
T-2 Toxin Induces Apoptotic Cell Death and Protective Autophagy in Mouse Microglia BV2 Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Measurement of Cell Viability
2.4. Measurement of Apoptosis
2.5. Measurement of ROS Production and Biomarkers of Oxidative Stress
2.6. Measurement of Caspase-3 Activities
2.7. Mitochondrial Membrane Potential Measurement
2.8. mRFP-GFP-LC3 Plasmid Transfection
2.9. Western Blotting
2.10. Statistical Analysis
3. Results
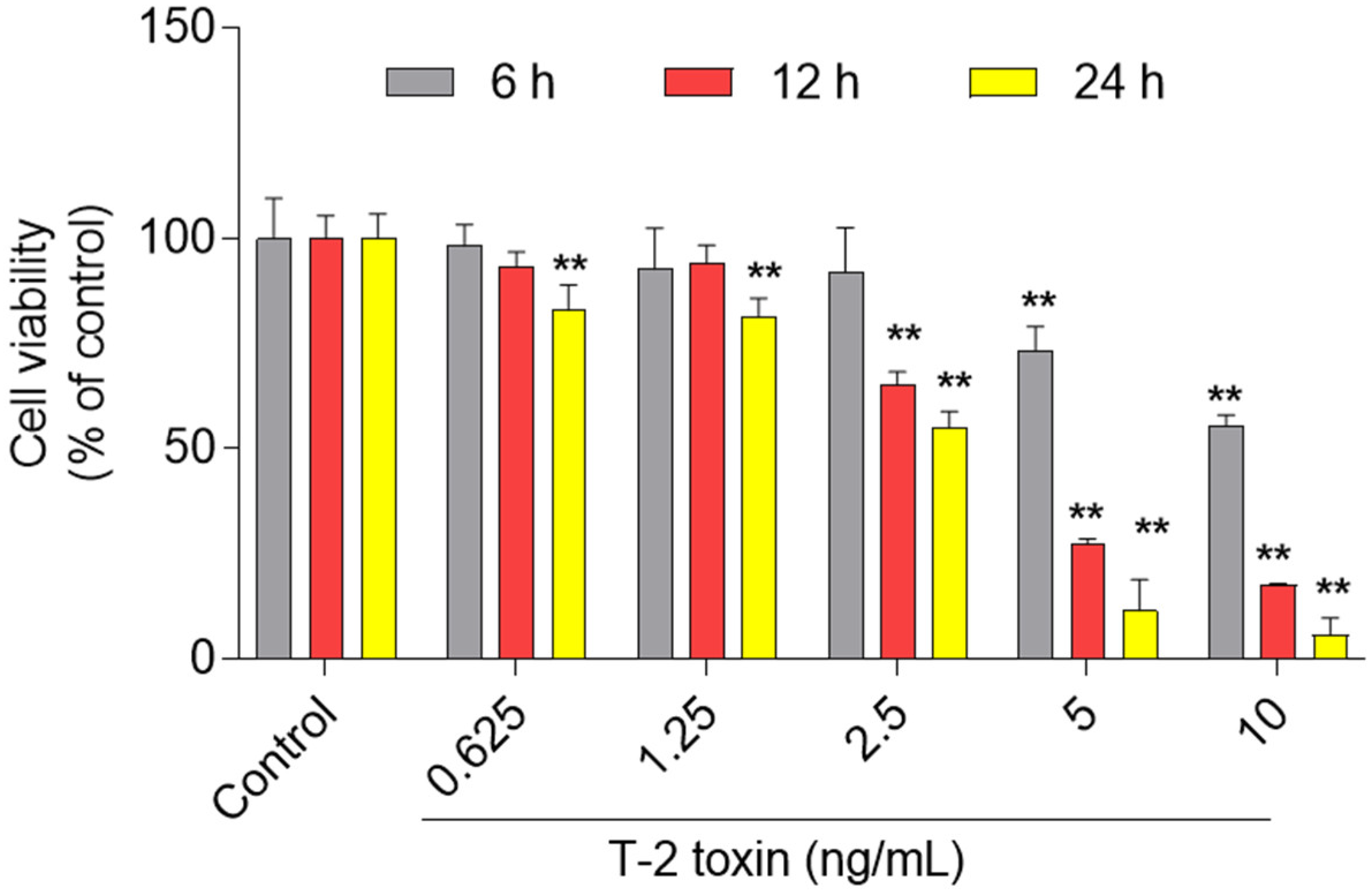
3.1. Cytotoxic Effect of T-2 Toxin in BV2 Cells
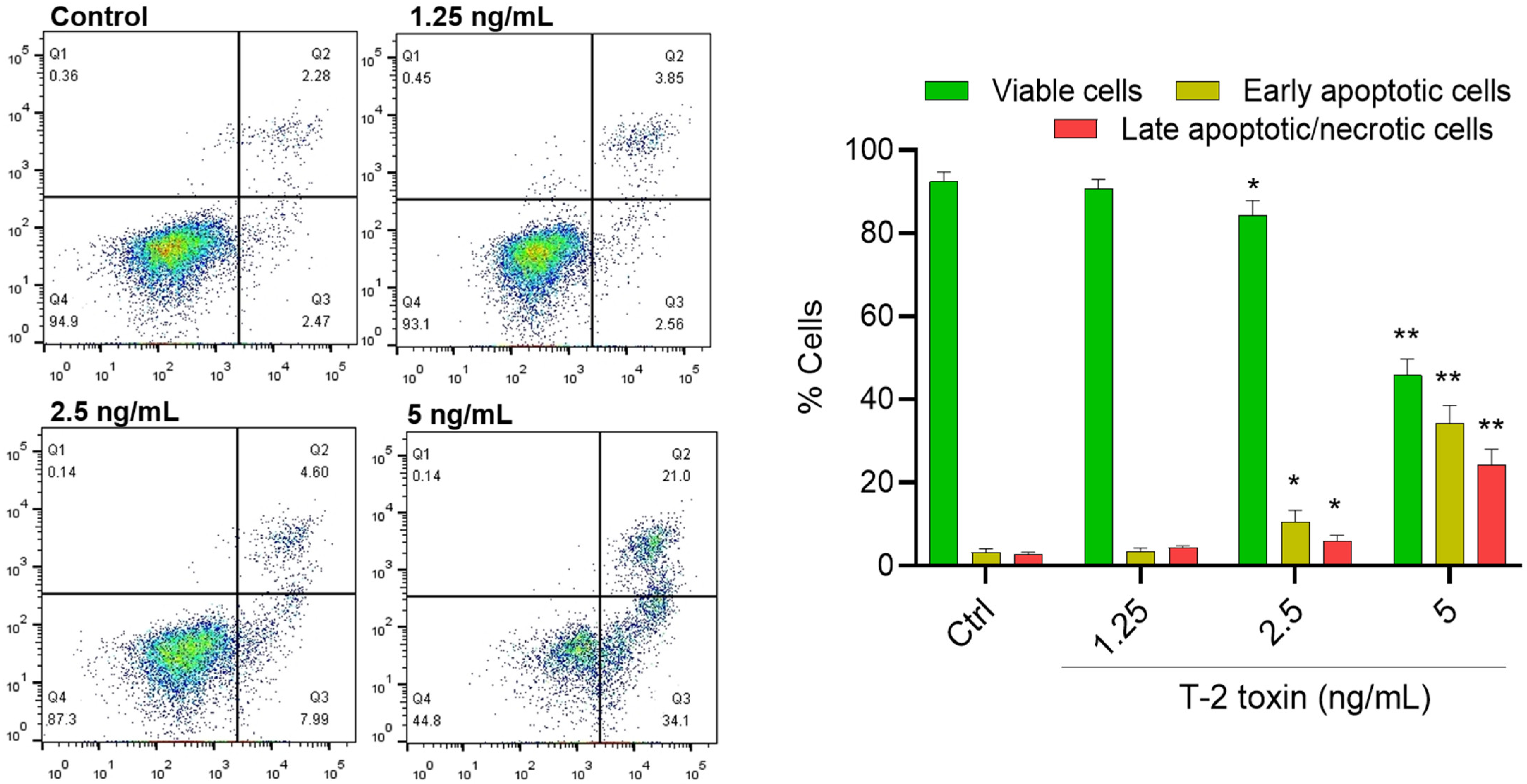
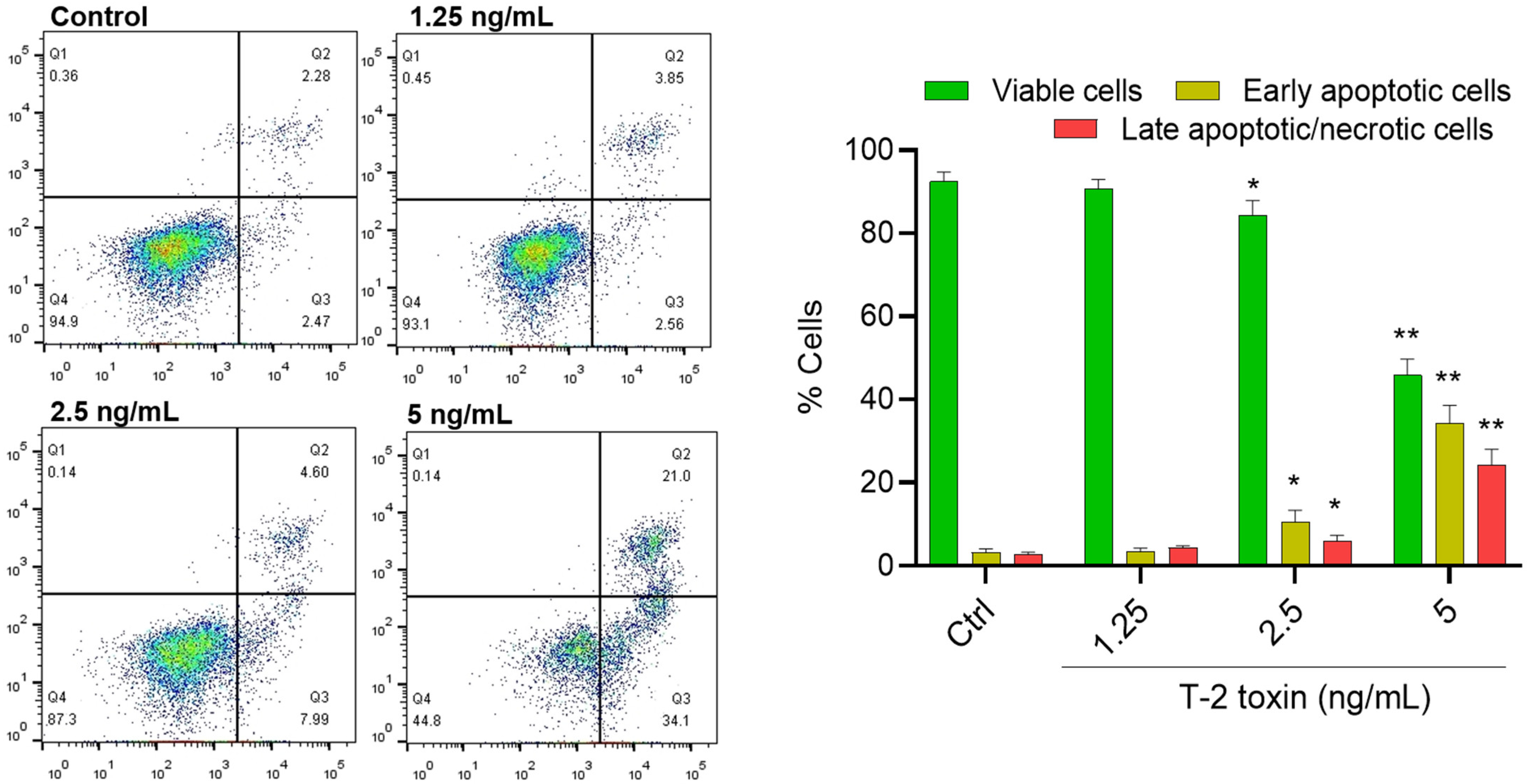
3.2. T-2 Toxin Induces Apoptotic Cell Death in BV2 Cells
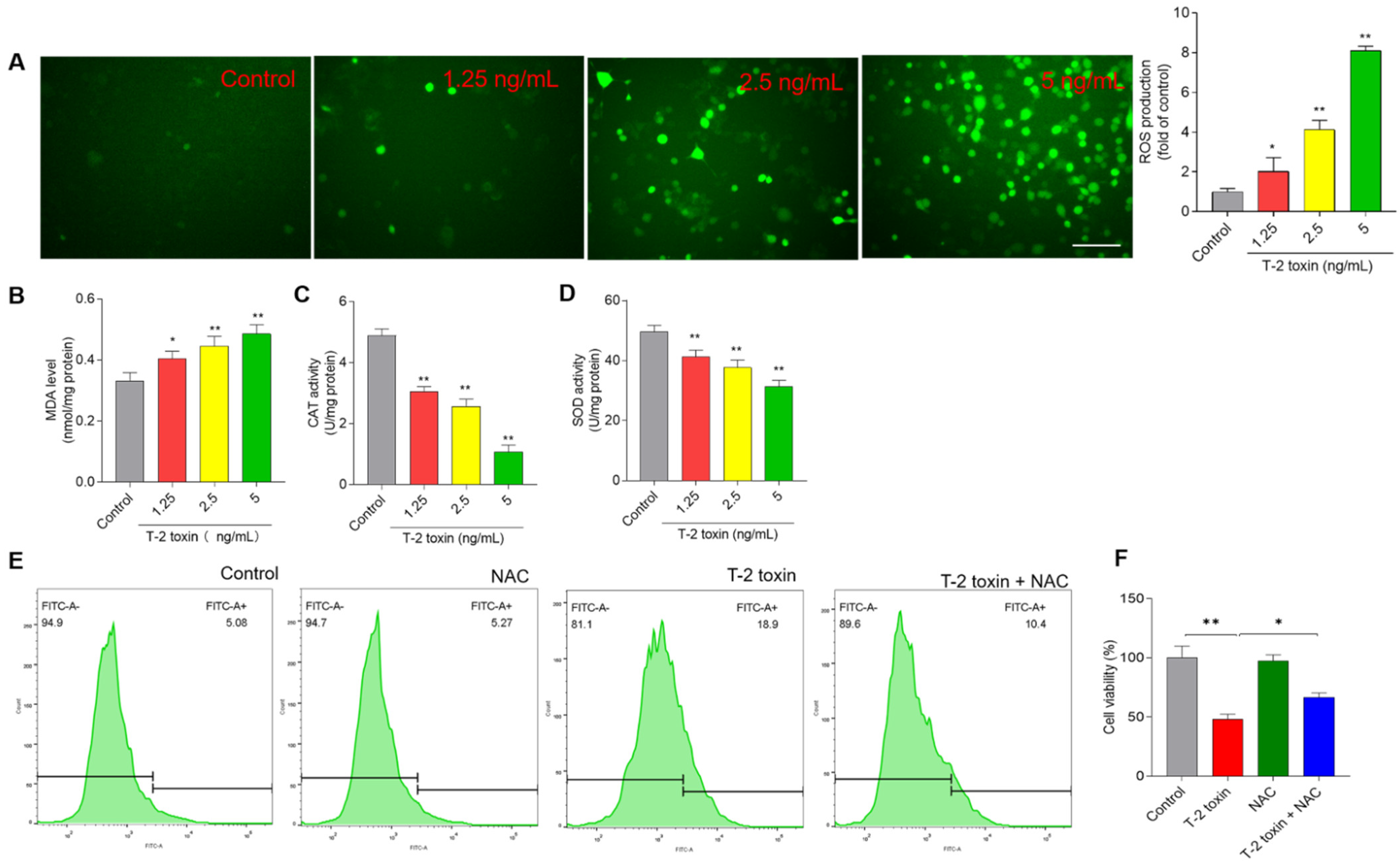
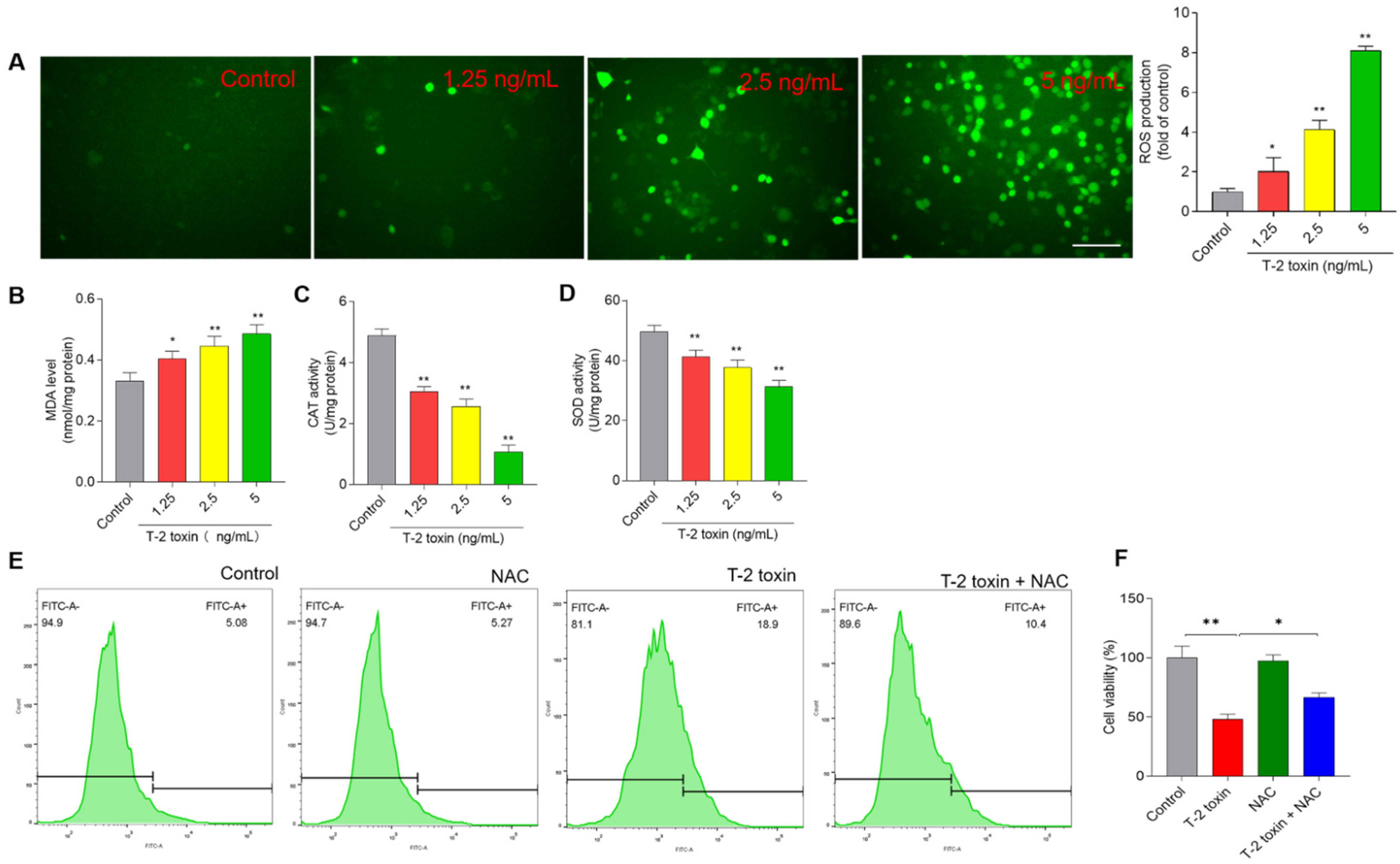
3.3. T-2 Toxin Induces Oxidative Stress in BV2 Cells
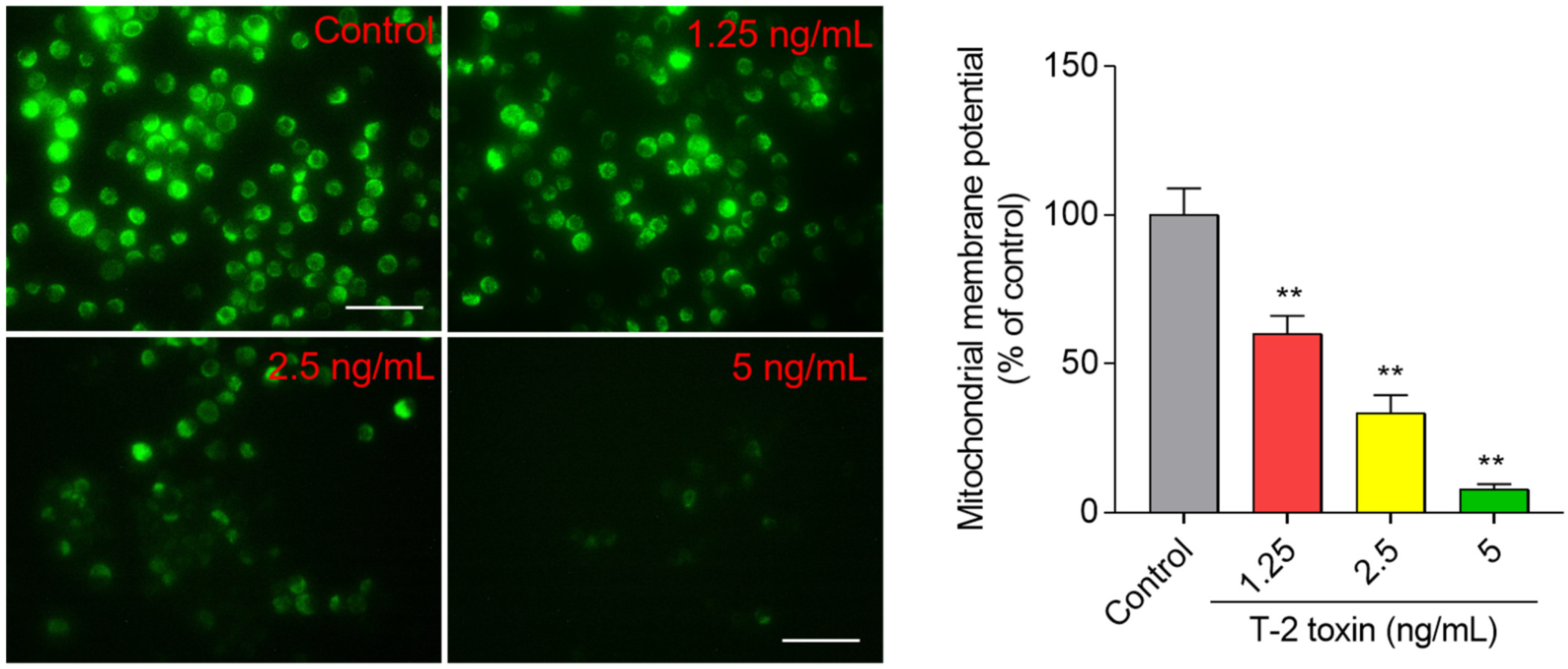
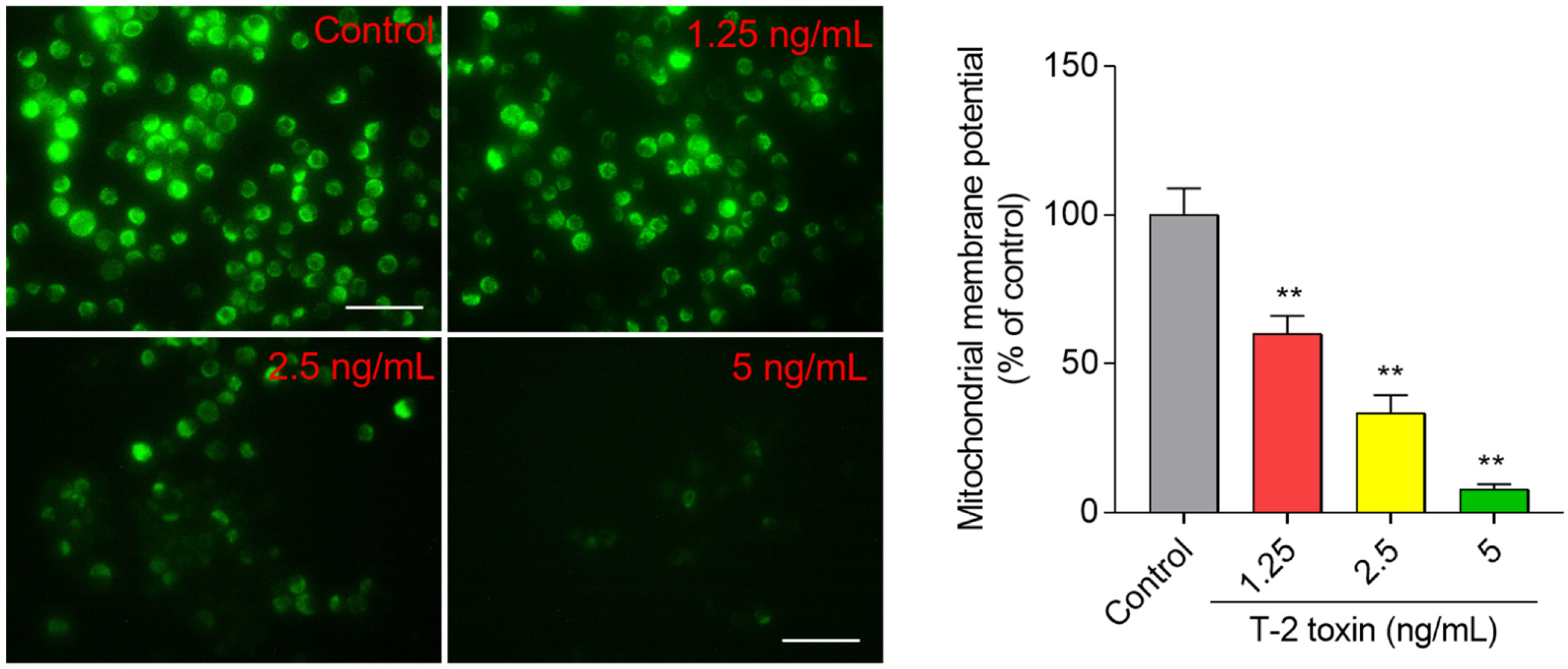
3.4. T-2 Toxin Induces Loss of Mitochondrial Membrane Potential in BV2 Cells
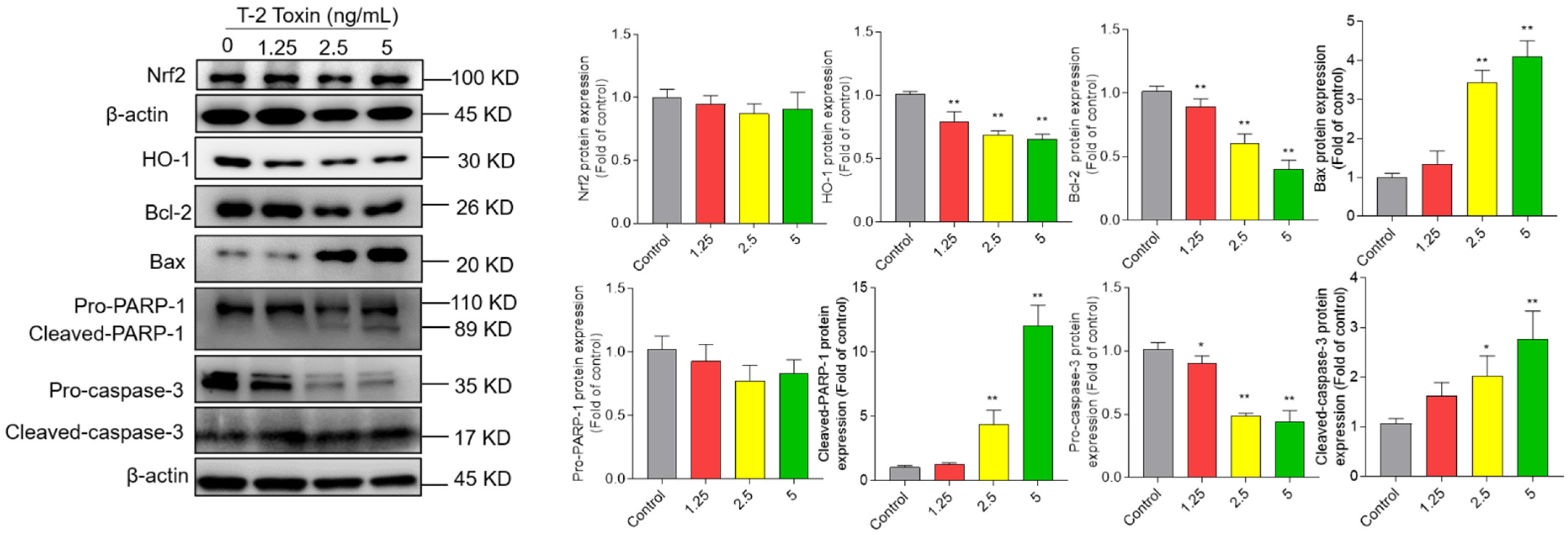
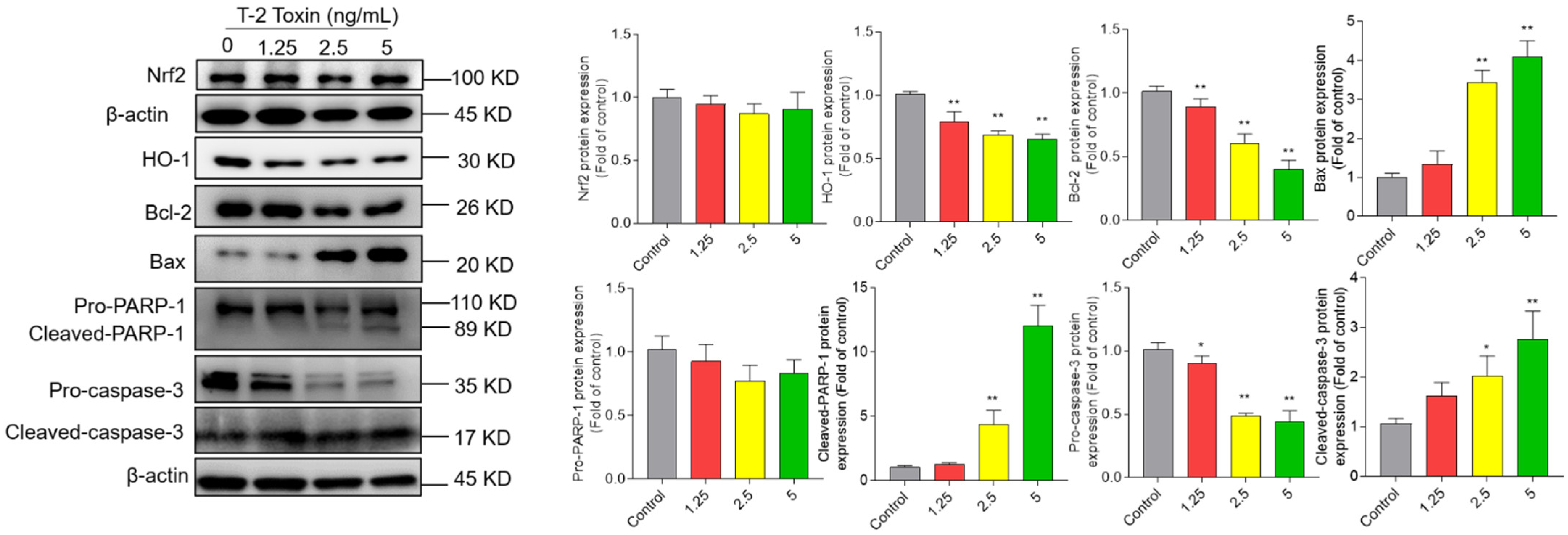
3.5. T-2 Toxin Treatment Activates Mitochondrial Apoptosis Pathway and Downregulated Nrf2/HO-1 Pathway in BV2 Cells
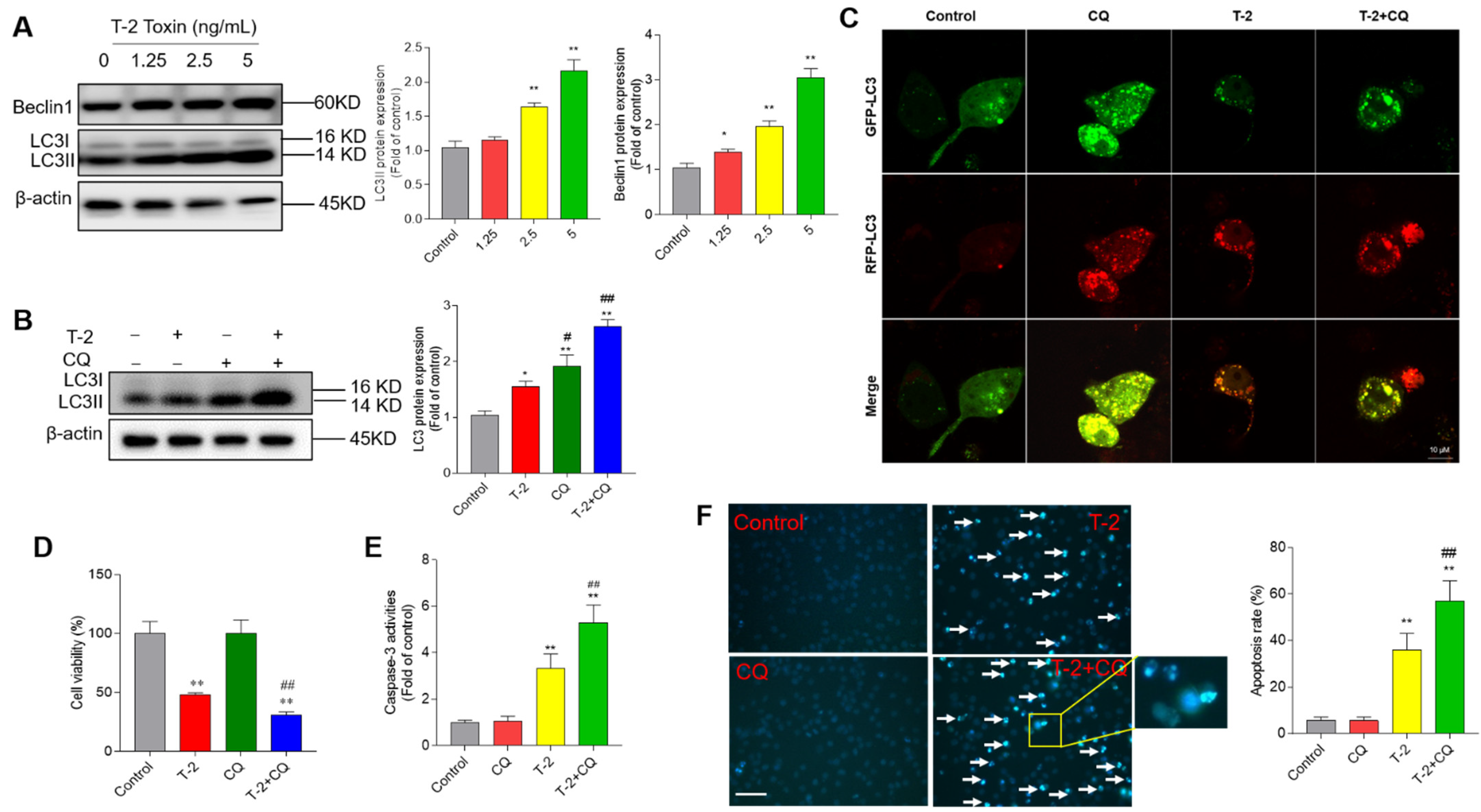
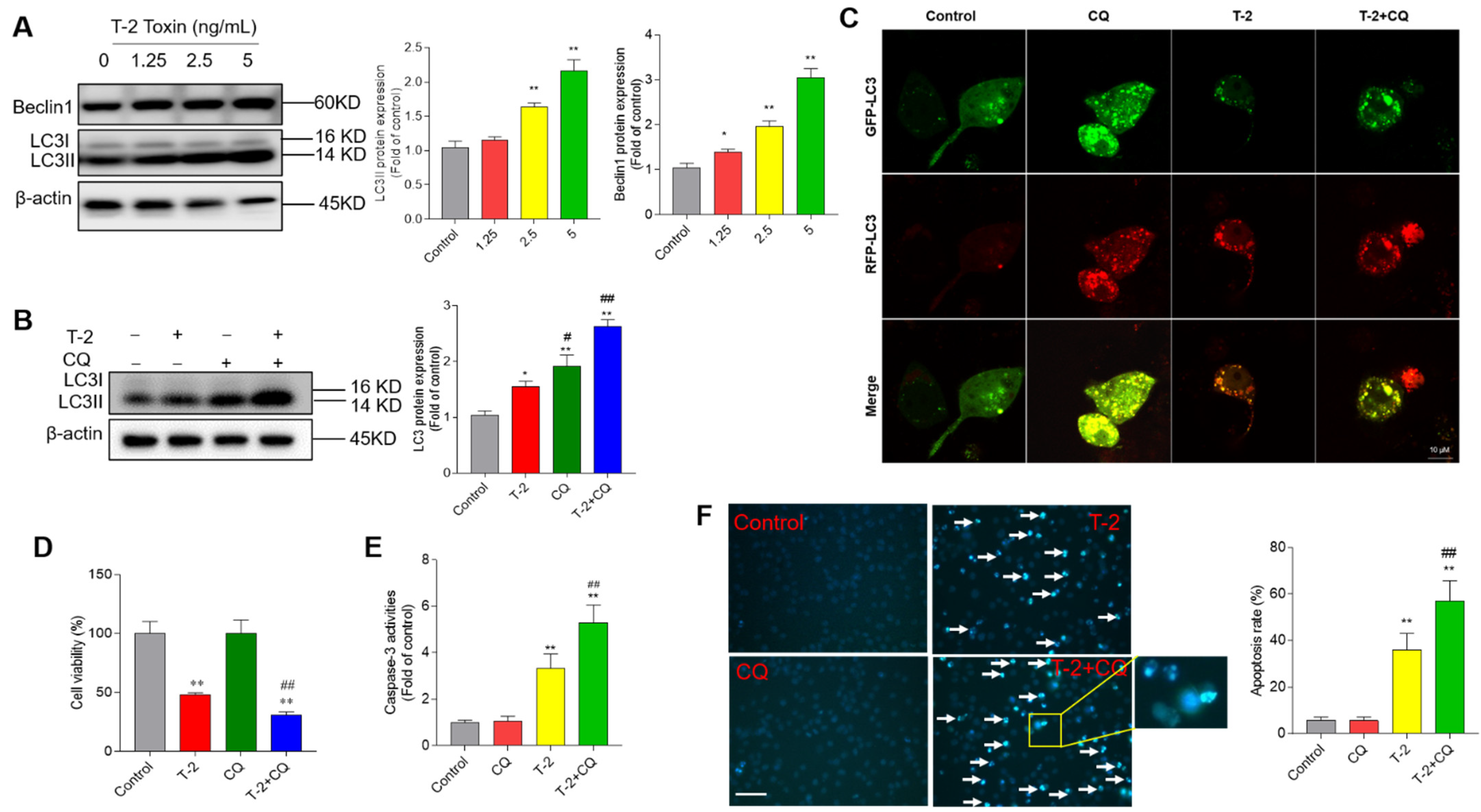
3.6. T-2 Toxin Treatment Activates Cell Autophagy and Autophagy Plays a Protective Role
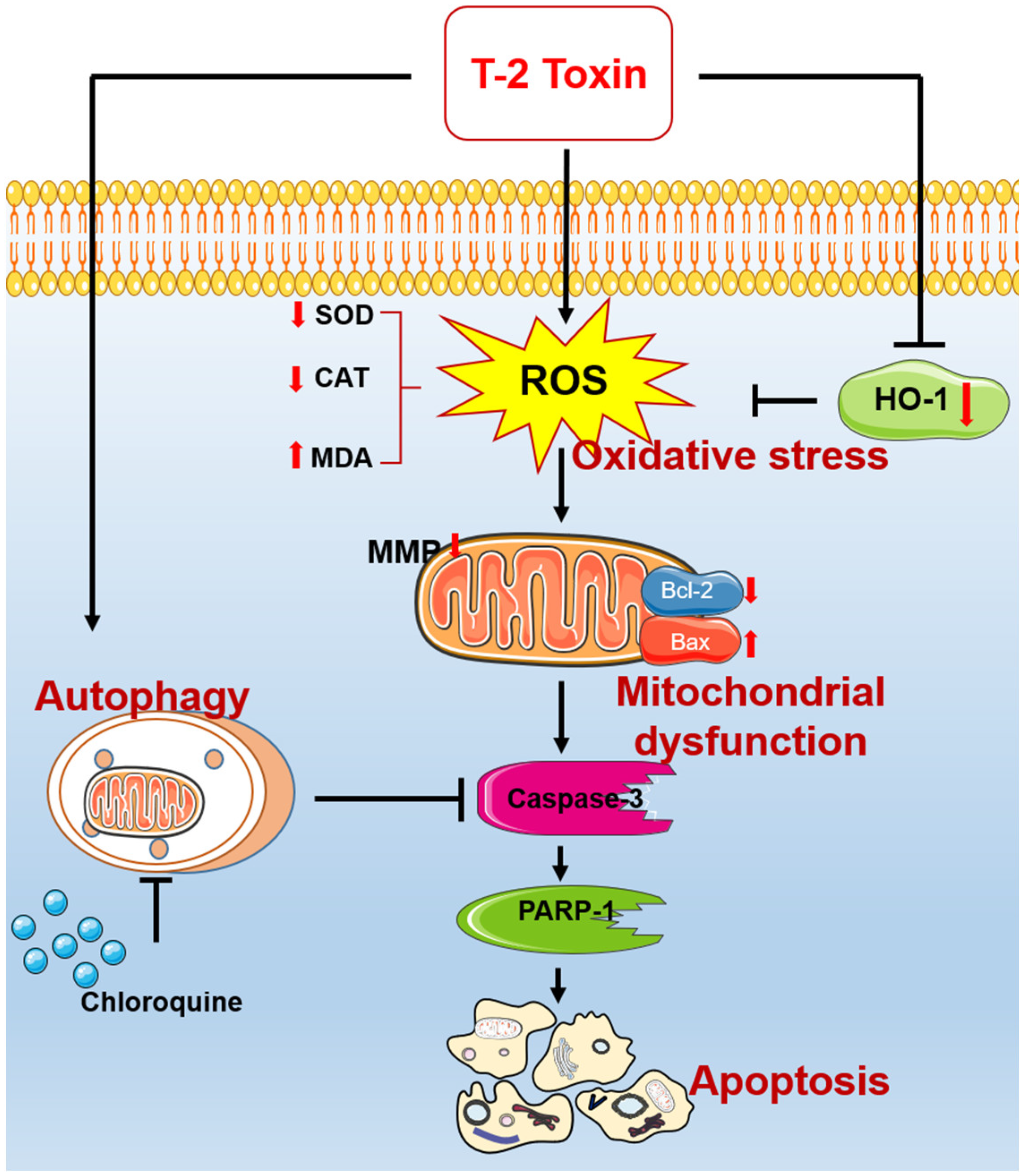
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, X.; Wang, Y.; Velkov, T.; Tang, S.; Dai, C. T-2 toxin-induced toxicity in neuroblastoma-2a cells involves the generation of reactive oxygen, mitochondrial dysfunction and inhibition of Nrf2/HO-1 pathway. Food Chem. Toxicol. 2018, 114, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Beier, R.C.; Shen, J.; De Smet, D.; De Saeger, S.; Zhang, S. T-2 toxin, a trichothecene mycotoxin: Review of toxicity, metabolism, and analytical methods. J. Agric. Food Chem. 2011, 59, 3441–3453. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.Y.; Li, Q.; Meng, F.G.; Fu, Y.; Zhao, Z.J.; Wang, L.H. T-2 toxin contamination in grains and selenium concentration in drinking water and grains in Kaschin-Beck disease endemic areas of Qinghai Province. Biol. Trace Elem. Res. 2012, 150, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Ge, B.; Zhao, K.; Wang, W.; Mi, J. Determination of 14 mycotoxins in Chinese herbs by liquid chromatography-tandem mass spectrometry with immunoaffinity purification. Se Pu Chin. J. Chromatogr. 2011, 29, 495–500. [Google Scholar] [CrossRef]
- Tan, Y.; Kuang, Y.; Zhao, R.; Chen, B.; Wu, J. Determination of T-2 and HT-2 Toxins in Traditional Chinese Medicine Marketed in China by LC–ELSD after Sample Clean-Up by Two Solid-Phase Extractions. Chromatographia 2011, 73, 407–410. [Google Scholar] [CrossRef]
- De Ruyck, K.; De Boevre, M.; Huybrechts, I.; De Saeger, S. Dietary mycotoxins, co-exposure, and carcinogenesis in humans: Short review. Mutat. Res. Rev. Mutat. Res. 2015, 766, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, X.; Yao, Q.; Song, M.; Han, Y.; Shao, B.; Li, Y. T-2 toxin impairs male fertility by disrupting hypothalamic-pituitary-testis axis and declining testicular function in mice. Chemosphere 2019, 234, 909–916. [Google Scholar] [CrossRef]
- Yang, X.; Liu, P.; Cui, Y.; Xiao, B.; Liu, M.; Song, M.; Huang, W.; Li, Y. Review of the Reproductive Toxicity of T-2 Toxin. J. Agric. Food Chem. 2020, 68, 727–734. [Google Scholar] [CrossRef]
- Lu, Q.; Hu, S.; Guo, P.; Zhu, X.; Ren, Z.; Wu, Q.; Wang, X. PPAR-γ with its anti-fibrotic action could serve as an effective therapeutic target in T-2 toxin-induced cardiac fibrosis of rats. Food Chem. Toxicol. 2021, 152, 112183. [Google Scholar] [CrossRef]
- Yang, J.; Guo, W.; Wang, J.; Yang, X.; Zhang, Z.; Zhao, Z. T-2 Toxin-Induced Oxidative Stress Leads to Imbalance of Mitochondrial Fission and Fusion to Activate Cellular Apoptosis in the Human Liver 7702 Cell Line. Toxins 2020, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, H.; Zhang, M.; Wang, J.; Zhang, Z.; Wang, Y.; Sun, Y.; Zhang, Z. Protective effect of selenomethionine on T-2 toxin-induced liver injury in New Zealand rabbits. BMC Vet. Res. 2021, 17, 153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.; Yang, X.; Liu, M.; Huang, W.; Zhang, J.; Song, M.; Shao, B.; Li, Y. The nephrotoxicity of T-2 toxin in mice caused by oxidative stress-mediated apoptosis is related to Nrf2 pathway. Food Chem. Toxicol. 2021, 149, 112027. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhu, L.; Yi, X.; Huang, Y.; Zhao, H.; Chen, Y.; Yuan, Z.; Wen, L.; Wu, J.; Yi, J. Betulinic Acid Alleviates Spleen Oxidative Damage Induced by Acute Intraperitoneal Exposure to T-2 Toxin by Activating Nrf2 and Inhibiting MAPK Signaling Pathways. Antioxidants 2021, 10, 158. [Google Scholar] [CrossRef]
- Mackei, M.; Matis, G.; Neogrady, Z. The effects of T-2 toxin on animal health, focusing especially on poultry. Magyar Allatorvosok Lapja 2018, 140, 475–483. [Google Scholar]
- Yang, S.P.; De Boevre, M.; Zhang, H.Y.; De Ruyck, K.; Sun, F.F.; Zhang, J.Z.; Jim, Y.; Li, Y.S.; Wang, Z.H.; Zhang, S.X.; et al. Metabolism of T-2 Toxin in Farm Animals and Human In Vitro and in Chickens In Vivo Using Ultra High-Performance Liquid Chromatography- Quadrupole/Time-of-Flight Hybrid Mass Spectrometry Along with Online Hydrogen/Deuterium Exchange Technique. J. Agric. Food Chem. 2017, 65, 7217–7227. [Google Scholar] [CrossRef]
- Arcella, D.; Gergelova, P.; Innocenti, M.L.; Steinkellner, H.; EFS, E.F.S.A. Human and animal dietary exposure to T-2 and HT-2 toxin. EFSA J. 2017, 15, e04972. [Google Scholar] [CrossRef]
- Aksoy, A.; Yavuz, O.; Das, Y.K.; Guvenc, D.; Muglali, O.H. Occurrence of Aflatoxin B1, T-2 Toxin and Zearalenone in Compound Animal Feed. J. Anim. Vet. Adv. 2009, 8, 403–407. [Google Scholar]
- Ohta, M.; Ishii, K.; Ueno, Y. Metabolism of Trichothecene Mycotoxins. 1. Microsomal Deacetylation of T-2 Toxin in Animal-Tissues. J. Biochem. 1977, 82, 1591–1598. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Yang, J.Y.; Meng, X.P.; Qiao, X.L. l-arginine protects against T-2 toxin-induced male reproductive impairments in mice. Theriogenology 2018, 126, 249–253. [Google Scholar] [CrossRef]
- Hemmati, A.A.; Kalantari, H.; Jalali, A.; Rezai, S.; Zadeh, H.H. Healing effect of quince seed mucilage on T-2 toxin-induced dermal toxicity in rabbit. Exp. Toxicol. Pathol. 2012, 64, 181–186. [Google Scholar] [CrossRef]
- Agrawal, M.; Yadav, P.; Lomash, V.; Bhaskar, A.S.; Lakshmana Rao, P.V. T-2 toxin induced skin inflammation and cutaneous injury in mice. Toxicology 2012, 302, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Maria, W.; Sabine, H.; Franziska, E.; Tanja, S.; Hans-Joachim, G.; Hans-Ulrich, H.; Stefan, S. Influence of T-2 and HT-2 Toxin on the Blood-Brain Barrier In Vitro: New Experimental Hints for Neurotoxic Effects. PLoS ONE 2013, 8, e60484. [Google Scholar]
- Ravindran, J.; Agrawal, M.; Gupta, N.; Rao, P.V.L. Alteration of blood brain barrier permeability by T-2 toxin: Role of MMP-9 and inflammatory cytokines. Toxicology 2011, 280, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Weidner, M.; Lenczyk, M.; Schwerdt, G.; Gekle, M.; Humpf, H.U. Neurotoxic potential and cellular uptake of T-2 toxin in human astrocytes in primary culture. Chem. Res. Toxicol. 2013, 26, 347–355. [Google Scholar] [CrossRef]
- Fairhurst, S.; Marrs, T.C.; Parker, H.C.; Scawin, J.W.; Swanston, D.W. Acute toxicity of T2 toxin in rats, mice, guinea pigs, and pigeons. Toxicology 1987, 43, 31–49. [Google Scholar] [CrossRef]
- Sheng, K.; Lu, X.; Yue, J.; Gu, W.; Gu, C.; Zhang, H.; Wu, W. Role of neurotransmitters 5-hydroxytryptamine and substance P in anorexia induction following oral exposure to the trichothecene T-2 toxin. Food Chem. Toxicol. 2018, 123, 1–8. [Google Scholar] [CrossRef]
- Agrawal, M.; Bhaskar, A.S.B.; Lakshmana Rao, P.V. Involvement of mitogen-activated protein kinase pathway in T-2 toxin-induced cell cycle alteration and apoptosis in human neuroblastoma cells. Mol. Neurobiol. 2015, 51, 1379–1394. [Google Scholar] [CrossRef]
- Pei, X.; Jiang, H.; Liu, X.; Li, L.; Li, C.; Xiao, X.; Li, D.; Tang, S. Targeting HMGB1 inhibits T-2 toxin-induced neurotoxicity via regulation of oxidative stress, neuroinflammation and neuronal apoptosis. Food Chem. Toxicol. 2021, 151, 112134. [Google Scholar] [CrossRef]
- Liu, X.; Huang, D.; Guo, P.; Wu, Q.; Dai, M.; Cheng, G.; Hao, H.; Xie, S.; Yuan, Z.; Wang, X. PKA/CREB and NF-κB pathway regulates AKNA transcription: A novel insight into T-2 toxin-induced inflammation and GH deficiency in GH3 cells. Toxicology 2017, 392, 81–95. [Google Scholar] [CrossRef]
- Dai, C.; Xiao, X.; Sun, F.; Zhang, Y.; Hoyer, D.; Shen, J.; Tang, S.; Velkov, T. T-2 toxin neurotoxicity: Role of oxidative stress and mitochondrial dysfunction. Arch. Toxicol. 2019, 93, 3041–3056. [Google Scholar] [CrossRef]
- Damiano, S.; Andretta, E.; Longobardi, C.; Prisco, F.; Paciello, O.; Squillacioti, C.; Mirabella, N.; Florio, S.; Ciarcia, R. Effects of Curcumin on the Renal Toxicity Induced by Ochratoxin A in Rats. Antioxidants 2020, 9, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiano, S.; Jarriyawattanachaikul, W.; Girolami, F.; Longobardi, C.; Nebbia, C.; Andretta, E.; Lauritano, C.; Dabbou, S.; Avantaggiato, G.; Schiavone, A.; et al. Curcumin Supplementation Protects Broiler Chickens Against the Renal Oxidative Stress Induced by the Dietary Exposure to Low Levels of Aflatoxin B1. Front. Vet. Sci. 2021, 8, 822227. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Nabekura, J. Functions of microglia in the central nervous system—Beyond the immune response. Neuron Glia Biol. 2011, 7, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes-Glioblastoma Crosstalk: Crucial Molecular Mechanisms and Microenvironmental Factors. Front. Cell. Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef] [Green Version]
- He, K.; Liang, X.; Wei, T.; Liu, N.; Wang, Y.; Zou, L.; Lu, J.; Yao, Y.; Kong, L.; Zhang, T.; et al. DNA damage in BV-2 cells: An important supplement to the neurotoxicity of CdTe quantum dots. J. Appl. Toxicol. JAT 2019, 39, 525–539. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, Q.; Song, E.; Song, Y. Polybrominated diphenyl ethers quinone exhibits neurotoxicity by inducing DNA damage, cell cycle arrest, apoptosis and p53-driven adaptive response in microglia BV2 cells. Toxicology 2021, 457, 152807. [Google Scholar] [CrossRef]
- Novo, J.P.; Martins, B.; Raposo, R.S.; Pereira, F.C.; Oriá, R.B.; Malva, J.O.; Fontes-Ribeiro, C. Cellular and Molecular Mechanisms Mediating Methylmercury Neurotoxicity and Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 3101. [Google Scholar] [CrossRef]
- Taroncher, M.; Rodríguez-Carrasco, Y.; Ruiz, M.J. T-2 toxin and its metabolites: Characterization, cytotoxic mechanisms and adaptive cellular response in human hepatocarcinoma (HepG2) cells. Food Chem. Toxicol. 2020, 145, 111654. [Google Scholar] [CrossRef]
- Königs, M.; Mulac, D.; Schwerdt, G.; Gekle, M.; Humpf, H.U. Metabolism and cytotoxic effects of T-2 toxin and its metabolites on human cells in primary culture. Toxicology 2009, 258, 106–115. [Google Scholar] [CrossRef]
- Dai, C.; Tang, S.; Velkov, T.; Xiao, X. Colistin-Induced Apoptosis of Neuroblastoma-2a Cells Involves the Generation of Reactive Oxygen Species, Mitochondrial Dysfunction, and Autophagy. Mol. Neurobiol. 2016, 53, 4685–4700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.; Li, D.; Gong, L.; Xiao, X.; Tang, S. Curcumin Ameliorates Furazolidone-Induced DNA Damage and Apoptosis in Human Hepatocyte L02 Cells by Inhibiting ROS Production and Mitochondrial Pathway. Molecules 2016, 21, 1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.; Xiao, X.; Li, D.; Tun, S.; Wang, Y.; Velkov, T.; Tang, S. Chloroquine ameliorates carbon tetrachloride-induced acute liver injury in mice via the concomitant inhibition of inflammation and induction of apoptosis. Cell Death Dis. 2018, 9, 1164. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Ciccotosto, G.D.; Cappai, R.; Tang, S.; Li, D.; Xie, S.; Xiao, X.; Velkov, T. Curcumin Attenuates Colistin-Induced Neurotoxicity in N2a Cells via Anti-inflammatory Activity, Suppression of Oxidative Stress, and Apoptosis. Mol. Neurobiol. 2018, 55, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Li, B.; Zhou, Y.; Li, D.; Zhang, S.; Li, H.; Xiao, X.; Tang, S. Curcumin attenuates quinocetone induced apoptosis and inflammation via the opposite modulation of Nrf2/HO-1 and NF-kB pathway in human hepatocyte L02 cells. Food Chem. Toxicol. 2016, 95, 52–63. [Google Scholar] [CrossRef]
- Fan, K.; Xu, J.; Jiang, K.; Liu, X.; Meng, J.; Di Mavungu, J.D.; Guo, W.; Zhang, Z.; Jing, J.; Li, H.; et al. Determination of multiple mycotoxins in paired plasma and urine samples to assess human exposure in Nanjing, China. Environ. Pollut. 2019, 248, 865–873. [Google Scholar] [CrossRef]
- Guo, P.; Liu, A.; Huang, D.; Wu, Q.; Fatima, Z.; Tao, Y.; Cheng, G.; Wang, X.; Yuan, Z. Brain damage and neurological symptoms induced by T-2 toxin in rat brain. Toxicol. Lett. 2018, 286, 96–107. [Google Scholar] [CrossRef]
- Chang, X.; Li, J.; Niu, S.; Xue, Y.; Tang, M. Neurotoxicity of metal-containing nanoparticles and implications in glial cells. J. Appl. Toxicol. JAT 2021, 41, 65–81. [Google Scholar] [CrossRef]
- Marinelli, S.; Basilico, B.; Marrone, M.C.; Ragozzino, D. Microglia-neuron crosstalk: Signaling mechanism and control of synaptic transmission. Semin. Cell Dev. Biol. 2019, 94, 138–151. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria: Back to the future. Nat. Rev. Mol. Cell. Biol. 2018, 19, 76. [Google Scholar] [CrossRef]
- Fu, X.Y.; Yang, M.F.; Cao, M.Z.; Li, D.W.; Sun, B.-L. Strategy to Suppress Oxidative Damage-Induced Neurotoxicity in PC12 Cells by Curcumin: The Role of ROS-Mediated DNA Damage and the MAPK and AKT Pathways. Mol. Neurobiol. 2016, 53, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Wu, Y.; Guo, J.; Rong, J.; Ma, L.; Zhao, Z.; Zuo, D.; Peng, S. T-2 toxin induces apoptosis in differentiated murine embryonic stem cells through reactive oxygen species-mediated mitochondrial pathway. Apoptosis 2012, 17, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Deyu, H.; Luqing, C.; Xianglian, L.; Pu, G.; Qirong, L.; Xu, W.; Zonghui, Y. Protective mechanisms involving enhanced mitochondrial functions and mitophagy against T-2 toxin-induced toxicities in GH3 cells. Toxicol. Lett. 2018, 295, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Li, J.; Tang, S.; Li, J.; Xiao, X. Colistin-induced nephrotoxicity in mice involves the mitochondrial, death receptor, and endoplasmic reticulum pathways. Antimicrob. Agents Chemother. 2014, 58, 4075. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, Z.; Wang, X.; Yan, X.; He, Q.; Liu, S.; Ye, M.; Li, X.; Yuan, Z.; Wu, J.; et al. Involvement of endoplasmic reticulum stress-activated PERK-eIF2α-ATF4 signaling pathway in T-2 toxin-induced apoptosis of porcine renal epithelial cells. Toxicol. Appl. Pharmacol. 2021, 432, 115753. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, J.X.; He, J.H. T-2 toxin-induced DRP-1-dependent mitophagy leads to the apoptosis of mice Leydig cells (TM3). Food Chem. Toxicol. 2020, 136, 111082. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, Y.; Yuan, Z.; Yi, J.; Chen, J.; Wang, N.; Tian, Y. Autophagy and Apoptosis Interact to Modulate T-2 Toxin-Induced Toxicity in Liver Cells. Toxins 2019, 11, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Sairanen, T.; Szepesi, R.; Karjalainen-Lindsberg, M.L.; Saksi, J.; Paetau, A.; Lindsberg, P.J. Neuronal caspase-3 and PARP-1 correlate differentially with apoptosis and necrosis in ischemic human stroke. Acta Neuropathol. 2009, 118, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Yin, H.; Han, S.; Chen, Y.; Wang, Y.; Li, D.; Zhu, Q. T-2 Toxin Induces Oxidative Stress, Apoptosis and Cytoprotective Autophagy in Chicken Hepatocytes. Toxins 2020, 12, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Zhang, Y.F.; Meng, X.P.; Kong, X.F. Delayed effects of autophagy on T-2 toxin-induced apoptosis in mouse primary Leydig cells. Toxicol. Ind. Health 2019, 35, 256–263. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, T.; Zhang, Q.; Li, M.; Tang, S.; Dai, C. T-2 Toxin Induces Apoptotic Cell Death and Protective Autophagy in Mouse Microglia BV2 Cells. J. Fungi 2022, 8, 761. https://doi.org/10.3390/jof8080761
Sun T, Zhang Q, Li M, Tang S, Dai C. T-2 Toxin Induces Apoptotic Cell Death and Protective Autophagy in Mouse Microglia BV2 Cells. Journal of Fungi. 2022; 8(8):761. https://doi.org/10.3390/jof8080761
Chicago/Turabian StyleSun, Tun, Qinzhi Zhang, Meng Li, Shusheng Tang, and Chongshan Dai. 2022. "T-2 Toxin Induces Apoptotic Cell Death and Protective Autophagy in Mouse Microglia BV2 Cells" Journal of Fungi 8, no. 8: 761. https://doi.org/10.3390/jof8080761
APA StyleSun, T., Zhang, Q., Li, M., Tang, S., & Dai, C. (2022). T-2 Toxin Induces Apoptotic Cell Death and Protective Autophagy in Mouse Microglia BV2 Cells. Journal of Fungi, 8(8), 761. https://doi.org/10.3390/jof8080761