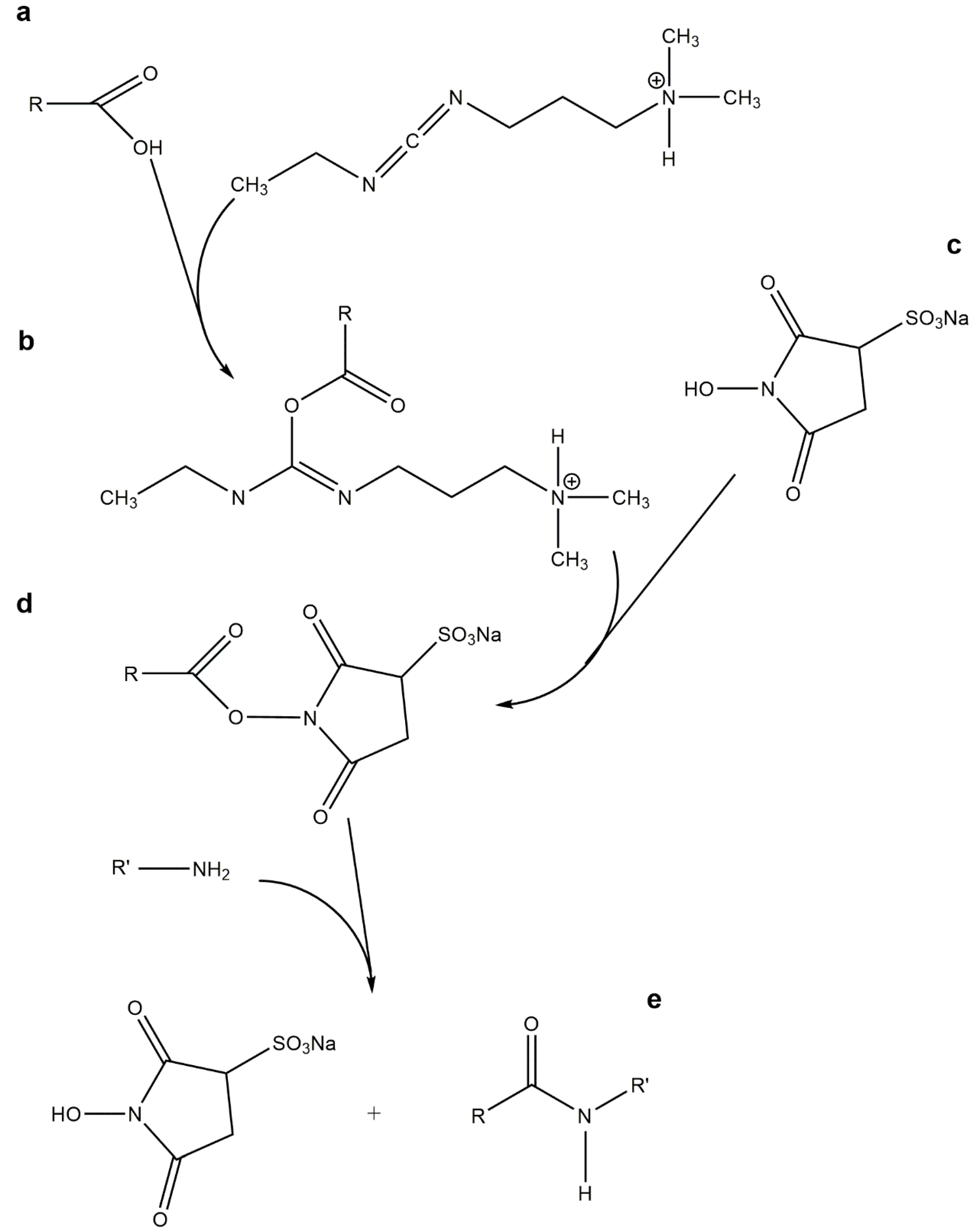
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Peptides Underlying a Paradigm Shift in Traditional Therapies
3. Peptide Delivery Systems
4. Hydrogels as Drug-Delivery Vehicles and Scaffolds for Tissue Regeneration
5. Peptide Tethering onto Hydrogels through “Click” Chemistry
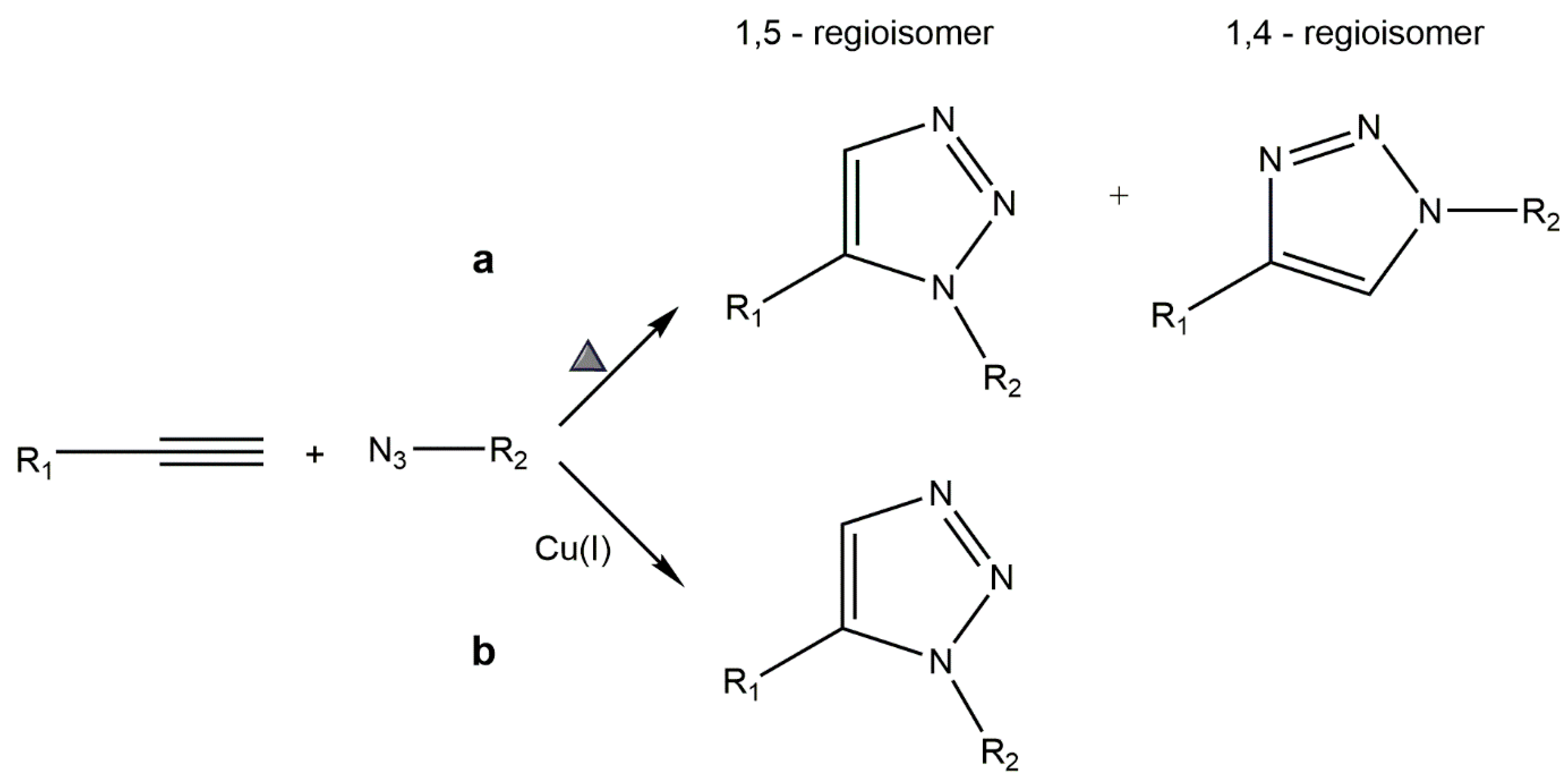
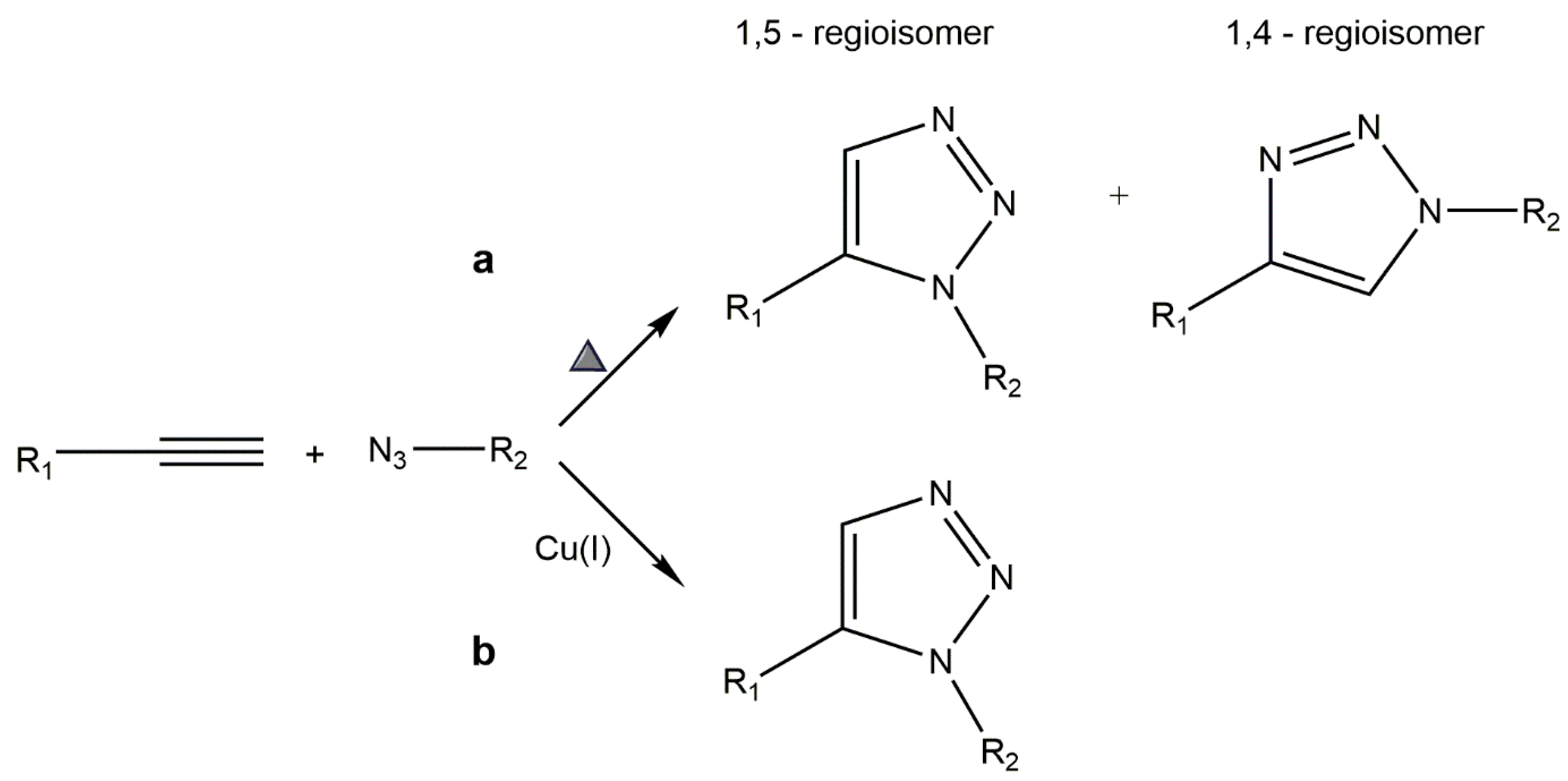
5.1. Copper-Catalyzed Azide–Alkyne Cycloaddition (CuAAC)
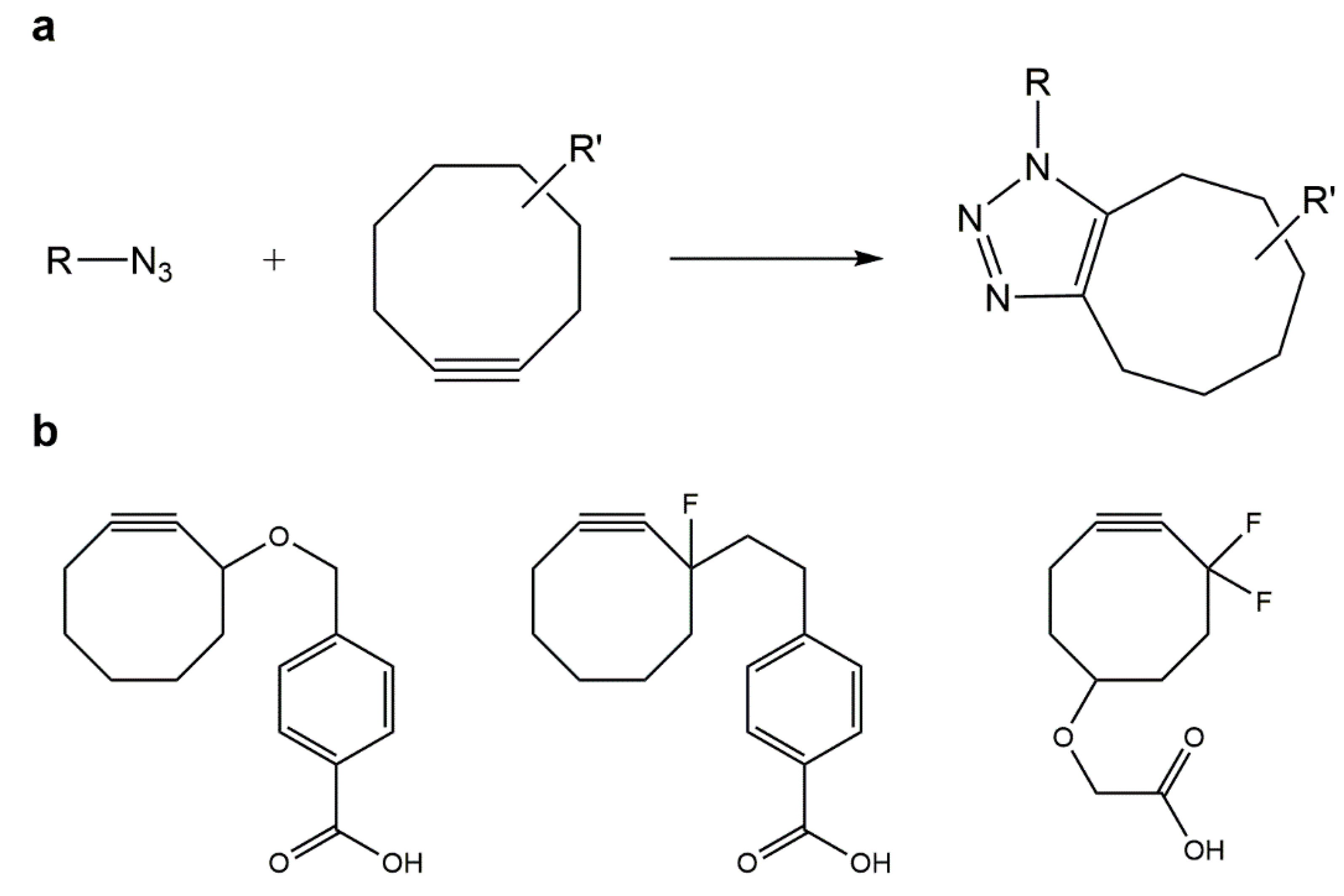
5.2. Strain-Promoted Azide–Alkyne Cycloaddition (SPAAC)
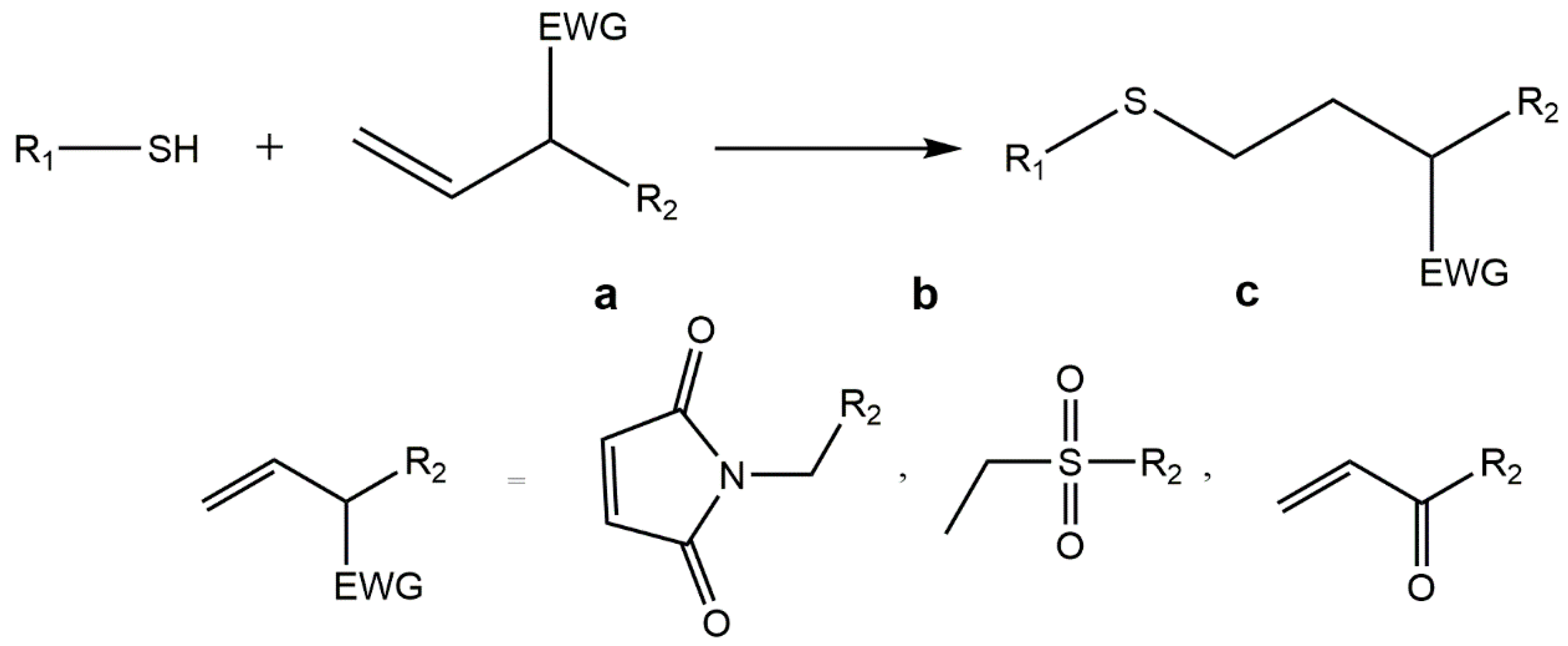
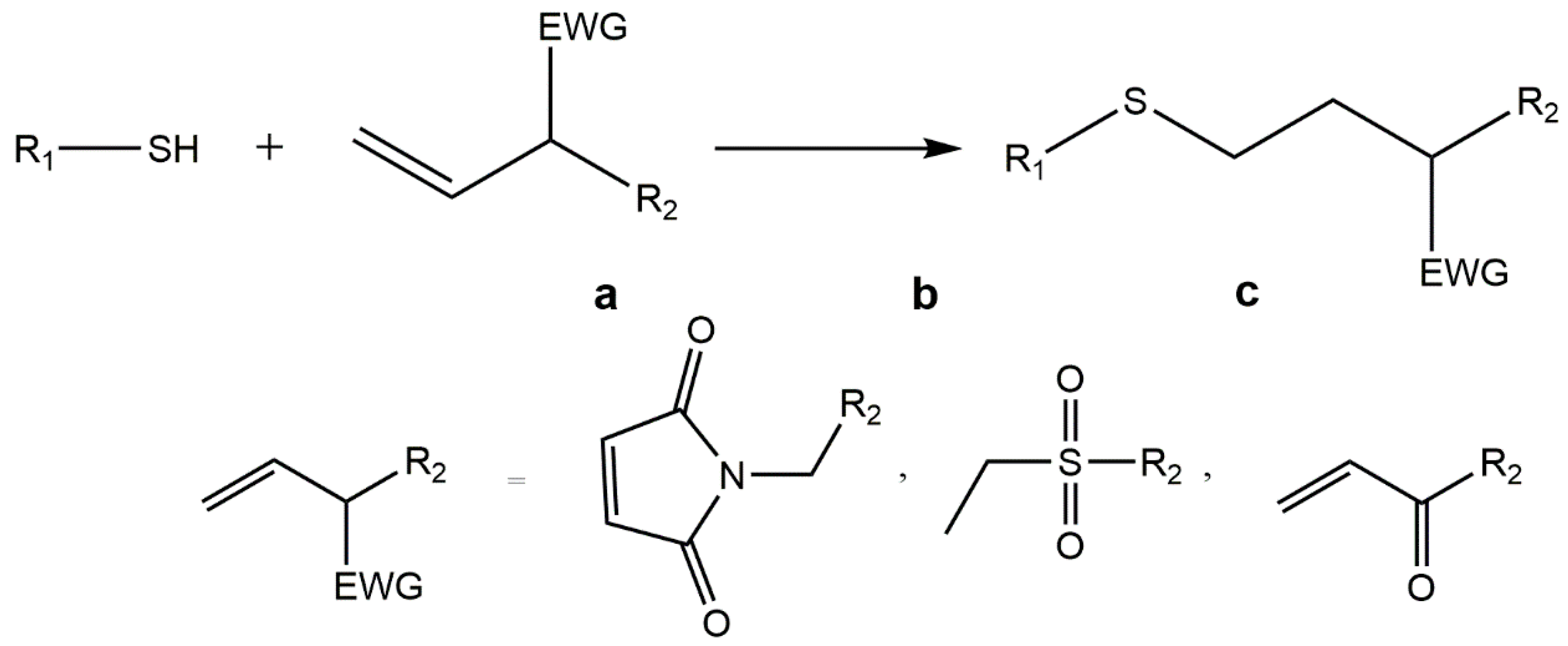
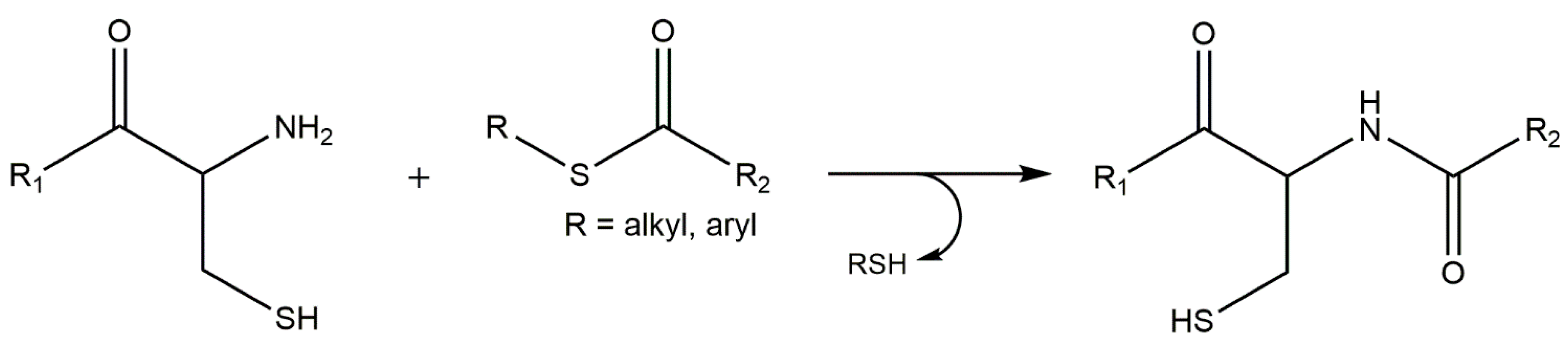
5.3. Thiol-ene “Click” Chemistry

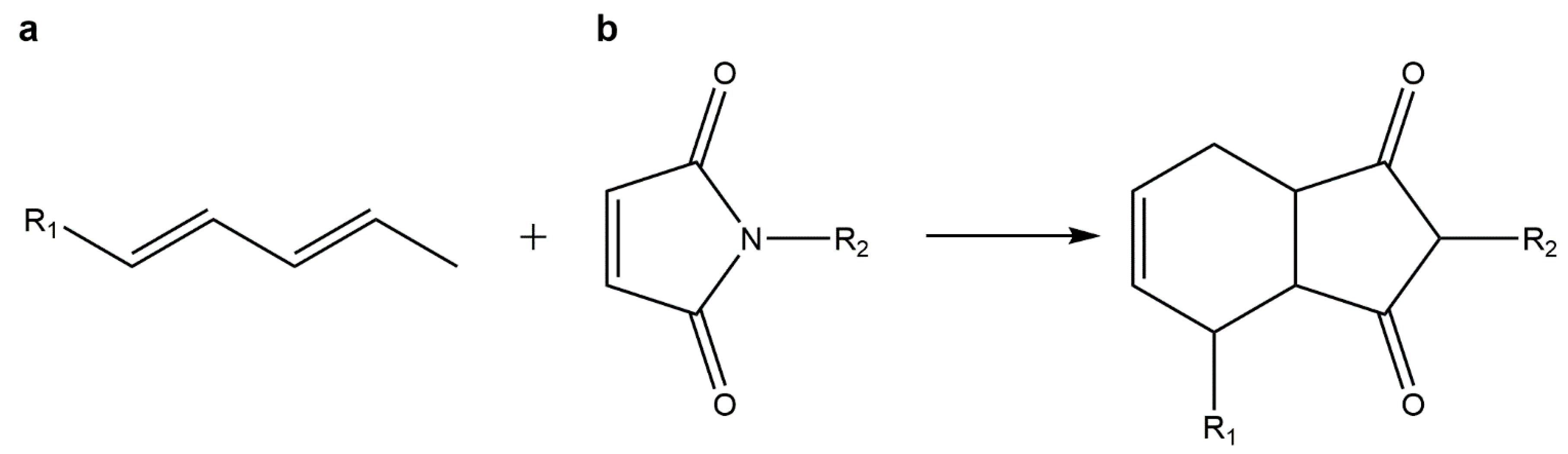
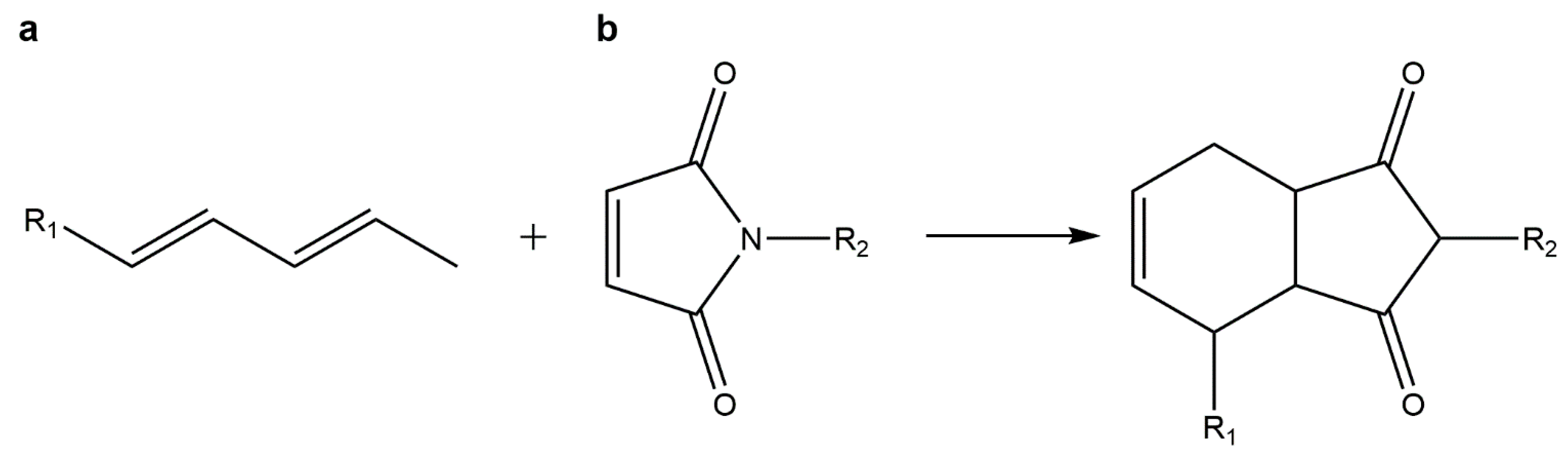
5.4. Diels–Alder Cycloadditions
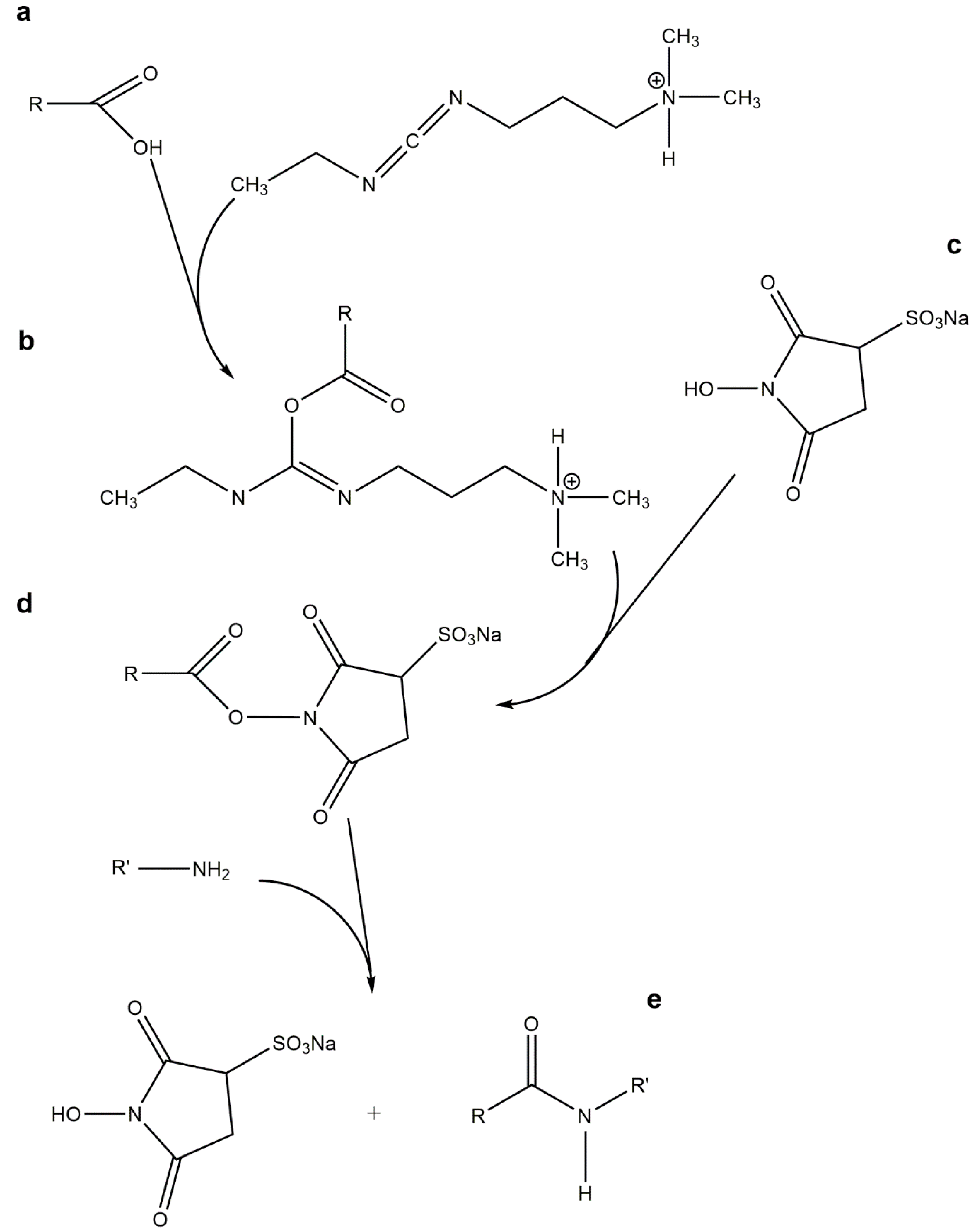
5.5. Oxime “Click” Chemistry
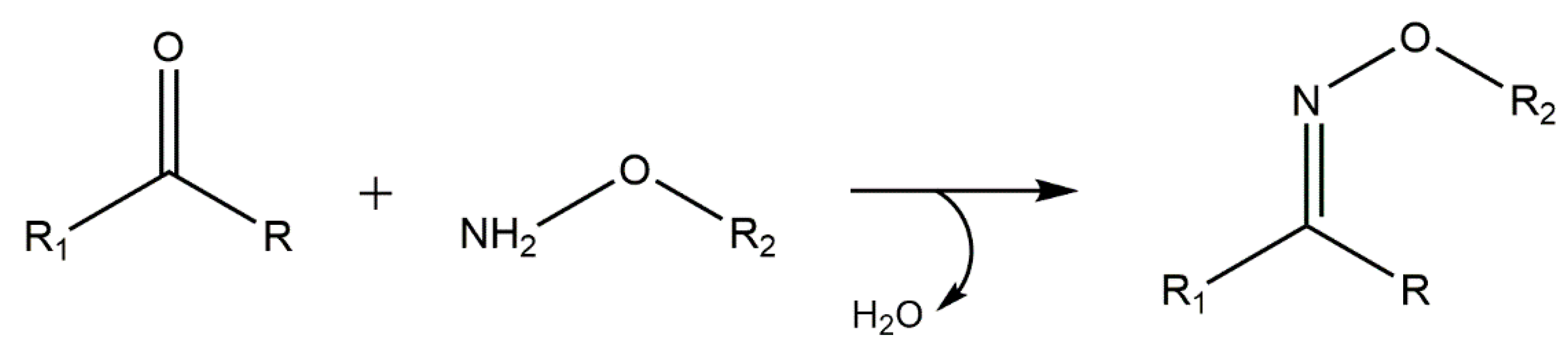
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, L.; Li, K.; Xiao, W.; Zheng, L.; Xiao, Y.; Fan, H.; Zhang, X. Preparation of collagen–chondroitin sulfate–hyaluronic acid hybrid hydrogel scaffolds and cell compatibility in vitro. Carbohydr. Polym. 2011, 84, 118–125. [Google Scholar] [CrossRef]
- Orsi, S.; de Capua, A.; Guarnieri, D.; Marasco, D.; Netti, P.A. Cell recruitment and transfection in gene activated collagen matrix. Biomaterials 2010, 31, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Cameron, N.; Deming, T. Peptide-based materials for nanomedicine. Macromol. Biosci. 2015, 15, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; Fournier, A.; Szarpak-Jankowska, A.; Block, M.R.; Auzély-Velty, R. Type, density, and presentation of grafted adhesion peptides on polysaccharide-based hydrogels control preosteoblast behavior and differentiation. Biomacromolecules 2015, 16, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, D.; de Capua, A.; Ventre, M.; Borzacchiello, A.; Pedone, C.; Marasco, D.; Ruvo, M.; Netti, P.A. Covalently immobilized RGD gradient on PEG hydrogel scaffold influences cell migration parameters. Acta Biomater. 2010, 6, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y. Covalently immobilized biosignal molecule materials for tissue engineering. Soft Matter 2008, 4, 46–56. [Google Scholar] [CrossRef]
- Albericio, F.; Kruger, H.G. Therapeutic peptides. Future Med. Chem. 2012, 4, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Becker, M.L. “Click” reactions: A versatile toolbox for the synthesis of peptide-conjugates. Chem. Soc. Rev. 2014, 43, 7013–7039. [Google Scholar] [CrossRef] [PubMed]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Góngora-Benítez, M.; Tulla-Puche, J.; Albericio, F. Handles for Fmoc solid-phase synthesis of protected peptides. ACS Comb. Sci. 2013, 15, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Loffet, A. Peptides as drugs: Is there a market? J. Pept. Sci. 2002, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.M.B.; Cohen, M.A.; Bloom, S.R. Peptides as drugs. QJM 1999, 92, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.; Bemquerer, M.; Nascimento, C. Some mechanistic aspects on Fmoc solid phase peptide synthesis. Int. J. Pept. Res. Ther. 2014, 20, 53–69. [Google Scholar] [CrossRef]
- Chow, D.; Nunalee, M.L.; Lim, D.W.; Simnick, A.J.; Chilkoti, A. Peptide-based biopolymers in biomedicine and biotechnology. Mater. Sci. Eng. R Rep. 2008, 62, 125–155. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yang, J.; Sega, E. Issues related to targeted delivery of proteins and peptides. AAPS J. 2006, 8, E466–E478. [Google Scholar] [CrossRef] [PubMed]
- McGregor, D.P. Discovering and improving novel peptide therapeutics. Curr. Opin. Pharmacol. 2008, 8, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.K.; Viswanathan, M.; Kent, R.B.; Wood, C.R. Therapeutic peptides: Technological advances driving peptides into development. Curr. Opin. Biotechnol. 2006, 17, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Chandrudu, S.; Simerska, P.; Toth, I. Chemical methods for peptide and protein production. Molecules 2013, 18, 4373–4388. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Coin, I.; Beyermann, M.; Bienert, M. Solid-phase peptide synthesis: From standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2007, 2, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, A.A.; Reichert, J.M. Future directions for peptide therapeutics development. Drug Discov. Today 2013, 18, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.H.; Segura, T. Evolving the use of peptides as components of biomaterials. Biomaterials 2011, 32, 4198–4204. [Google Scholar] [CrossRef] [PubMed]
- Du, A.W.; Stenzel, M.H. Drug carriers for the delivery of therapeutic peptides. Biomacromolecules 2014, 15, 1097–1114. [Google Scholar] [CrossRef] [PubMed]
- Kogan, M.J.; Olmedo, I.; Hosta, L.; Guerrero, A.R.; Cruz, L.J.; Albericio, F. Peptides and metallic nanoparticles for biomedical applications. Nanomedicine 2007, 2, 287–306. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Anseth, K. PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm. Res. 2009, 26, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Stephanopoulos, N.; Ortony, J.H.; Stupp, S.I. Self-assembly for the synthesis of functional biomaterials. Acta Mater. 2013, 61, 912–930. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.B.; Zha, R.H.; Stupp, S.I. Peptide self-assembly for crafting functional biological materials. Curr. Opin. Solid State Mater. Sci. 2011, 15, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Sur, S.; Dankers, P.Y.W.; da Silva, R.M.P.; Boekhoven, J.; Poor, T.A.; Stupp, S.I. Post-assembly functionalization of supramolecular nanostructures with bioactive peptides and fluorescent proteins by native chemical ligation. Bioconjugate Chem. 2014, 25, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.J.; Tongers, J.; Renault, M.-A.; Roncalli, J.G.; Losordo, D.W.; Stupp, S.I. Reprint of: Development of bioactive peptide amphiphiles for therapeutic cell delivery. Acta Biomater. 2015, 23, S42–S51. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Zhang, Y.; Wang, C.-F.; Zhang, C.; Wang, X.-M.; Wang, D.-L.; Bai, X.-D.; Wen, T.-Y.; Xin, H.-K.; Wu, J.-H.; et al. Biological evaluation of human degenerated nucleus pulposus cells in functionalized self-assembling peptide nanofiber hydrogel scaffold. Tissue Eng. A 2014, 20, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.C.; Schneider, J.P. Self-assembling materials for therapeutic delivery. Acta Biomater. 2009, 5, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Kopeček, J.; Yang, J. Peptide-directed self-assembly of hydrogels. Acta Biomater. 2009, 5, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Antosova, Z.; Mackova, M.; Kral, V.; Macek, T. Therapeutic application of peptides and proteins: Parenteral forever? Trends Biotechnol. 2009, 27, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Censi, R.; di Martino, P.; Vermonden, T.; Hennink, W.E. Hydrogels for protein delivery in tissue engineering. J. Control. Release 2012, 161, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Casault, S.; Kenward, M.; Slater, G.W. Combinatorial design of passive drug delivery platforms. Int. J. Pharm. 2007, 339, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Vandermeulen, G.W.M.; Klok, H.-A. Peptide/protein hybrid materials: Enhanced control of structure and improved performance through conjugation of biological and synthetic polymers. Macromol. Biosci. 2004, 4, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Furth, M.E.; Atala, A.; van Dyke, M.E. Smart biomaterials design for tissue engineering and regenerative medicine. Biomaterials 2007, 28, 5068–5073. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Yuk, S.H. Polymeric protein delivery systems. Prog. Polym. Sci. 2007, 32, 669–697. [Google Scholar] [CrossRef]
- Tessmar, J.K.; Göpferich, A.M. Matrices and scaffolds for protein delivery in tissue engineering. Adv. Drug Deliv. Rev. 2007, 59, 274–291. [Google Scholar] [CrossRef] [PubMed]
- Lalatsa, A.; Schätzlein, A.G.; Mazza, M.; Le, T.B.H.; Uchegbu, I.F. Amphiphilic poly(l-amino acids)—New materials for drug delivery. J. Control. Release 2012, 161, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Saravanakumar, G.; Kim, K.; Kwon, I.C. Targeted delivery of low molecular drugs using chitosan and its derivatives. Adv. Drug Deliv. Rev. 2010, 62, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Sokolsky-Papkov, M.; Agashi, K.; Olaye, A.; Shakesheff, K.; Domb, A.J. Polymer carriers for drug delivery in tissue engineering. Adv. Drug Deliv. Rev. 2007, 59, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Almany, L.; Seliktar, D. Biosynthetic hydrogel scaffolds made from fibrinogen and polyethylene glycol for 3D cell cultures. Biomaterials 2005, 26, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Strehin, I.; Nahas, Z.; Arora, K.; Nguyen, T.; Elisseeff, J. A versatile pH sensitive chondroitin sulfate-PEG tissue adhesive and hydrogel. Biomaterials 2010, 31, 2788–2797. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Mooney, D.J. Hydrogels for tissue engineering. Chem. Rev. 2001, 101, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Marchant, R.E. Design properties of hydrogel tissue-engineering scaffolds. Expert Rev. Med. Devices 2011, 8, 607–626. [Google Scholar] [CrossRef] [PubMed]
- Kopeček, J. Hydrogel biomaterials: A smart future? Biomaterials 2007, 28, 5185–5192. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, B.V.; Khurshid, S.S.; Fisher, O.Z.; Khademhosseini, A.; Peppas, N.A. Hydrogels in regenerative medicine. Adv. Mater. 2009, 21, 3307–3329. [Google Scholar] [CrossRef] [PubMed]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Deligkaris, K.; Tadele, T.S.; Olthuis, W.; van den Berg, A. Hydrogel-based devices for biomedical applications. Sens. Actuators B Chem. 2010, 147, 765–774. [Google Scholar] [CrossRef]
- Hu, X.; Li, D.; Zhou, F.; Gao, C. Biological hydrogel synthesized from hyaluronic acid, gelatin and chondroitin sulfate by click chemistry. Acta Biomater. 2011, 7, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Bidarra, S.J.; Barrias, C.C.; Fonseca, K.B.; Barbosa, M.A.; Soares, R.A.; Granja, P.L. Injectable in situ crosslinkable RGD-modified alginate matrix for endothelial cells delivery. Biomaterials 2011, 32, 7897–7904. [Google Scholar] [CrossRef] [PubMed]
- Augst, A.D.; Kong, H.J.; Mooney, D.J. Alginate hydrogels as biomaterials. Macromol. Biosci. 2006, 6, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Bae, K.H.; Wang, L.-S.; Kurisawa, M. Injectable biodegradable hydrogels: Progress and challenges. J. Mater. Chem. B 2013, 1, 5371–5388. [Google Scholar] [CrossRef]
- Buwalda, S.J.; Boere, K.W.M.; Dijkstra, P.J.; Feijen, J.; Vermonden, T.; Hennink, W.E. Hydrogels in a historical perspective: From simple networks to smart materials. J. Control. Release 2014, 190, 254–273. [Google Scholar] [CrossRef] [PubMed]
- Samchenko, Y.; Ulberg, Z.; Korotych, O. Multipurpose smart hydrogel systems. Adv. Colloid Interface Sci. 2011, 168, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Hennink, W.E.; van Nostrum, C.F. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2012, 64, S223–S236. [Google Scholar] [CrossRef]
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. Bioactive modification of poly(ethylene glycol) hydrogels for tissue engineering. Biomaterials 2010, 31, 4639–4656. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012, 64, S18–S23. [Google Scholar] [CrossRef]
- Branco, M.C.; Pochan, D.J.; Wagner, N.J.; Schneider, J.P. The effect of protein structure on their controlled release from an injectable peptide hydrogel. Biomaterials 2010, 31, 9527–9534. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.Y.; Shinde, U.P.; Yeon, B.; Jeong, B. Recent progress of in situ formed gels for biomedical applications. Prog. Polym. Sci. 2013, 38, 672–701. [Google Scholar] [CrossRef]
- Drury, J.L.; Mooney, D.J. Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Appelman, T.P.; Mizrahi, J.; Elisseeff, J.H.; Seliktar, D. The influence of biological motifs and dynamic mechanical stimulation in hydrogel scaffold systems on the phenotype of chondrocytes. Biomaterials 2011, 32, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed]
- Boontheekul, T.; Kong, H.-J.; Mooney, D.J. Controlling alginate gel degradation utilizing partial oxidation and bimodal molecular weight distribution. Biomaterials 2005, 26, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.; Powell, C.; Ahmed, S.M.; Alsberg, E. Biodegradable, photocrosslinked alginate hydrogels with independently tailorable physical properties and cell adhesivity. Tissue Eng. A 2010, 16, 2915–2925. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, J.-H.; Jeon, O.; Kwon, I.C.; Park, K. Engineered polymers for advanced drug delivery. Eur. J. Pharm. Biopharm. 2009, 71, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Silva, E.A.; Mooney, D.J. Growth factor delivery-based tissue engineering: General approaches and a review of recent developments. J. R. Soc. Interface 2011, 8, 153–170. [Google Scholar] [PubMed]
- Qiu, Y.; Park, K. Environment-sensitive hydrogels for drug delivery. Adv. Drug Deliv. Rev. 2012, 64, S49–S60. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Kim, S.W. Drug release from biodegradable injectable thermosensitive hydrogel of PEG–PLGA–PEG triblock copolymers. J. Control. Release 2000, 63, 155–163. [Google Scholar] [CrossRef]
- Cha, M.-H.; Choi, J.; Choi, B.G.; Park, K.; Kim, I.H.; Jeong, B.; Han, D.K. Synthesis and characterization of novel thermo-responsive f68 block copolymers with cell-adhesive RGD peptide. J. Colloid Interface Sci. 2011, 360, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Joo, M.K.; Bahk, K.H.; Choi, Y.Y.; Kim, H.-T.; Kim, W.-K.; Jeong Lee, H.; Sohn, Y.S.; Jeong, B. Enzymatically degradable temperature-sensitive polypeptide as a new in situ gelling biomaterial. J. Control. Release 2009, 137, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.G.; Park, M.H.; Cho, S.-H.; Joo, M.K.; Oh, H.J.; Kim, E.H.; Park, K.; Han, D.K.; Jeong, B. In situ thermal gelling polypeptide for chondrocytes 3D culture. Biomaterials 2010, 31, 9266–9272. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Joo, M.K.; Oh, H.; Sohn, Y.S.; Jeong, B. Injectable gel: Poly(ethylene glycol)-sebacic acid polyester. Polymer 2006, 47, 3760–3766. [Google Scholar] [CrossRef]
- Jeong, B.; Kim, S.W.; Bae, Y.H. Thermosensitive sol-gel reversible hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 154–162. [Google Scholar] [CrossRef]
- Huang, J.; Heise, A. Stimuli responsive synthetic polypeptides derived from N-carboxyanhydride (NCA) polymerisation. Chem. Soc. Rev. 2013, 42, 7373–7390. [Google Scholar] [CrossRef] [PubMed]
- DuBose, J.W.; Cutshall, C.; Metters, A.T. Controlled release of tethered molecules via engineered hydrogel degradation: Model development and validation. J. Biomed. Mater. Res. Part A 2005, 74A, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Hubbell, J.A. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 2005, 23, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, K.B.; Granja, P.L.; Barrias, C.C. Engineering proteolytically-degradable artificial extracellular matrices. Prog. Polym. Sci. 2014, 39, 2010–2029. [Google Scholar] [CrossRef]
- Weber, L.M.; Hayda, K.N.; Haskins, K.; Anseth, K.S. The effects of cell-matrix interactions on encapsulated β-cell function within hydrogels functionalized with matrix-derived adhesive peptides. Biomaterials 2007, 28, 3004–3011. [Google Scholar] [CrossRef] [PubMed]
- DeLong, S.A.; Gobin, A.S.; West, J.L. Covalent immobilization of RGDs on hydrogel surfaces to direct cell alignment and migration. J. Control. Release 2005, 109, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Tsurkan, M.V.; Chwalek, K.; Schoder, M.; Freudenberg, U.; Werner, C. Chemoselective peptide functionalization of starPEG-GAG hydrogels. Bioconjugate Chem. 2014, 25, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Yeo, Y.; Geng, W.; Ito, T.; Kohane, D.S.; Burdick, J.A.; Radisic, M. Photocrosslinkable hydrogel for myocyte cell culture and injection. J. Biomed. Mater. Res. Part B Appl. Biomater. 2007, 81, 312–322. [Google Scholar] [CrossRef] [PubMed]
- DeLong, S.A.; Moon, J.J.; West, J.L. Covalently immobilized gradients of BFGF on hydrogel scaffolds for directed cell migration. Biomaterials 2005, 26, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.; Nicodemus, G.; Villanueva, I. Designing 3D photopolymer hydrogels to regulate biomechanical cues and tissue growth for cartilage tissue engineering. Pharm. Res. 2008, 25, 2379–2386. [Google Scholar] [CrossRef] [PubMed]
- Grover, G.N.; Lam, J.; Nguyen, T.H.; Segura, T.; Maynard, H.D. Biocompatible hydrogels by oxime click chemistry. Biomacromolecules 2012, 13, 3013–3017. [Google Scholar] [CrossRef] [PubMed]
- Maia, F.R.; Barbosa, M.; Gomes, D.B.; Vale, N.; Gomes, P.; Granja, P.L.; Barrias, C.C. Hydrogel depots for local co-delivery of osteoinductive peptides and mesenchymal stem cells. J. Control. Release 2014, 189, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Romano, N.H.; Sengupta, D.; Chung, C.; Heilshorn, S.C. Protein-engineered biomaterials: Nanoscale mimics of the extracellular matrix. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-A.; Yeom, J.; Hwang, B.W.; Hoffman, A.S.; Hahn, S.K. In situ-forming injectable hydrogels for regenerative medicine. Prog. Polym. Sci. 2014, 39, 1973–1986. [Google Scholar] [CrossRef]
- Nicodemus, G.D.; Bryant, S.J. Cell encapsulation in biodegradable hydrogels for tissue engineering applications. Tissue Eng. Part B Rev. 2008, 14, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.A.; Madlambayan, G.; Mooney, D.J. Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials 1999, 20, 45–53. [Google Scholar] [CrossRef]
- Bartczak, D.; Kanaras, A.G. Preparation of peptide-functionalized gold nanoparticles using one pot EDC/sulfo-NHS coupling. Langmuir 2011, 27, 10119–10123. [Google Scholar] [CrossRef] [PubMed]
- Sinz, A. Chemical cross-linking and mass spectrometry for mapping three-dimensional structures of proteins and protein complexes. J. Mass Spectrom. 2003, 38, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Connelly, J.T.; García, A.J.; Levenston, M.E. Inhibition of in vitro chondrogenesis in RGD-modified three-dimensional alginate gels. Biomaterials 2007, 28, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, U.; Hermann, A.; Welzel, P.B.; Stirl, K.; Schwarz, S.C.; Grimmer, M.; Zieris, A.; Panyanuwat, W.; Zschoche, S.; Meinhold, D.; et al. A star-PEG-heparin hydrogel platform to aid cell replacement therapies for neurodegenerative diseases. Biomaterials 2009, 30, 5049–5060. [Google Scholar] [CrossRef] [PubMed]
- Tsurkan, M.V.; Chwalek, K.; Levental, K.R.; Freudenberg, U.; Werner, C. Modular starPEG-heparin gels with bifunctional peptide linkers. Macromol. Rapid Commun. 2010, 31, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.Z.; Tian, W.M.; Hou, S.P.; Xu, Q.Y.; Lee, I.S. Hyaluronic acid hydrogel immobilized with RGD peptides for brain tissue engineering. J. Mater. Sci. Mater. Med. 2006, 17, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Iha, R.K.; Wooley, K.L.; Nyström, A.M.; Burke, D.J.; Kade, M.J.; Hawker, C.J. Applications of orthogonal “click” chemistries in the synthesis of functional soft materials. Chem. Rev. 2009, 109, 5620–5686. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Nwe, K.; Brechbiel, M.W. Growing applications of “click chemistry” for bioconjugation in contemporary biomedical research. Cancer Biother. Radiopharm. 2009, 24, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Zeglis, B.M.; Lewis, J.S.; Anderson, C.J. The growing impact of bioorthogonal click chemistry on the development of radiopharmaceuticals. J. Nucl. Med. 2013, 54, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Amblard, F.; Cho, J.H.; Schinazi, R.F. Cu(I)-catalyzed huisgen azide–alkyne 1, 3-dipolar cycloaddition reaction in nucleoside, nucleotide, and oligonucleotide chemistry. Chem. Rev. 2009, 109, 4207–4220. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, J.; Deng, C.; Suuronen, E.J.; Zhong, Z. Click hydrogels, microgels and nanogels: Emerging platforms for drug delivery and tissue engineering. Biomaterials 2014, 35, 4969–4985. [Google Scholar] [CrossRef] [PubMed]
- Truong, V.X.; Ablett, M.P.; Gilbert, H.T.J.; Bowen, J.; Richardson, S.M.; Hoyland, J.A.; Dove, A.P. In situ-forming robust chitosan-poly(ethylene glycol) hydrogels prepared by copper-free azide–alkyne click reaction for tissue engineering. Biomater. Sci. 2014, 2, 167–175. [Google Scholar] [CrossRef]
- Such, G.K.; Johnston, A.P.R.; Liang, K.; Caruso, F. Synthesis and functionalization of nanoengineered materials using click chemistry. Prog. Polym. Sci. 2012, 37, 985–1003. [Google Scholar] [CrossRef]
- Becer, C.R.; Hoogenboom, R.; Schubert, U.S. Click chemistry beyond metal-catalyzed cycloaddition. Angew. Chem. Int. Ed. 2009, 48, 4900–4908. [Google Scholar] [CrossRef] [PubMed]
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide–alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click chemistry reactions in medicinal chemistry: Applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2008, 28, 278–308. [Google Scholar] [CrossRef] [PubMed]
- Jagasia, R.; Holub, J.M.; Bollinger, M.; Kirshenbaum, K.; Finn, M.G. Peptide cyclization and cyclodimerization by CuI-mediated azide–alkyne cycloaddition. J. Org. Chem. 2009, 74, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Castro, V.; Rodriguez, H.; Albericio, F. Wang linker free of side reactions. Org. Lett. 2012, 15, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Zampano, G.; Bertoldo, M.; Ciardelli, F. Defined chitosan-based networks by C-6-azide–alkyne “click” reaction. React. Funct. Polym. 2010, 70, 272–281. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase:[1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, V.; Cornelio, L.; di Meo, C.; Nardecchia, S.; Lamanna, R. Novel hydrogels via click chemistry: Synthesis and potential biomedical applications. Biomacromolecules 2007, 8, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.R.; Martins, M.C.L.; Mafra, L.; Gomes, P. Synthesis of an O-alkynyl-chitosan and its chemoselective conjugation with a PEG-like amino-azide through click chemistry. Carbohydr. Polym. 2012, 87, 240–249. [Google Scholar] [CrossRef]
- He, X.; Ma, J.; Jabbari, E. Effect of grafting RGD and BMP-2 protein-derived peptides to a hydrogel substrate on osteogenic differentiation of marrow stromal cells. Langmuir 2008, 24, 12508–12516. [Google Scholar] [CrossRef] [PubMed]
- Lallana, E.; Riguera, R.; Fernandez-Megia, E. Reliable and efficient procedures for the conjugation of biomolecules through huisgen azide–alkyne cycloadditions. Angew. Chem. Int. Ed. 2011, 50, 8794–8804. [Google Scholar] [CrossRef] [PubMed]
- Sachin, K.; Jadhav, V.H.; Kim, E.-M.; Kim, H.L.; Lee, S.B.; Jeong, H.-J.; Lim, S.T.; Sohn, M.-H.; Kim, D.W. F-18 labeling protocol of peptides based on chemically orthogonal strain-promoted cycloaddition under physiologically friendly reaction conditions. Bioconjugate Chem. 2012, 23, 1680–1686. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.-F. Copper-free azide–alkyne cycloadditions: New insights and perspectives. Angew. Chem. Int. Ed. 2008, 47, 2182–2184. [Google Scholar] [CrossRef] [PubMed]
- DeForest, C.A.; Anseth, K.S. Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat. Chem. 2011, 3, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, C.E.; Bowman, C.N. Thiol-ene click chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar]
- Singh, S.P.; Schwartz, M.P.; Lee, J.Y.; Fairbanks, B.D.; Anseth, K.S. A peptide functionalized poly(ethylene glycol) (PEG) hydrogel for investigating the influence of biochemical and biophysical matrix properties on tumor cell migration. Biomater. Sci. 2014, 2, 1024–1034. [Google Scholar] [PubMed]
- Lowe, A.B. Thiol-ene “click” reactions and recent applications in polymer and materials synthesis. Polym. Chem. 2010, 1, 17–36. [Google Scholar] [CrossRef]
- DeForest, C.A.; Sims, E.A.; Anseth, K.S. Peptide-functionalized click hydrogels with independently tunable mechanics and chemical functionality for 3D cell culture. Chem. Mater. 2010, 22, 4783–4790. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, B.D.; Schwartz, M.P.; Halevi, A.E.; Nuttelman, C.R.; Bowman, C.N.; Anseth, K.S. A versatile synthetic extracellular matrix mimic via thiol-norbornene photopolymerization. Adv. Mater. (Deerfield Beach, FL) 2009, 21, 5005–5010. [Google Scholar] [CrossRef] [PubMed]
- DeForest, C.A.; Anseth, K.S. Photoreversible patterning of biomolecules within click-based hydrogels. Angew. Chem. Int. Ed. 2012, 51, 1816–1819. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Yu, J.; Tang, W.; Zheng, J.; Defante, A.; Guo, K.; Wesdemiotis, C.; Becker, M.L. Peptide-functionalized oxime hydrogels with tunable mechanical properties and gelation behavior. Biomacromolecules 2013, 14, 3749–3758. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.M.; Koshy, S.T.; Hilderbrand, S.A.; Mooney, D.J.; Joshi, N.S. Versatile click alginate hydrogels crosslinked via tetrazine–norbornene chemistry. Biomaterials 2015, 50, 30–37. [Google Scholar] [PubMed]
- El-Sagheer, A.H.; Brown, T. Click chemistry with DNA. Chem. Soc. Rev. 2010, 39, 1388–1405. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Tirelli, N.; Cerritelli, S.; Cavalli, L.; Hubbell, J.A. Systematic modulation of michael-type reactivity of thiols through the use of charged amino acids. Bioconjugate Chem. 2001, 12, 1051–1056. [Google Scholar] [CrossRef]
- Lutolf, M.P.; Hubbell, J.A. Synthesis and physicochemical characterization of end-linked poly(ethylene glycol)-co-peptide hydrogels formed by michael-type addition. Biomacromolecules 2003, 4, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Salinas, C.N.; Cole, B.B.; Kasko, A.M.; Anseth, K.S. Chondrogenic differentiation potential of human mesenchymal stem cells photoencapsulated within poly(ethylene glycol)-arginine-glycine-aspartic acid-serine thiol-methacrylate mixed-mode networks. Tissue Eng. 2007, 13, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.P.; Sprangers, A.J.; Byce, J.R.; Su, J.; Squirrell, J.M.; Messersmith, P.B.; Eliceiri, K.W.; Ogle, B.M. ECM-incorporated hydrogels cross-linked via native chemical ligation to engineer stem cell microenvironments. Biomacromolecules 2013, 14, 3102–3111. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.-H.; Su, J.; Messersmith, P.B. Hydrogels cross-linked by native chemical ligation. Biomacromolecules 2009, 10, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Nimmo, C.M.; Owen, S.C.; Shoichet, M.S. Diels–Alder click cross-linked hyaluronic acid hydrogels for tissue engineering. Biomacromolecules 2011, 12, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Herrera, M.; Gandini, A.; Goycoolea, F.; Jacobsen, N.; Lizardi-Mendoza, J.; Recillas-Mota, M.; Argüelles-Monal, W. Furan–chitosan hydrogels based on click chemistry. Iran. Polym. J. 2015, 24, 349–357. [Google Scholar] [CrossRef]
- Alge, D.L.; Azagarsamy, M.A.; Donohue, D.F.; Anseth, K.S. Synthetically tractable click hydrogels for three-dimensional cell culture formed using tetrazine–norbornene chemistry. Biomacromolecules 2013, 14, 949–953. [Google Scholar] [CrossRef] [PubMed]
- DeForest, C.A.; Polizzotti, B.D.; Anseth, K.S. Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nat. Mater. 2009, 8, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Polizzotti, B.D.; Fairbanks, B.D.; Anseth, K.S. Three-dimensional biochemical patterning of click-based composite hydrogels via thiolene photopolymerization. Biomacromolecules 2008, 9, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa, M.; Martins, M.C.L.; Gomes, P. Grafting Techniques towards Production of Peptide-Tethered Hydrogels, a Novel Class of Materials with Biomedical Interest. Gels 2015, 1, 194-218. https://doi.org/10.3390/gels1020194
Barbosa M, Martins MCL, Gomes P. Grafting Techniques towards Production of Peptide-Tethered Hydrogels, a Novel Class of Materials with Biomedical Interest. Gels. 2015; 1(2):194-218. https://doi.org/10.3390/gels1020194
Chicago/Turabian StyleBarbosa, Mariana, M. Cristina L. Martins, and Paula Gomes. 2015. "Grafting Techniques towards Production of Peptide-Tethered Hydrogels, a Novel Class of Materials with Biomedical Interest" Gels 1, no. 2: 194-218. https://doi.org/10.3390/gels1020194
APA StyleBarbosa, M., Martins, M. C. L., & Gomes, P. (2015). Grafting Techniques towards Production of Peptide-Tethered Hydrogels, a Novel Class of Materials with Biomedical Interest. Gels, 1(2), 194-218. https://doi.org/10.3390/gels1020194