
Does One Size Fit All? Variations in the DNA Barcode Gaps of Macrofungal Genera
 , , , and
, , , and
Abstract
1. Introduction
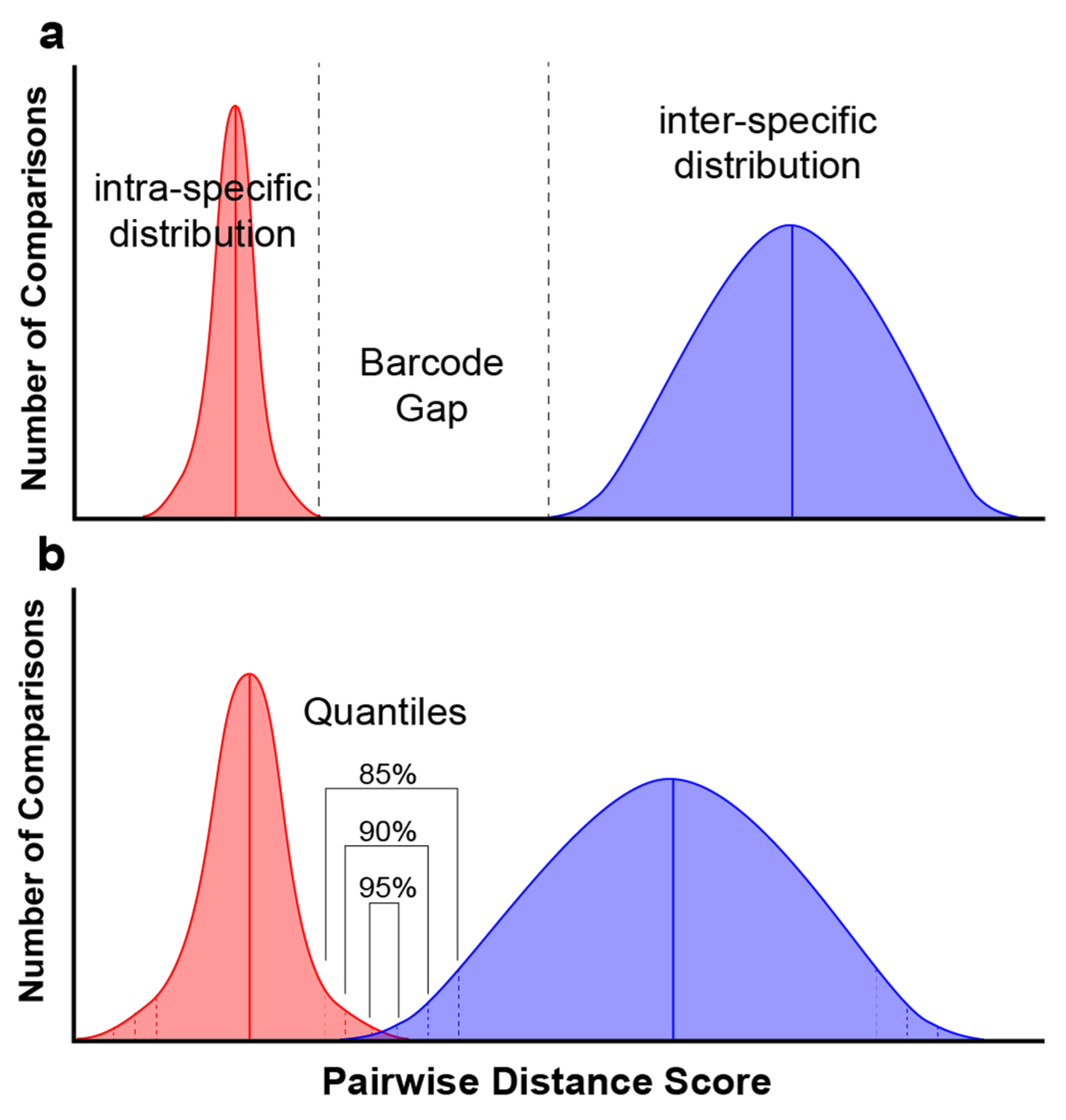
2. Materials and Methods
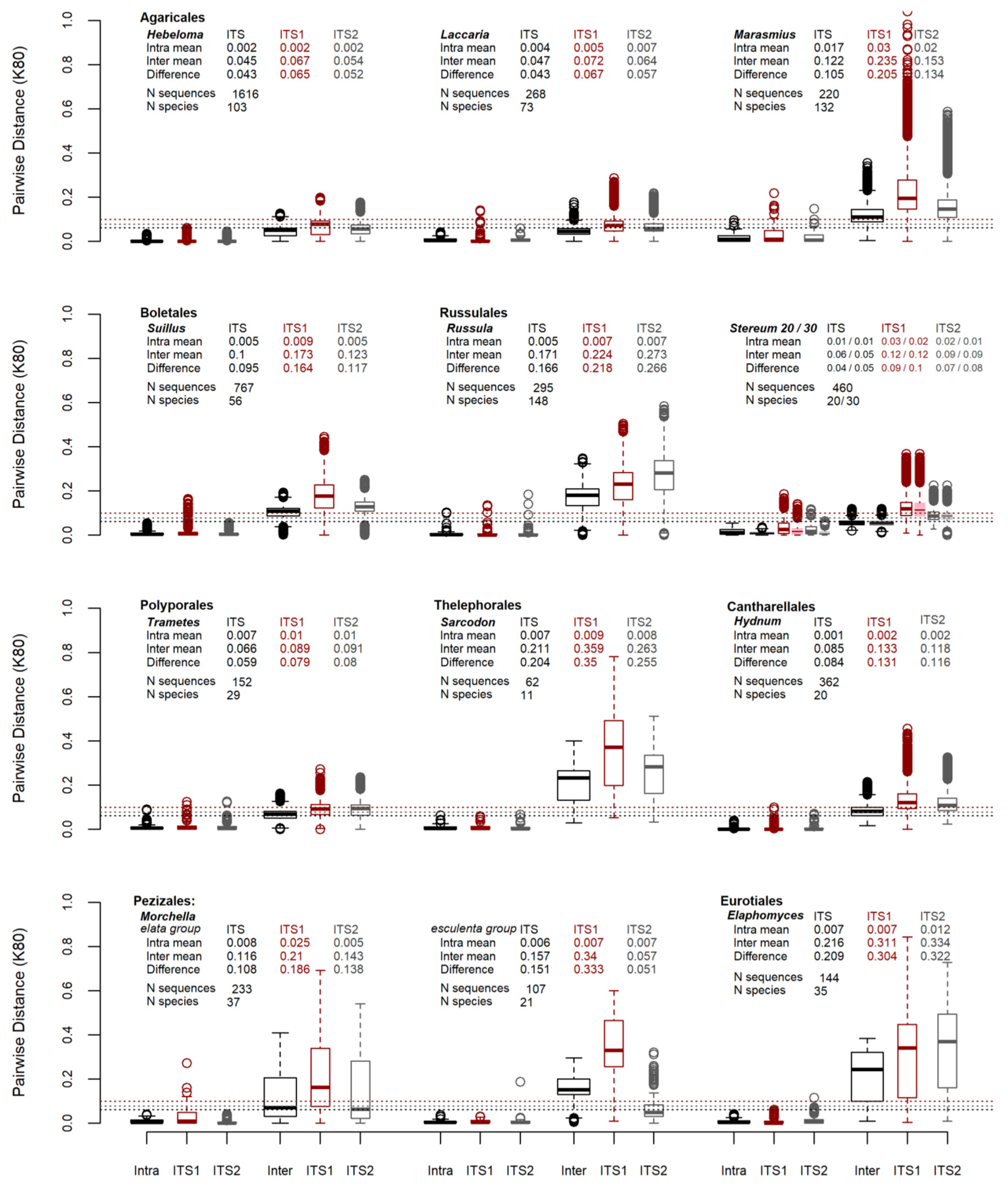
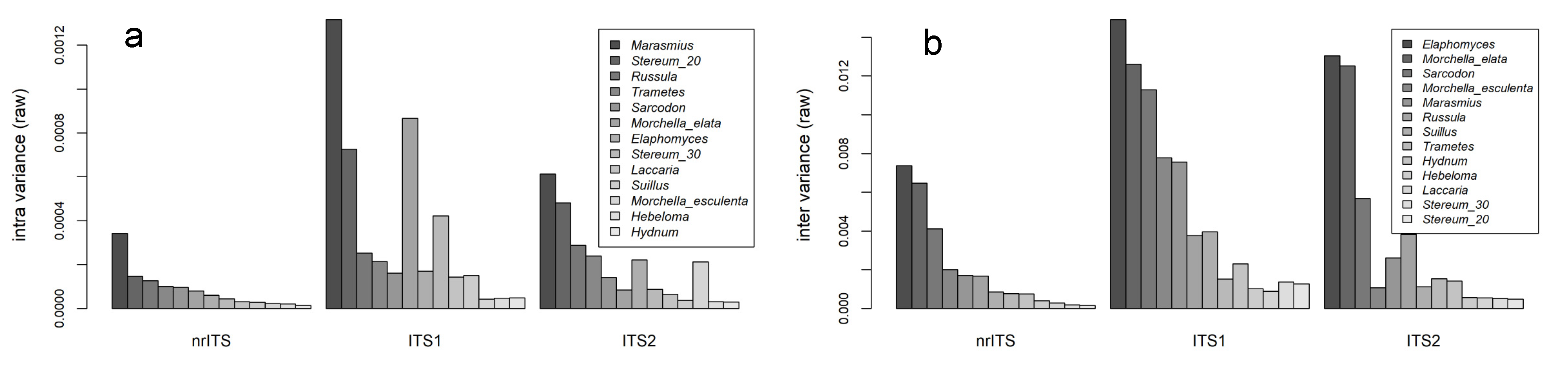
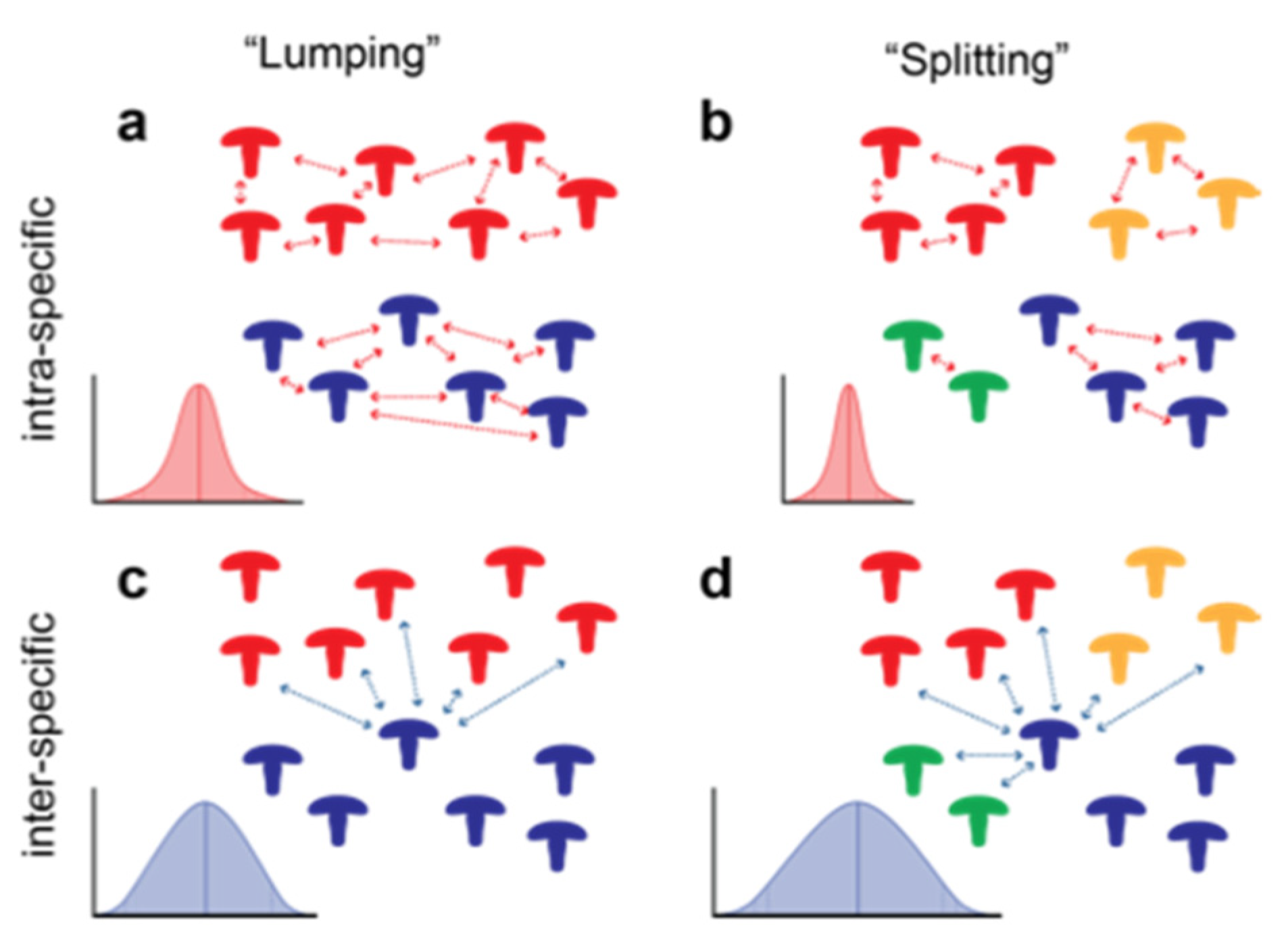
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Consortium, F.B. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Kristiansson, E.; Ryberg, M.; Hallenberg, N.; Larsson, K.-H. Intraspecific ITS variability in the Kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. 2008, 4, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.P.; Paulay, G. DNA barcoding: Error rates based on comprehensive sampling. PLoS Biol. 2005, 3, e422. [Google Scholar] [CrossRef]
- Moritz, C.; Cicero, C. DNA barcoding: Promise and pitfalls. PLoS Biol. 2004, 2, e354. [Google Scholar] [CrossRef]
- Phillips, J.D.; Gillis, D.J.; Hanner, R.H. Lack of Statistical Rigor in DNA Barcoding Likely Invalidates the Presence of a True Species’ Barcode Gap. Front. Ecol. Evol. 2022, 10, 859099. [Google Scholar] [CrossRef]
- Sa, W.; Qiao, J.; Gao, Q.; Li, Z.; Shang, Q. DNA Barcoding and Species Classification of Morchella. Genes 2022, 13, 1806. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.T.V.; Irinyi, L.; Chen, S.C.; Sorrell, T.C.; ISHAM Barcoding of Medical Fungi Working Group; Meyer, W. Dual DNA barcoding for the molecular identification of the agents of invasive fungal infections. Front. Microbiol. 2019, 10, 1647. [Google Scholar] [CrossRef]
- Badotti, F.; de Oliveira, F.S.; Garcia, C.F.; Vaz, A.B.M.; Fonseca, P.L.C.; Nahum, L.A.; Oliveira, G.; Góes-Neto, A. Effectiveness of ITS and sub-regions as DNA barcode markers for the identification of Basidiomycota (Fungi). BMC Microbiol. 2017, 17, 42. [Google Scholar] [CrossRef]
- Taylor, D.L.; Walters, W.A.; Lennon, N.J.; Bochicchio, J.; Krohn, A.; Caporaso, J.G.; Pennanen, T. Accurate estimation of fungal diversity and abundance through improved lineage-specific primers optimized for Illumina amplicon sequencing. Appl. Environ. Microbiol. 2016, 82, 7217–7226. [Google Scholar] [CrossRef]
- Blaalid, R.; Kumar, S.; Nilsson, R.H.; Abarenkov, K.; Kirk, P.; Kauserud, H. ITS 1 versus ITS 2 as DNA metabarcodes for fungi. Mol. Ecol. Resour. 2013, 13, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Mello, A.; Napoli, C.; Murat, C.; Morin, E.; Marceddu, G.; Bonfante, P. ITS-1 versus ITS-2 pyrosequencing: A comparison of fungal populations in truffle grounds. Mycologia 2011, 103, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Bazzicalupo, A.L.; Bálint, M.; Schmitt, I. Comparison of ITS1 and ITS2 rDNA in 454 sequencing of hyperdiverse fungal communities. Fungal Ecol. 2013, 6, 102–109. [Google Scholar] [CrossRef]
- Hofstetter, V.; Buyck, B.; Eyssartier, G.; Schnee, S.; Gindro, K. The unbearable lightness of sequenced-based identification. Fungal Divers. 2019, 96, 243–284. [Google Scholar] [CrossRef]
- Hilário, S.; Santos, L.; Phillips, A.J.; Alves, A. Caveats of the internal transcribed spacer region as a barcode to resolve species boundaries in Diaporthe. Fungal Biol. 2021, 126, 54–74. [Google Scholar] [CrossRef]
- Lücking, R.; Truong, B.V.; Huong, D.T.T.; Le, N.H.; Nguyen, Q.D.; Nguyen, V.D.; Von Raab-Straube, E.; Bollendorff, S.; Govers, K.; Di Vincenzo, V. Caveats of fungal barcoding: A case study in Trametes s. lat.(Basidiomycota: Polyporales) in Vietnam reveals multiple issues with mislabelled reference sequences and calls for third-party annotations. Willdenowia 2020, 50, 383–403. [Google Scholar] [CrossRef]
- Osmundson, T.W.; Robert, V.A.; Schoch, C.L.; Baker, L.J.; Smith, A.; Robich, G.; Mizzan, L.; Garbelotto, M.M. Filling gaps in biodiversity knowledge for macrofungi: Contributions and assessment of an herbarium collection DNA barcode sequencing project. PLoS ONE 2013, 8, e62419. [Google Scholar] [CrossRef]
- Runnel, K.; Abarenkov, K.; Copoț, O.; Mikryukov, V.; Kõljalg, U.; Saar, I.; Tedersoo, L. DNA barcoding of fungal specimens using PacBio long-read high-throughput sequencing. Mol. Ecol. Resour. 2022, 22, 2871–2879. [Google Scholar] [CrossRef]
- Olds, C.G.; Berta-Thompson, J.W.; Loucks, J.J.; Levy, R.A.; Wilson, A.W. Applying a modified metabarcoding approach for the sequencing of macrofungal specimens from fungarium collections. Appl. Plant Sci. 2023, 11, e11508. [Google Scholar] [CrossRef]
- McRae, B.H.; Beier, P. Circuit theory predicts gene flow in plant and animal populations. Proc. Natl. Acad. Sci. USA 2007, 104, 19885–19890. [Google Scholar] [CrossRef]
- Miller, A.N.; Karakehian, J.; Raudabaugh, D.B. Next-Generation Sequencing of Ancient and Recent Fungarium Specimens. J. Fungi 2022, 8, 932. [Google Scholar] [CrossRef]
- Forin, N.; Nigris, S.; Voyron, S.; Girlanda, M.; Vizzini, A.; Casadoro, G.; Baldan, B. Next generation sequencing of ancient fungal specimens: The case of the Saccardo mycological herbarium. Front. Ecol. Evol. 2018, 6, 129. [Google Scholar] [CrossRef]
- Gueidan, C.; Li, L. A long-read amplicon approach to scaling up the metabarcoding of lichen herbarium specimens. MycoKeys 2022, 86, 195. [Google Scholar] [CrossRef] [PubMed]
- Kistenich, S.; Halvorsen, R.; Schrøder-Nielsen, A.; Thorbek, L.; Timdal, E.; Bendiksby, M. DNA sequencing historical lichen specimens. Front. Ecol. Evol. 2019, 7, 5. [Google Scholar] [CrossRef]
- Leavitt, S.D.; Kueler, R.; Newberry, C.C.; Rosentreter, R.; St Clair, L.L. Shotgun sequencing decades-old lichen specimens to resolve phylogenomic placement of type material. Plant Fungal Syst. 2019, 64, 237–247. [Google Scholar] [CrossRef]
- Wang, X.C.; Liu, C.; Huang, L.; Bengtsson-Palme, J.; Chen, H.; Zhang, J.H.; Cai, D.; Li, J.Q. ITS 1: A DNA barcode better than ITS 2 in eukaryotes? Mol. Ecol. Resour. 2015, 15, 573–586. [Google Scholar] [CrossRef]
- Garnica, S.; Schön, M.E.; Abarenkov, K.; Riess, K.; Liimatainen, K.; Niskanen, T.; Dima, B.; Soop, K.; Frøslev, T.G.; Jeppesen, T.S. Determining threshold values for barcoding fungi: Lessons from Cortinarius (Basidiomycota), a highly diverse and widespread ectomycorrhizal genus. FEMS Microbiol. Ecol. 2016, 92, fiw045. [Google Scholar] [CrossRef]
- Ihrmark, K.; Bödeker, I.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandström-Durling, M.; Clemmensen, K.E. New primers to amplify the fungal ITS2 region–evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef]
- Perotto, S.; Nepote-Fus, P.; Saletta, L.; Bandi, C.; Young, J.P.W. A diverse population of introns in the nuclear ribosomal genes of ericoid mycorrhizal fungi includes elements with sequence similarity to endonuclease-coding genes. Mol. Biol. Evol. 2000, 17, 44–59. [Google Scholar] [CrossRef][Green Version]
- Vrålstad, T.; Myhre, E.; Schumacher, T. Molecular diversity and phylogenetic affinities of symbiotic root-associated ascomycetes of the Helotiales in burnt and metal polluted habitats. New Phytol. 2002, 155, 131–148. [Google Scholar] [CrossRef]
- Feibelman, T.P.; Bayman, P.; Cibula, W.G. Length variation in the internal transcribed spacer of ribosomal DNA in chanterelles. Mycol. Res. 1994, 98, 614–618. [Google Scholar] [CrossRef]
- Rosenblad, M.A.; Martín, M.P.; Tedersoo, L.; Ryberg, M.; Larsson, E.; Wurzbacher, C.; Abarenkov, K.; Nilsson, R.H. Detection of signal recognition particle (SRP) RNAs in the nuclear ribosomal internal transcribed spacer 1 (ITS1) of three lineages of ectomycorrhizal fungi (Agaricomycetes, Basidiomycota). MycoKeys 2016, 13, 21–33. [Google Scholar]
- Irga, P.J.; Barker, K.; Torpy, F.R. Conservation mycology in Australia and the potential role of citizen science. Conserv. Biol. 2018, 32, 1031–1037. [Google Scholar] [CrossRef]
- Irga, P.J.; Dominici, L.; Torpy, F.R. The mycological social network a way forward for conservation of fungal biodiversity. Environ. Conserv. 2020, 47, 243–250. [Google Scholar] [CrossRef]
- Sheehan, B.; Stevenson, R.; Schwartz, J. Crowdsourcing Fungal Biodiversity: Approaches and standards used by an all-volunteer community science project. Biodivers. Inf. Sci. Stand. 2021, 5, e74225. [Google Scholar] [CrossRef]
- Eberhardt, U.; Schütz, N.; Bartlett, P.; Beker, H.J. 96 North American taxa sorted–Peck’s Hebeloma revisited. Mycologia 2022, 114, 337–387. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, U.; Schütz, N.; Bartlett, P.; Hosaka, K.; Kasuya, T.; Beker, H.J. Revisiting Hebeloma (Hymenogastraceae, Agaricales) in Japan: Four species recombined into other genera but three new species discovered. Mycol. Prog. 2022, 21, 447–472. [Google Scholar] [CrossRef]
- Eberhardt, U.; Beker, H.J.; Vesterholt, J. Decrypting the Hebeloma crustuliniforme complex: European species of Hebeloma section Denudata subsection Denudata (Agaricales). Pers.-Mol. Phylogeny Evol. Fungi 2015, 35, 101–147. [Google Scholar] [CrossRef]
- Eberhardt, U.; Beker, H.J.; Vesterholt, J.; Dukik, K.; Walther, G.; Vila, J.; Fernández Brime, S. European species of Hebeloma section Theobromina. Fungal Divers. 2013, 58, 103–126. [Google Scholar] [CrossRef]
- Eberhardt, U.; Beker, H.J.; Borgen, T.; Knudsen, H.; Schütz, N.; Elborne, S.A. A survey of Hebeloma (Hymenogastraceae) in Greenland. MycoKeys 2021, 79, 17. [Google Scholar] [CrossRef]
- Beker, H.J.; Eberhardt, U.; Schütz, N.; Gulden, G. A review of the genus Hebeloma in Svalbard. Mycoscience 2018, 59, 303–309. [Google Scholar] [CrossRef]
- Eberhardt, U.; Beker, H.J.; Schütz, N.; Pedersen, O.S.; Sysouphanthong, P.; Læssøe, T. Adventurous cuisine in Laos: Hebeloma parvisporum, a new species in Hebeloma section Porphyrospora. Mycologia 2020, 112, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Grilli, E.; Beker, H.J.; Eberhardt, U.; Schütz, N.; Leonardi, M.; Vizzini, A. Unexpected species diversity and contrasting evolutionary hypotheses in Hebeloma (Agaricales) sections Sinapizantia and Velutipes in Europe. Mycol. Prog. 2016, 15, 1–46. [Google Scholar] [CrossRef]
- Eberhardt, U.; Beker, H.J.; Schuetz, N.; Mikami, M.; Kasuya, T. Rooting Hebelomas: The Japanese ‘Hebeloma radicosum’ is a distinct species, Hebeloma sagarae sp. nov.(Hymenogastraceae, Agaricales). Phytotaxa 2020, 456, 125–144. [Google Scholar] [CrossRef]
- Eberhardt, U.; Schütz, N.; Beker, H.J.; Lee, S.S.; Horak, E. Hebeloma in the Malay Peninsula: Masquerading within Psathyrella. MycoKeys 2021, 77, 117. [Google Scholar] [CrossRef]
- Eberhardt, U.; Beker, H.J.; Vesterholt, J.; Schütz, N. The taxonomy of the European species of Hebeloma section Denudata subsections Hiemalia, Echinospora subsect. nov. and Clepsydroida subsect. nov. and five new species. Fungal Biol. 2016, 120, 72–103. [Google Scholar] [CrossRef]
- Wilson, A.W.; Hosaka, K.; Mueller, G.M. Evolution of ectomycorrhizae as a driver of diversification and biogeographic patterns in the model mycorrhizal mushroom genus Laccaria. New Phytol. 2017, 213, 1862–1873. [Google Scholar] [CrossRef]
- Wilson, A.W.; May, T.W.; Mueller, G.M. Biogeography of the ectomycorrhizal mushroom genus Laccaria. In Biogeography of Mycorrhizal Symbiosis; Springer: Berlin/Heidelberg, Germany, 2017; pp. 273–297. [Google Scholar]
- Nguyen, N.; Vellinga, E.C.; Bruns, T.D.; Kennedy, P. Phylogenetic assessment of global Suillus ITS sequences supports morphologically defined species and reveals synonymous and undescribed taxa. Mycologia 2016, 108, 1216–1228. [Google Scholar]
- DeLong-Duhon, S.; Bagley, R.K.; Forbes, A.A. DNA, Morphology, and Ecology Resurrect Previously Synonymized Species of North American Stereum and Suggest Extensive Undescribed Global Diversity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Justo, A.; Hibbett, D.S. Phylogenetic classification of Trametes (Basidiomycota, Polyporales) based on a five–marker dataset. Taxon 2011, 60, 1567–1583. [Google Scholar] [CrossRef]
- Paz, A.; Bellanger, J.-M.; Lavoise, C.; Molia, A.; Ławrynowicz, M.; Larsson, E.; Ibarguren, I.; Jeppson, M.; Læssøe, T.; Sauve, M. The genus Elaphomyces (Ascomycota, Eurotiales): A ribosomal DNA-based phylogeny and revised systematics of European ‘deer truffles’. Pers.-Mol. Phylogeny Evol. Fungi 2017, 38, 197–239. [Google Scholar] [CrossRef] [PubMed]
- Molia, A.; Larsson, E.; Jeppson, M.; Læssøe, T.; Larsson, K. Elaphomyces section Elaphomyces (Eurotiales, Ascomycota)—Taxonomy and phylogeny of North European taxa, with the introduction of three new species. Fungal Syst. Evol. 2020, 5, 283–300. [Google Scholar] [CrossRef]
- Maddison, W.P.; Maddison, D.R. Mesquite: A Modular System for Evolutionary Analysis. Version 2.75; Scientific Research: Atlanta, GA, USA, 2011.
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Miller, M.A.; Holder, M.T.; Vos, R.; Midford, P.E.; Liebowitz, T.; Chan, L.; Hoover, P.; Warnow, T. The CIPRES Portals. 2009. Available online: https://scholar.google.com/citations?view_op=view_citation&hl=en&user=BYZtDXEAAAAJ&cstart=20&pagesize=80&citation_for_view=BYZtDXEAAAAJ:_kc_bZDykSQC (accessed on 19 July 2023).
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef]
- Aime, M.C.; Miller, A.N.; Aoki, T.; Bensch, K.; Cai, L.; Crous, P.W.; Hawksworth, D.L.; Hyde, K.D.; Kirk, P.M.; Lücking, R. How to publish a new fungal species, or name, version 3.0. IMA Fungus 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Ryberg, M. Molecular operational taxonomic units as approximations of species in the light of evolutionary models and empirical data from Fungi. Mol. Ecol. 2015, 24, 5770–5777. [Google Scholar] [CrossRef]
- Quaedvlieg, W.; Binder, M.; Groenewald, J.; Summerell, B.; Carnegie, A.; Burgess, T.; Crous, P.W. Introducing the consolidated species concept to resolve species in the Teratosphaeriaceae. Pers.-Mol. Phylogeny Evol. Fungi 2014, 33, 1–40. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Lücking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Abarenkov, K.; Nilsson, R.H.; Schüssler, A.; Grelet, G.-A.; Kohout, P.; Oja, J.; Bonito, G.M.; Veldre, V.; Jairus, T. Tidying up international nucleotide sequence databases: Ecological, geographical and sequence quality annotation of ITS sequences of mycorrhizal fungi. PLoS ONE 2011, 6, e24940. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Ryberg, M.; Kristiansson, E.; Abarenkov, K.; Larsson, K.-H.; Kõljalg, U. Taxonomic reliability of DNA sequences in public sequence databases: A fungal perspective. PLoS ONE 2006, 1, e59. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Bahram, M.; Puusepp, R.; Nilsson, R.H.; James, T.Y. Novel soil-inhabiting clades fill gaps in the fungal tree of life. Microbiome 2017, 5, 42. [Google Scholar] [CrossRef]
- Ryberg, M.; Nilsson, R.H. New light on names and naming of dark taxa. MycoKeys 2018, 30, 31. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Brock, P.M.; Döring, H.; Bidartondo, M.I. How to know unknown fungi: The role of a herbarium. New Phytol. 2009, 181, 719–724. [Google Scholar] [CrossRef]
- Raja, H.A.; Oberlies, N.H.; Stadler, M. Occasional comment: Fungal identification to species-level can be challenging. Phytochemistry 2021, 190, 112855. [Google Scholar] [CrossRef]
- Parra, L.A.; Zamora, J.C.; Hawksworth, D.L.; Hibbett, D.S.; Kirk, P.M.; Lücking, R.; Rambold, G.; Bensch, K.; Yao, Y.-J.; Robert, V. Proposals for consideration at IMC11 to modify provisions related solely to fungi in the International Code of Nomenclature for algae, fungi, and plants. IMA Fungus 2018, 9, i–vii. [Google Scholar] [CrossRef]
- Lücking, R.; Kirk, P.M.; Hawksworth, D.L. Sequence-based nomenclature: A reply to Thines et al. and Zamora et al. and provisions for an amended proposal “from the floor” to allow DNA sequences as types of names. IMA Fungus 2018, 9, 185–198. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Ryberg, M.; Wurzbacher, C.; Tedersoo, L.; Anslan, S.; Põlme, S.; Spirin, V.; Mikryukov, V.; Svantesson, S.; Hartmann, M. How, not if, is the question mycologists should be asking about DNA-based typification. MycoKeys 2023, 96, 143–157. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 85% Quantiles | 90% Quantiles | 95% Quantiles | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nrITS | ITS1 | ITS2 | nrITS | ITS1 | ITS2 | nrITS | ITS1 | ITS2 | ||||||||||||
| Name | Ntaxa | Nseq | Gap? | Size | Gap? | Size | Gap? | Size | Gap? | Size | Gap? | Size | Gap? | Size | Gap? | Size | Gap? | Size | Gap? | Size |
| Hebeloma | 103 | 1616 | TRUE | 0.0051 | TRUE | 0.0043 | TRUE | 0.0052 | TRUE | 0.0016 | FALSE | TRUE | 0.0000 | FALSE | FALSE | FALSE | ||||
| Laccaria | 73 | 268 | TRUE | 0.0121 | TRUE | 0.0094 | TRUE | 0.0105 | TRUE | 0.0063 | FALSE | TRUE | 0.0100 | TRUE | 0.0018 | FALSE | TRUE | 0.0000 | ||
| Marasmius | 132 | 220 | TRUE | 0.0152 | TRUE | 0.0128 | TRUE | 0.0046 | TRUE | 0.0075 | FALSE | FALSE | FALSE | FALSE | FALSE | |||||
| Suillus | 56 | 767 | TRUE | 0.0230 | TRUE | 0.0153 | TRUE | 0.0331 | TRUE | 0.0142 | FALSE | TRUE | 0.0235 | TRUE | 0.0013 | FALSE | TRUE | 0.0039 | ||
| Russula | 148 | 295 | TRUE | 0.0771 | TRUE | 0.0720 | TRUE | 0.1192 | TRUE | 0.0648 | TRUE | 0.0531 | TRUE | 0.0900 | TRUE | 0.0380 | TRUE | 0.0274 | TRUE | 0.0515 |
| Stereum | ||||||||||||||||||||
| “lumping” | 20 | 460 | TRUE | 0.0036 | FALSE | FALSE | FALSE | FALSE | FALSE | FALSE | FALSE | |||||||||
| “splitting” | 30 | 460 | TRUE | 0.0156 | FALSE | TRUE | 0.0322 | TRUE | 0.0114 | FALSE | TRUE | 0.0230 | TRUE | 0.0058 | FALSE | TRUE | 0.0125 | |||
| Trametes | 29 | 152 | FALSE | FALSE | TRUE | 1E-04 | FALSE | FALSE | FALSE | FALSE | FALSE | FALSE | ||||||||
| Sarcodon | 11 | 62 | TRUE | 0.0413 | TRUE | 0.0472 | TRUE | 0.0527 | TRUE | 0.0152 | TRUE | 0.0423 | TRUE | 0.0134 | FALSE | TRUE | 0.0277 | TRUE | 0.0000 | |
| Hydnum | 20 | 362 | TRUE | 0.0425 | TRUE | 0.0552 | TRUE | 0.0503 | TRUE | 0.0349 | TRUE | 0.0418 | TRUE | 0.0450 | TRUE | 0.0267 | TRUE | 0.0258 | TRUE | 0.0313 |
| Morchella | ||||||||||||||||||||
| elata group | 37 | 233 | FALSE | FALSE | FALSE | FALSE | FALSE | FALSE | FALSE | FALSE | FALSE | |||||||||
| esculenta group | 21 | 107 | TRUE | 0.0306 | TRUE | 0.0607 | TRUE | 0.0043 | TRUE | 0.0196 | TRUE | 0.0407 | FALSE | TRUE | 0.0029 | TRUE | 0.0082 | FALSE | ||
| Elaphomyces | 35 | 144 | TRUE | 0.0397 | TRUE | 0.0380 | TRUE | 0.0393 | TRUE | 0.0103 | FALSE | TRUE | 0.0199 | FALSE | FALSE | FALSE | ||||
| Count | AVE | Count | AVE | Count | AVE | Count | AVE | Count | AVE | Count | AVE | Count | AVE | Count | AVE | Count | AVE | |||
| 11 | 0.0278 | 9 | 0.0350 | 11 | 0.0320 | 10 | 0.0186 | 4 | 0.0445 | 8 | 0.0281 | 6 | 0.0127 | 4 | 0.0223 | 6 | 0.0166 | |||
| ITS | ITS1 | ITS2 | ||||
|---|---|---|---|---|---|---|
| Names | Gap? | Mean | Gap? | Mean | gap? | Mean |
| Hebeloma | FALSE | 0.7% | FALSE | 1.1% | FALSE | 1.2% |
| Laccaria | TRUE | 1.8% | FALSE | 2.5% | FALSE | 2.5% |
| Marasmius | FALSE | 5.0% | FALSE | 10.1% | FALSE | 5.9% |
| Suillus | TRUE | 1.7% | FALSE | 2.9% | TRUE | 2.1% |
| Russula | TRUE | 4.0% | TRUE | 4.5% | TRUE | 5.7% |
| Stereum | ||||||
| “lumping” | FALSE | 3.5% | FALSE | 6.9% | FALSE | 5.4% |
| “splitting” | TRUE | 2.5% | FALSE | 5.8% | TRUE | 3.3% |
| Trametes | FALSE | 1.8% | FALSE | 2.4% | FALSE | 2.9% |
| Sarcodon | FALSE | 3.7% | TRUE | 5.5% | TRUE | 3.7% |
| Hydnum | TRUE | 2.0% | TRUE | 2.8% | TRUE | 2.9% |
| Morchella | ||||||
| elata group | FALSE | 1.8% | FALSE | 5.0% | FALSE | 1.8% |
| esculenta group | TRUE | 1.6% | TRUE | 2.4% | FALSE | 1.1% |
| Elaphomyces | FALSE | 2.4% | FALSE | 4.0% | FALSE | 4.2% |
| Count | 6 | 4 | 5 | |||
| Max | 4.0% | 5.5% | 5.7% | |||
| Min | 1.6% | 2.4% | 2.1% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, A.W.; Eberhardt, U.; Nguyen, N.; Noffsinger, C.R.; Swenie, R.A.; Loucks, J.L.; Perry, B.A.; Herrera, M.; Osmundson, T.W.; DeLong-Duhon, S.; et al. Does One Size Fit All? Variations in the DNA Barcode Gaps of Macrofungal Genera. J. Fungi 2023, 9, 788. https://doi.org/10.3390/jof9080788
Wilson AW, Eberhardt U, Nguyen N, Noffsinger CR, Swenie RA, Loucks JL, Perry BA, Herrera M, Osmundson TW, DeLong-Duhon S, et al. Does One Size Fit All? Variations in the DNA Barcode Gaps of Macrofungal Genera. Journal of Fungi. 2023; 9(8):788. https://doi.org/10.3390/jof9080788
Chicago/Turabian StyleWilson, Andrew W., Ursula Eberhardt, Nhu Nguyen, Chance R. Noffsinger, Rachel A. Swenie, Justin L. Loucks, Brian A. Perry, Mariana Herrera, Todd W. Osmundson, Sarah DeLong-Duhon, and et al. 2023. "Does One Size Fit All? Variations in the DNA Barcode Gaps of Macrofungal Genera" Journal of Fungi 9, no. 8: 788. https://doi.org/10.3390/jof9080788
APA StyleWilson, A. W., Eberhardt, U., Nguyen, N., Noffsinger, C. R., Swenie, R. A., Loucks, J. L., Perry, B. A., Herrera, M., Osmundson, T. W., DeLong-Duhon, S., Beker, H. J., & Mueller, G. M. (2023). Does One Size Fit All? Variations in the DNA Barcode Gaps of Macrofungal Genera. Journal of Fungi, 9(8), 788. https://doi.org/10.3390/jof9080788