

Genomic Comparison of Two Species of Samsoniella with Other Genera in the Family Cordycipitaceae

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Test Materials
2.2. Strain Culture
2.3. Genome Sequencing and Assembly
2.4. Gene Prediction and Annotation
2.4.1. Repeat Sequence Analysis
2.4.2. Prediction of Non-Coding RNA
2.4.3. Prediction of Protein-Coding Genes
2.4.4. Carbohydrate Active Enzyme (CAZy) Analysis
2.4.5. Evolutionary Genealogy of Genes: Non-Supervised Orthologous Groups Database (eggNOG) Analysis
2.4.6. Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis
2.4.7. Swiss-Prot Annotation of Protein-Coding Genes
2.4.8. Gene Ontology (GO) Analysis
2.4.9. Cytochrome P450 Analysis
2.5. Analysis of Secondary Metabolite Biosynthesis Gene Cluster
2.6. Synteny Analysis
2.7. Cluster Analysis
3. Results
3.1. Basic Genomic Characteristics of S. hepiali ICMM 82-2 and S. yunnanensis YFCC 1527
3.1.1. Genome Sequencing and Assembly
3.1.2. Genome Annotation
3.1.3. Additional Annotation
Pathogen Host Interactions (PHIs)
Carbohydrate Genes
3.2. Basic Characteristics of 14 Genomes of Cordycipitaceae Species
3.3. Analysis of Secondary Metabolite Biosynthesis Gene Clusters in 13 Cordycipitaceae Species
3.3.1. Overview of 14 Genomic BGCs of Cordycipitaceae Species
3.3.2. Difference Analysis of 14 Genomic Domains in Cordycipitaceae Species
3.3.3. Analysis of SM BGCs of 13 Species of Cordycipitaceae
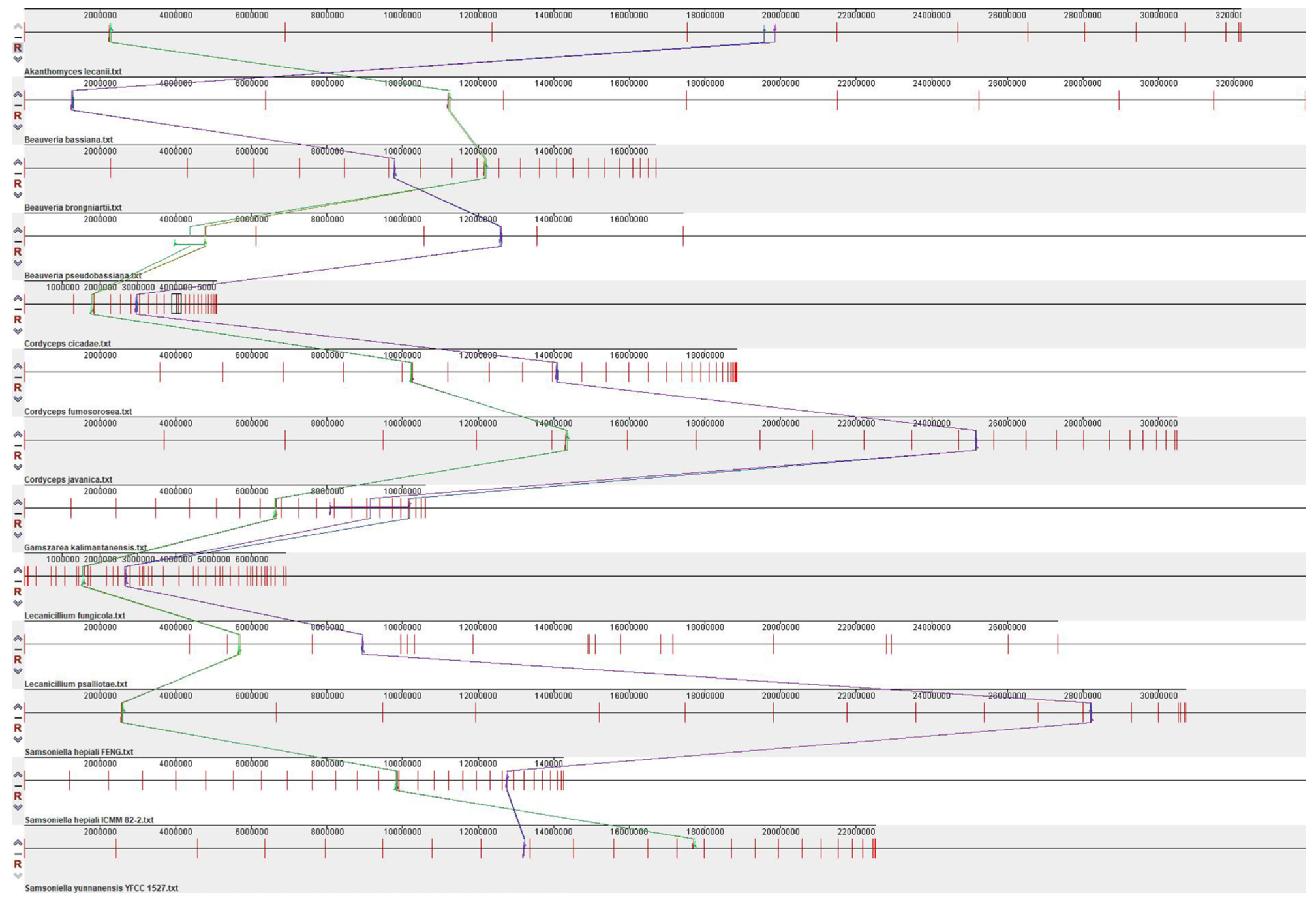
3.4. Synteny Analysis of 13 Species of Cordycipitaceae
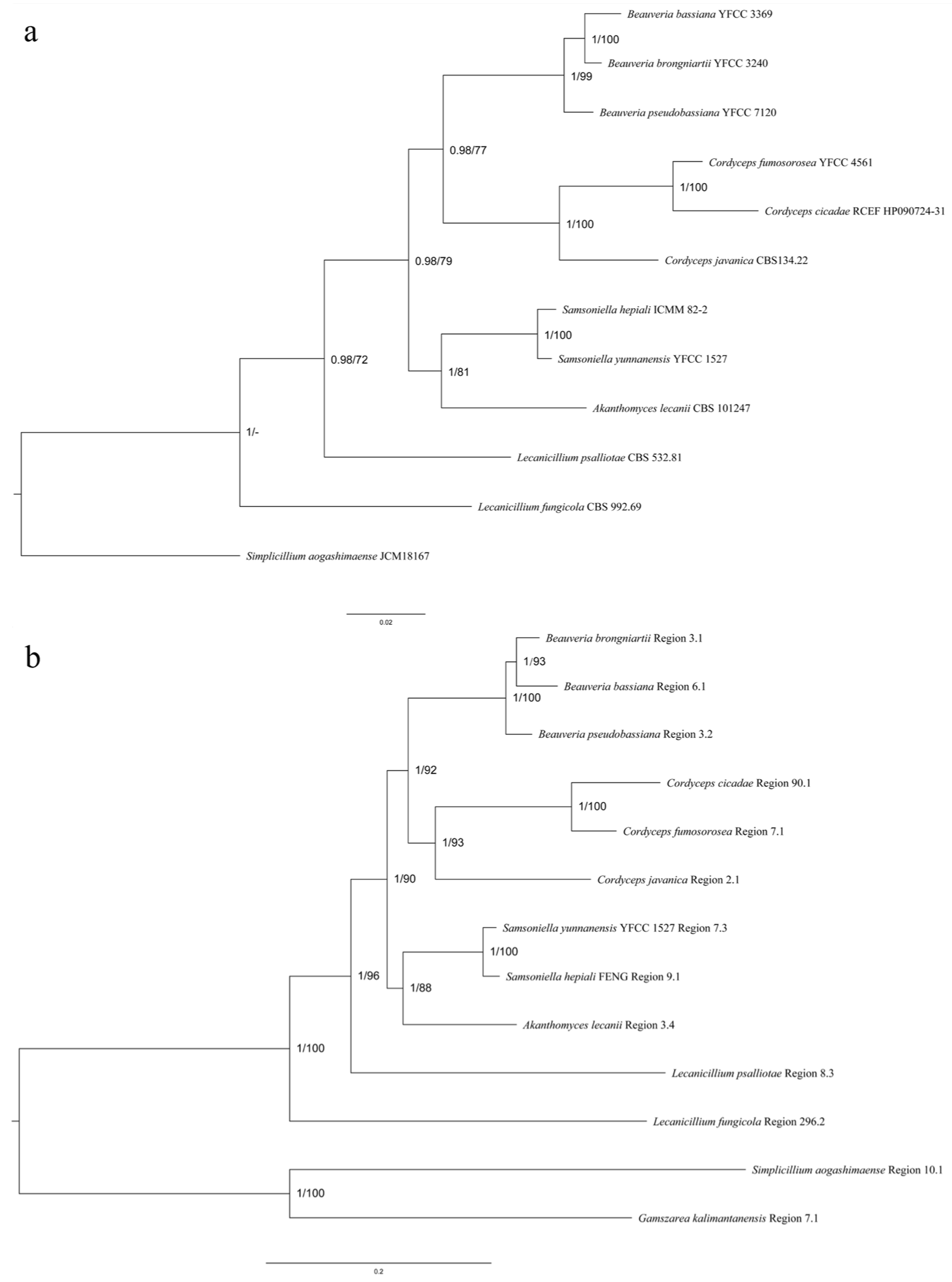
3.5. Cluster Analysis
3.5.1. SM BGCs Cluster Analysis
3.5.2. Comparative Analysis of HR-PKS Homologous Region Cluster Tree and Multi-Gene Genetic Distance Tree
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.-M.; Zhi, J.-R.; Qu, J.-J.; Zou, X. Estimated divergence times of Lecanicillium in the family Cordycipitaceae provide insights into the attribution of Lecanicillium. Front. Microbiol. 2022, 13, 859886. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.B.; Wang, Y.; Fan, Q.; Duan, D.E.; Zhang, G.D.; Dai, R.Q.; Dai, Y.D.; Zeng, W.B.; Chen, Z.H.; Li, D.D.; et al. Multigene phylogeny of the family Cordycipitaceae (hypocreales): New taxa and the new systematic position of the Chinese cordycipitoid fungus Paecilomyces hepialid. Fungal Divers. 2020, 103, 1–46. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Dong, Q.; Fan, Q.; Dao, V.-M.; Yu, H. Morphological and phylogenetic characterization reveals five new species of Samsoniella (Cordycipitaceae, Hypocreales). J. Fungi 2022, 8, 747. [Google Scholar] [CrossRef]
- Qasim, M.; Islam, S.U.; Islam, W.; Noman, A.; Khan, K.A.; Hafeez, M.; Hussain, D.; Dash, C.K.; Bamisile, B.S.; Akutse, K.S.; et al. Characterization of mycotoxins from entomopathogenic fungi (Cordyceps fumosorosea) and their toxic effects to the development of asian citrus psyllid reared on healthy and diseased citrus plants. Toxicon 2020, 188, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Weng, Q.; Zhang, X.; Chen, W.; Hu, Q. Secondary metabolites and the risks of Isaria fumosorosea and Isaria farinose. Molecules 2019, 24, 664. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Zhang, Z.; Liu, X.; Wang, F. The transcriptome analysis on urea response mechanism in the process of ergosterol synthesis by Cordyceps cicadae. Sci. Rep. 2021, 11, 10927. [Google Scholar] [CrossRef]
- Asai, T.; Yamamoto, T.; Oshima, Y. Aromatic polyketide production in Cordyceps indigotica, an entomopathogenic fungus, induced by exposure to a histone deacetylase inhibitor. Org. Lett. 2012, 14, 2006–2009. [Google Scholar] [CrossRef]
- Sun, J.; Xu, J.; Wang, S.; Hou, Z.; Lu, X.; An, L.; Du, P. A new cerebroside from Cordyceps militaris with anti-PTP1B activity. Fitoterapia 2019, 138, 104342. [Google Scholar] [CrossRef]
- Rukachaisirikul, V.; Pramjit, S.; Pakawatchai, C.; Isaka, M.; Supothina, S. 10-membered macrolides from the insect pathogenic fungus Cordyceps militaris BCC 2816. J. Nat. Prod. 2004, 67, 1953–1955. [Google Scholar] [CrossRef]
- Zhao, C.; Bu, H.; Zhu, J.; Wang, Y.; Oliver, K.M.; Hu, F.; Huang, B.; Li, Z.; Peng, F. Integration of untargeted metabolomics with transcriptomics provides insights into beauvericin biosynthesis in Cordyceps chanhua under H2O2-induced oxidative stress. J. Fungi 2022, 8, 484. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Y.L.; Zhang, M.L.; Zhang, H.D.; Huang, J.Z.; Li, L. Genome mining and biosynthesis of the Acyl-CoA: Cholesterol acyltransferase inhibitor beauveriolide I and III in Cordyceps militaris. J. Biotechnol. 2020, 309, 85–91. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, Q.; Weng, Q. Secondary metabolites (SMs) of Isaria cicadae and Isaria tenuipes. RSC Adv. 2018, 9, 172–184. [Google Scholar] [CrossRef]
- He, Y.; Zhang, W.; Peng, F.; Lu, R.; Zhou, H.; Bao, G.; Wang, B.; Huang, B.; Li, Z.; Hu, F. Metabolomic variation in wild and cultured Cordyceps and mycelia of Isaria cicadae. Biomed. Chromatogr. 2019, 33, e4478. [Google Scholar] [CrossRef] [PubMed]
- Kneifel, H.; König, W.A.; Loeffler, W.; Müller, R. Ophiocordin, an antifungal antibiotic of Cordyceps ophioglossoides. Arch. Microbiol. 1977, 113, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Tianzhu, Z.; Shihai, Y.; Juan, D. Antidepressant-like effects of cordycepin in a mice model of chronic unpredictable mild stress. Evid. Based Complement. Altern. Med. 2014, 2014, 438506. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Shang, Y.; Cen, K.; Wang, C. Fungal biosynthesis of the bibenzoquinone oosporein to evade insect immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 11365–11370. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Song, W.; Gao, J.; Yan, S.; Guo, C.; Zhang, T. Enhanced production of cordycepic acid from Cordyceps cicadae isolated from a wild environment. Braz. J. Microbiol. 2022, 53, 673–688. [Google Scholar] [CrossRef]
- Arulprakasam, K.R.; Dharumadurai, D. Genome mining of biosynthetic gene clusters intended for secondary metabolites conservation in actinobacteria. Microb. Pathog. 2021, 161, 105252. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef]
- Musiol-Kroll, E.M.; Wohlleben, W. Maßgeschneiderte polyketidsynthasen zur herstellung von polyketid-derivaten. Biospektrum 2020, 26, 437–439. [Google Scholar] [CrossRef]
- Sigrist, R.; Luhavaya, H.; McKinnie, S.M.K.; da Silva, A.F.; Jurberg, I.D.; Moore, B.S.; de Oliveira, L.G. Nonlinear biosynthetic assembly of alpiniamide by a hybrid cis/trans-AT PKS-NRPS. ACS Chem. Biol. 2020, 15, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.E.; Ishikawa, F.; Re, R.N.; Suzuki, T.; Dohmae, N.; Kakeya, H.; Tanabe, G.; Burkart, M.D. Developing crosslinkers specific for epimerization domain in NRPS initiation modules to evaluate mechanism. RSC Chem. Biol. 2022, 3, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Hur, G.H.; Vickery, C.R.; Burkart, M.D. Explorations of catalytic domains in non-ribosomal peptide synthetase enzymology. Nat. Prod. Rep. 2012, 29, 1074–1098. [Google Scholar] [CrossRef]
- Yuan, X.L.; Hua, M.; Chen, J.; Wang, J.; Wang, Y. Identification and cloning of a polyketide synthase III gene in the Lichenized-fungi Nephrmopsis pallescens (Chinese). Acta Bot. Boreal. Occodent. Sin. 2017, 37, 2146–2152. [Google Scholar]
- Sayari, M.; Steenkamp, E.T.; van der Nest, M.A.; Wingfield, B.D. Diversity and evolution of polyketide biosynthesis gene clusters in the Ceratocystidaceae. Fungal Biol. 2018, 122, 856–866. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, J.; Chen, C.; Teng, J.; Wang, C.; Luo, D. Structure and biosynthesis of fumosorinone, a new protein tyrosine phosphatase 1B inhibitor firstly isolated from the entomogenous fungus Isaria fumosorosea. Fungal Genet. Biol. 2015, 81, 191–200. [Google Scholar] [CrossRef]
- Xu, Y.; Orozco, R.; Wijeratne, E.K.; Espinosa-Artiles, P.; Gunatilaka, A.L.; Stock, S.P.; Molnár, I. Biosynthesis of the cyclooligomer depsipeptide bassianolide, an insecticidal virulence factor of Beauveria bassiana. Fungal Genet. Biol. 2009, 46, 353–364. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Y.; Yuan, X.; Huang, O.; Dong, Q.; Li, D.; Ding, S.; Ma, F.; Yu, H. Genomic Comparative Analysis of Cordyceps pseudotenuipes with Other Species from Cordyceps. Metabolites 2022, 12, 844. [Google Scholar] [CrossRef]
- Tempel, S. Using and understanding RepeatMasker. Methods Mol. Biol. 2012, 859, 29–51. [Google Scholar]
- Kapitonov, V.V.; Jurka, J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat. Rev. Genet. 2008, 9, 411–412. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S. Annotating non-coding RNAs with Rfam. Curr. Protoc. Bioinform. 2005, 9, 12.5.1–12.5.12. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef] [PubMed]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using Evidence Modeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; Sahu, J.; Iyer, S.V.; Khamari, L.; De Silva, N.; et al. PHI-base in 2022: A multi-species phenotype database for Pathogen-Host Interactions. Nucleic Acids Res. 2022, 50, D837–D847. [Google Scholar] [CrossRef]
- Garron, M.L.; Henrissat, B. The continuing expansion of CAZymes and their families. Curr. Opin. Chem. Biol. 2019, 53, 82–87. [Google Scholar] [CrossRef]
- Drula, E.; Garron, M.L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.M.; Szewczyk, E.; Davidson, A.D.; Keller, N.; Oakley, B.R.; Wang, C.C. A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, asperfuranone, in Aspergillus nidulans. J. Am. Chem. Soc. 2009, 131, 2965–2970. [Google Scholar] [CrossRef] [PubMed]
- von Bargen, K.W.; Niehaus, E.-M.; Krug, I.; Bergander, K.; Würthwein, E.-U.; Tudzynski, B.; Humpf, H.-U. Isolation and structure elucidation of fujikurins A-D: Products of the PKS19 gene cluster in Fusarium fujikuroi. J. Nat. Prod. 2015, 78, 1809–1815. [Google Scholar] [CrossRef]
- Latkowska, E.; Bober, B.; Chrapusta, E.; Adamski, M.; Kaminski, A.; Bialczyk, J. Secondary metabolites of the lichen Hypogymnia physodes (L.) Nyl. and their presence in spruce (Picea abies (L.) H. Karst.) bark. Phytochemistry 2015, 118, 116–123. [Google Scholar] [CrossRef]
- Caloni, F.; Fossati, P.; Anadón, A.; Bertero, A. Beauvericin: The beauty and the beast. Environ. Toxicol. Pharmacol. 2020, 75, 103349. [Google Scholar] [CrossRef]
- Wu, Q.; Patocka, J.; Kuca, K. Beauvericin, a Fusarium mycotoxin: Anticancer activity, mechanisms, and human exposure risk assessment. Mini Rev. Med. Chem. 2019, 19, 206–214. [Google Scholar] [CrossRef]
- Xu, Y.; Orozco, R.; Wijeratne, E.M.; Gunatilaka, A.A.; Stock, S.P.; Molnár, I. Biosynthesis of the cyclooligomer depsipeptide beauvericin, a virulence factor of the entomopathogenic fungus Beauveria bassiana. Chem. Biol. 2008, 15, 898–907. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Wang, Z.; Wang, Y.; Chen, Y.; Tang, D.; Yu, H. Genomic Comparison of Two Species of Samsoniella with Other Genera in the Family Cordycipitaceae. J. Fungi 2023, 9, 1146. https://doi.org/10.3390/jof9121146
Lu Y, Wang Z, Wang Y, Chen Y, Tang D, Yu H. Genomic Comparison of Two Species of Samsoniella with Other Genera in the Family Cordycipitaceae. Journal of Fungi. 2023; 9(12):1146. https://doi.org/10.3390/jof9121146
Chicago/Turabian StyleLu, Yingling, Zhiqin Wang, Yi Wang, Yue Chen, Dexiang Tang, and Hong Yu. 2023. "Genomic Comparison of Two Species of Samsoniella with Other Genera in the Family Cordycipitaceae" Journal of Fungi 9, no. 12: 1146. https://doi.org/10.3390/jof9121146
APA StyleLu, Y., Wang, Z., Wang, Y., Chen, Y., Tang, D., & Yu, H. (2023). Genomic Comparison of Two Species of Samsoniella with Other Genera in the Family Cordycipitaceae. Journal of Fungi, 9(12), 1146. https://doi.org/10.3390/jof9121146