
Evaluation of Aspergillus niger Six Constitutive Strong Promoters by Fluorescent-Auxotrophic Selection Coupled with Flow Cytometry: A Case for Citric Acid Production

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Cultivation Conditions
2.2. Plasmids Construction
2.3. A. Niger Strains Construction
2.4. Fluorescence Microscopic Analysis
2.5. Flow Cytometry Analysis
2.6. Citric Acid Fermentation and Detection
2.7. RNA Sequencing and Transcriptomics Analysis
3. Results
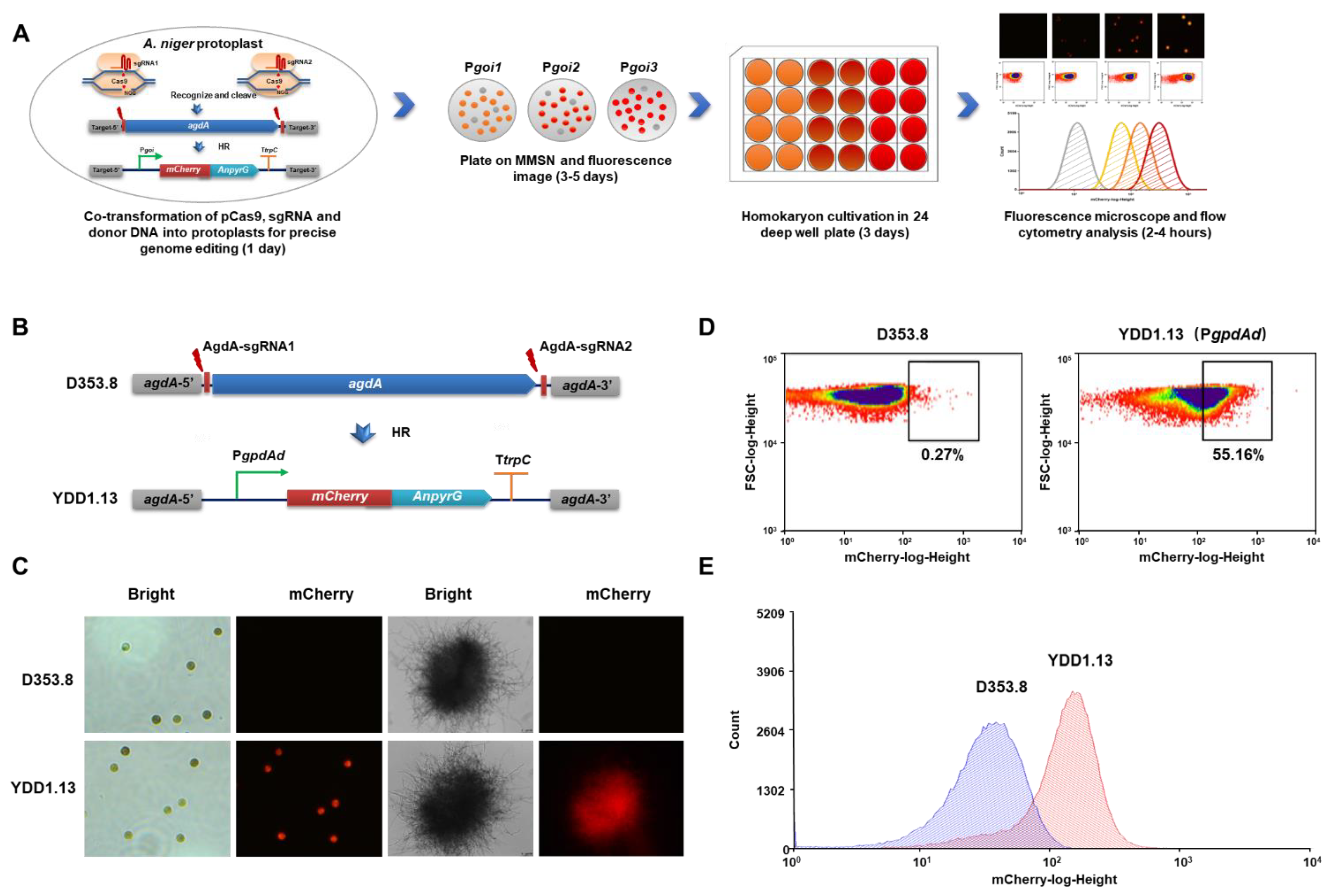
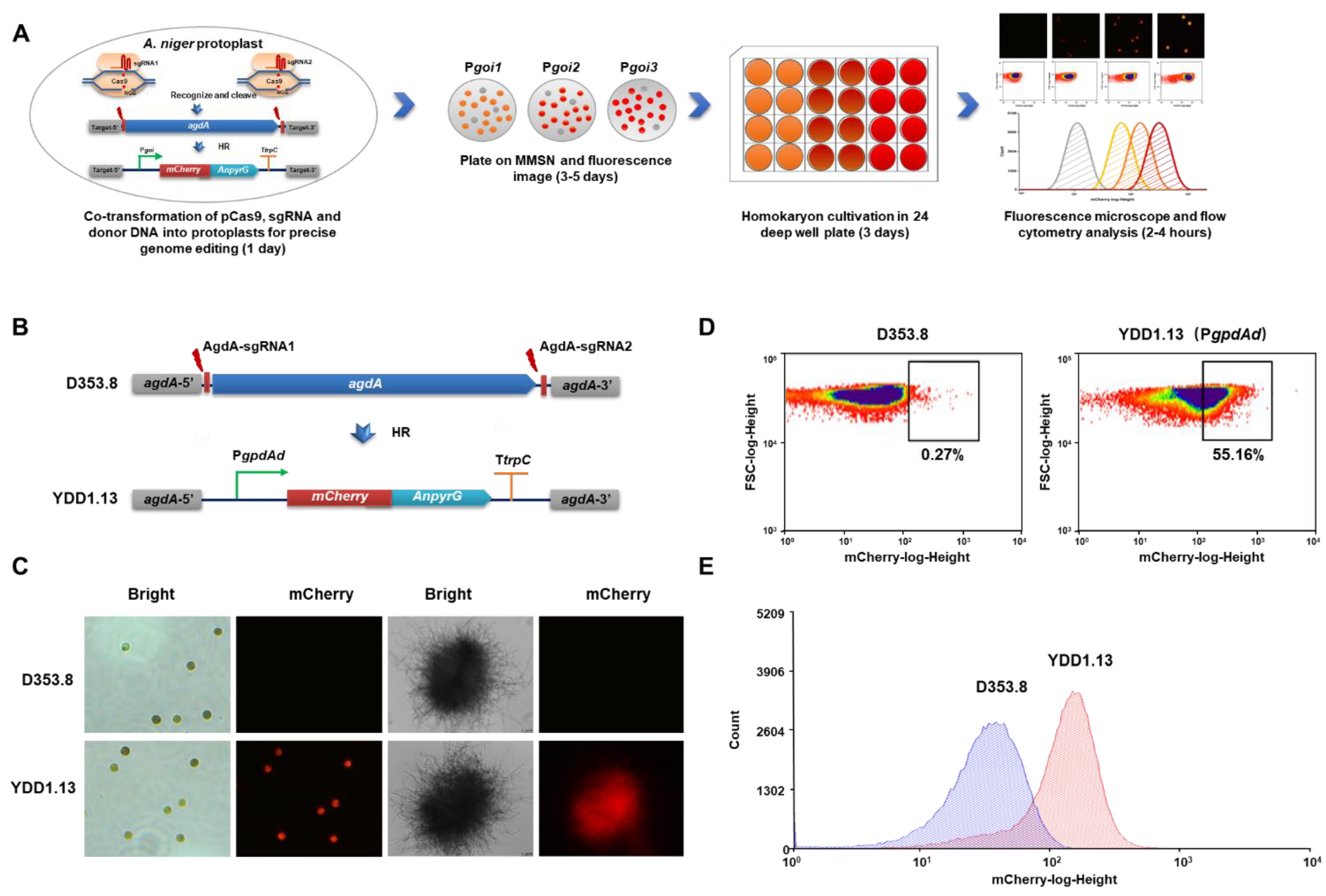
3.1. Fluorescent-Auxotrophic Double Selection Coupling with Flow Cytometry Workflow
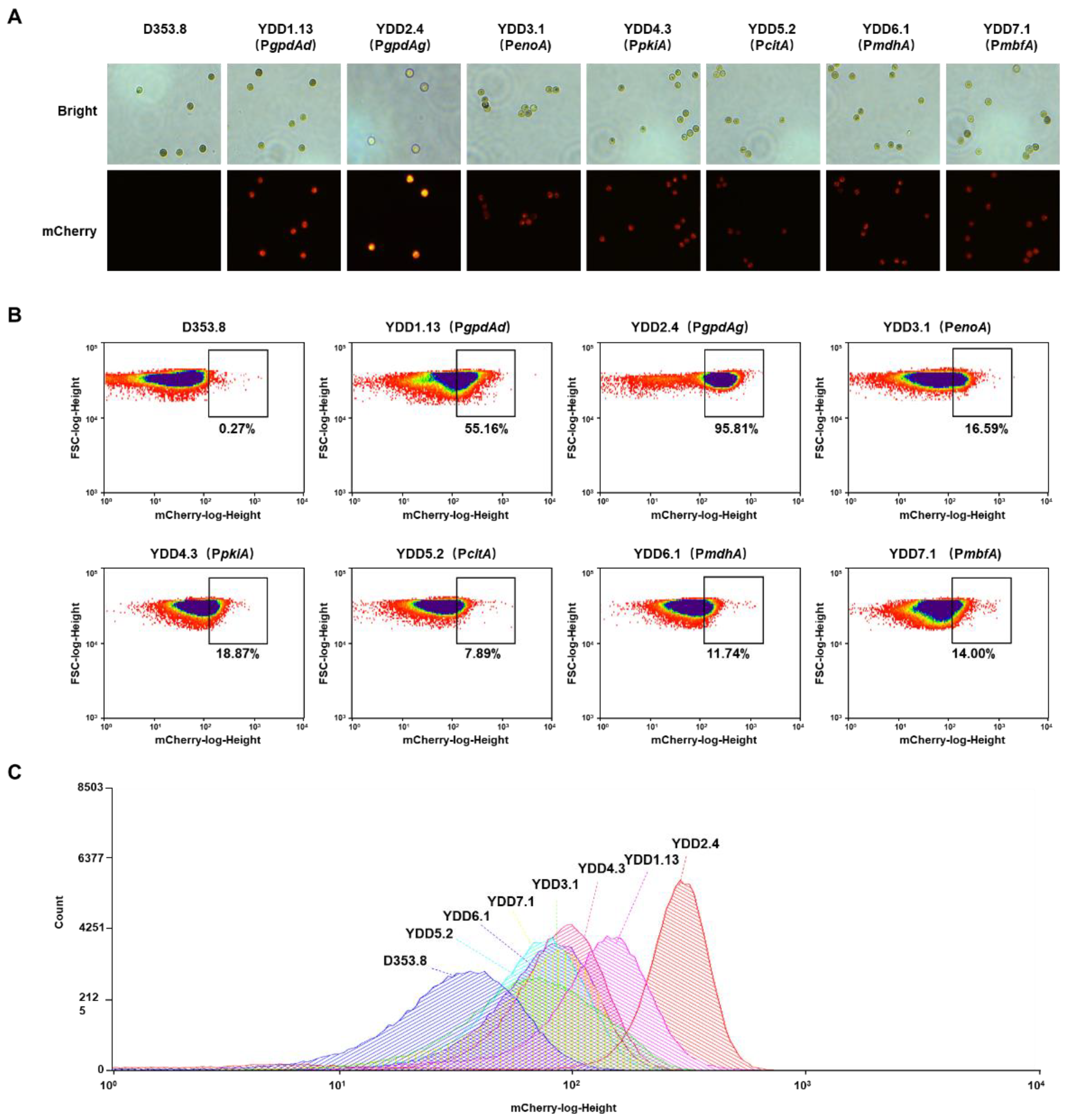
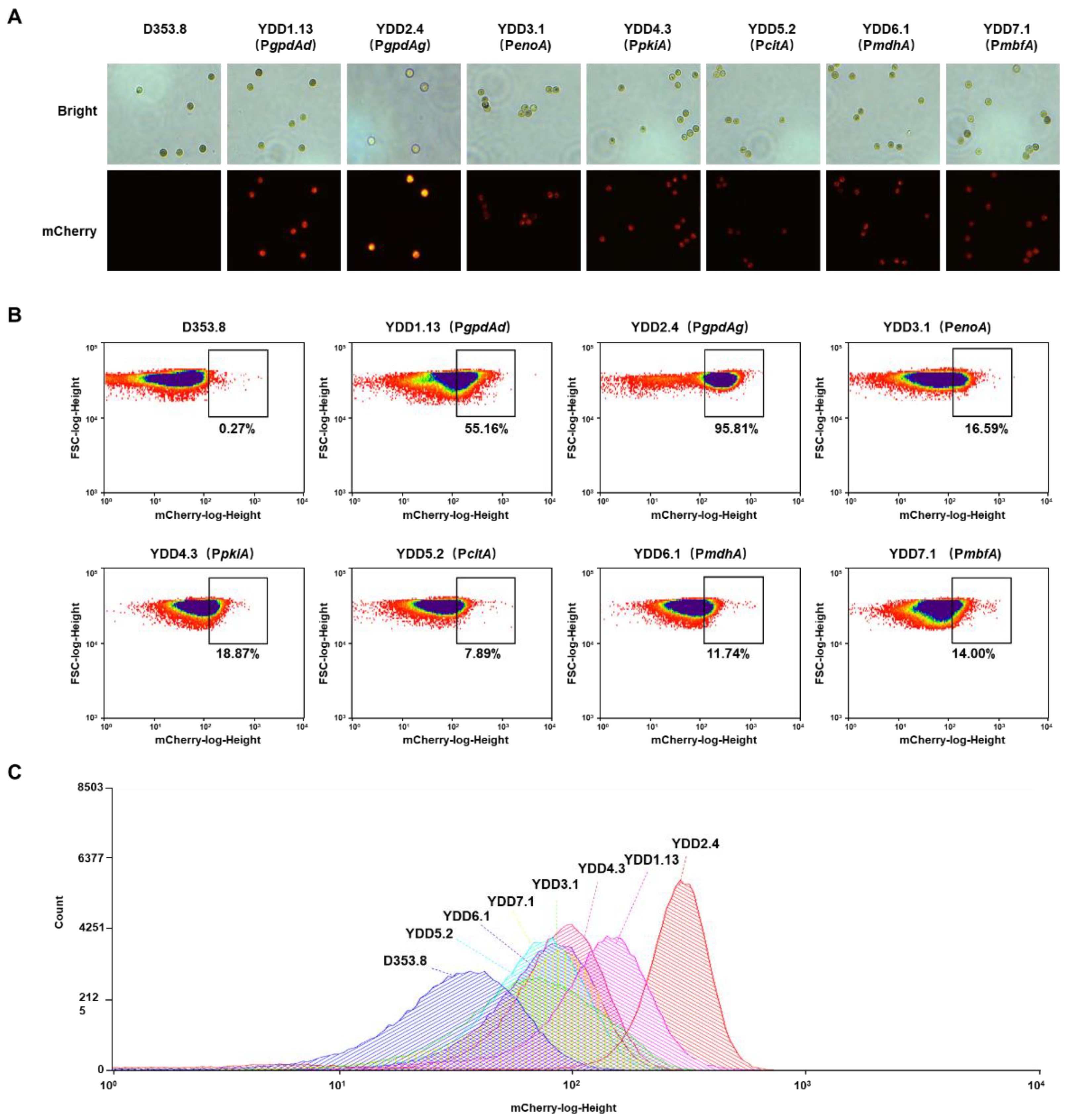
3.2. Evaluation of Endogenous Constitutive Promoters Using the Fluorescent-Auxotrophic Selection Workflow
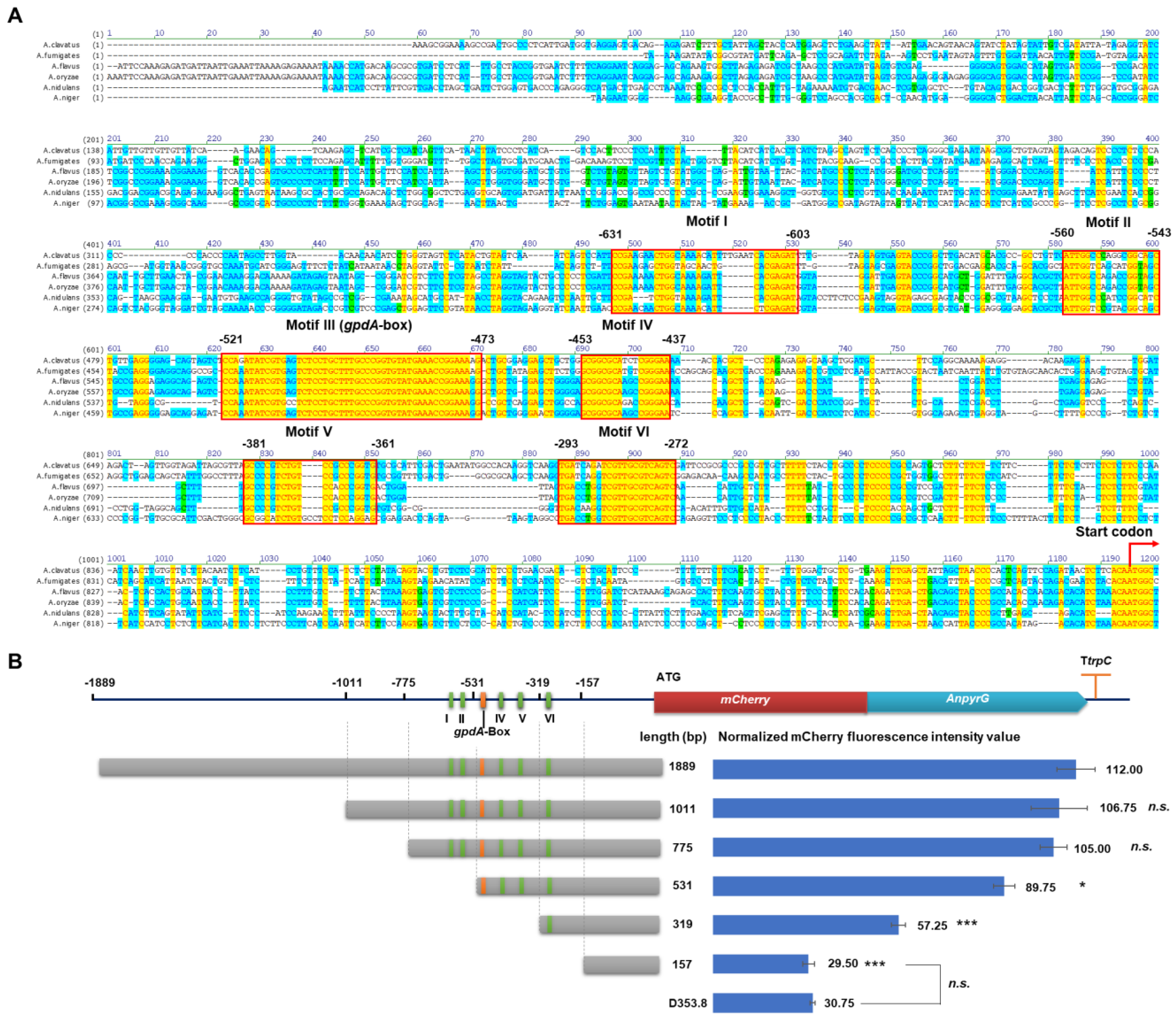
3.3. Characterization of Essential Elements in the PgpdAg Promoter
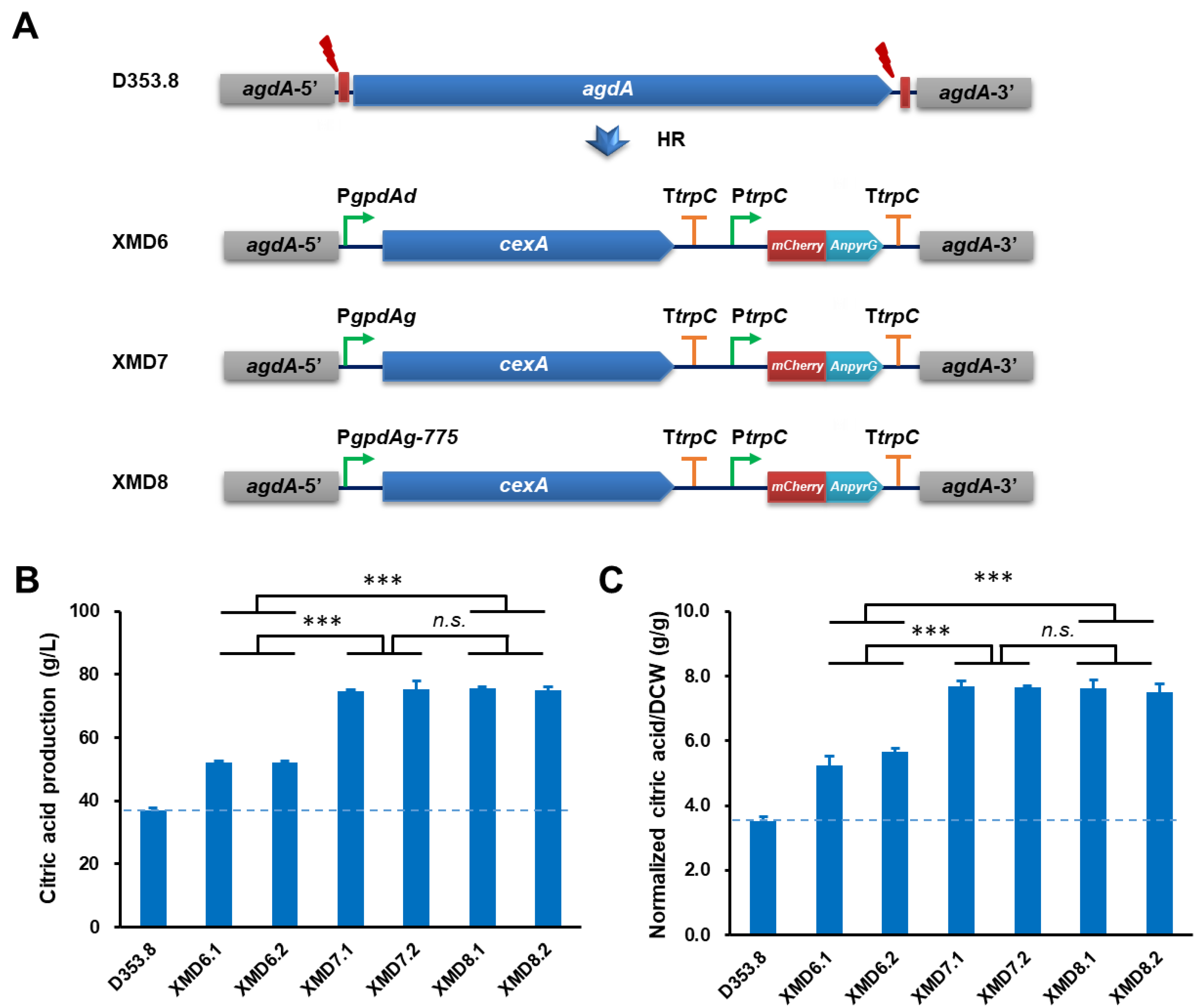
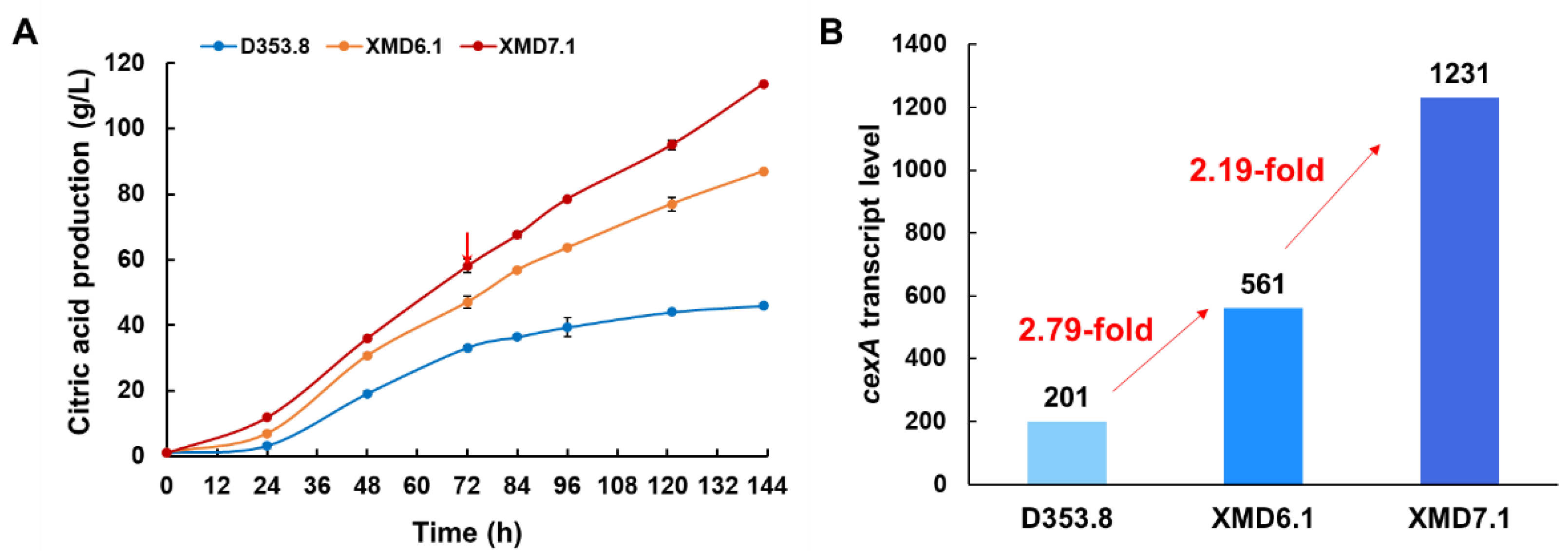
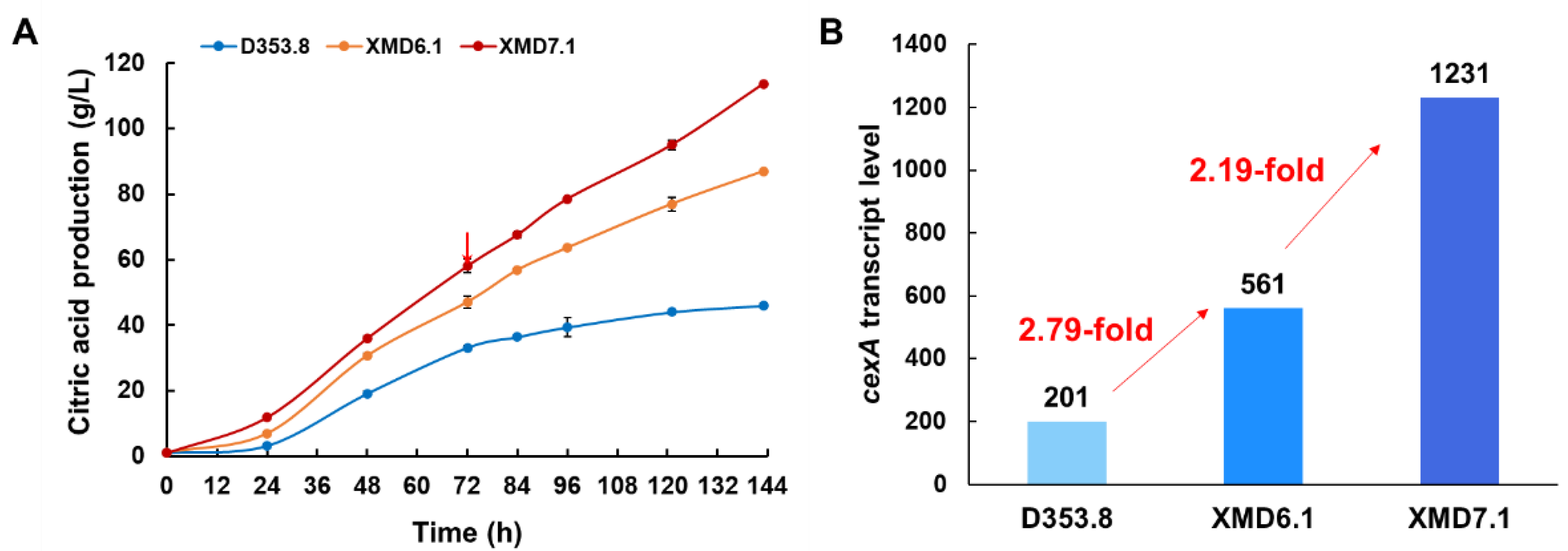
3.4. Application of PgpdAg Dramatically Improved Citric Acid Production in A. Niger
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meyer, V.; Wu, B.; Ram, A.F. Aspergillus as a multi-purpose cell factory: Current status and perspectives. Biotechnol. Lett. 2011, 33, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Z.; Zheng, X.; Tong, Y.; Shi, Y.C.; Sun, J. Systems metabolic engineering for citric acid production by Aspergillus niger in the post-genomic era. Microb. Cell. Fact. 2019, 18, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, V.; Cairns, T.; Barthel, L.; King, R.; Kunz, P.; Schmideder, S.; Muller, H.; Briesen, H.; Dinius, A.; Krull, R. Understanding and controlling filamentous growth of fungal cell factories: Novel tools and opportunities for targeted morphology engineering. Fungal. Biol. Biotechnol. 2021, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhou, Y.; Cao, W.; Liu, H. Improved Production of malic acid in Aspergillus niger by abolishing citric acid accumulation and enhancing glycolytic flux. ACS Synth. Biol. 2020, 9, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Steiger, M.G.; Rassinger, A.; Mattanovich, D.; Sauer, M. Engineering of the citrate exporter protein enables high citric acid production in Aspergillus niger. Metab. Eng. 2019, 52, 224–231. [Google Scholar] [CrossRef]
- Zheng, X.; Zheng, P.; Zhang, K.; Cairns, T.C.; Meyer, V.; Sun, J.; Ma, Y. 5S rRNA promoter for guide rna expression enabled highly efficient CRISPR/Cas9 genome editing in Aspergillus niger. ACS Synth. Biol. 2019, 8, 1568–1574. [Google Scholar] [CrossRef]
- Rozhkova, A.M.; Kislitsin, V.Y. CRISPR/Cas Genome Editing in Filamentous Fungi. Biochemistry 2021, 86, S120–S139. [Google Scholar] [CrossRef]
- Zheng, X.; Zheng, P.; Sun, J. CRISPR/Cas-based genome editing in Aspergillus niger. Sheng Wu Gong Cheng Xue Bao 2021, 37, 980–990. [Google Scholar]
- Ganzlin, M.; Rinas, U. In-depth analysis of the Aspergillus niger glucoamylase (glaA) promoter performance using high-throughput screening and controlled bioreactor cultivation techniques. J. Biotechnol. 2008, 135, 266–271. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, A.L.S.; Even, S.; Muller, C.; Punt, P.J.; van den Hondel, C.; Nielsen, J. Study of the glucoamylase promoter in Aspergillus niger using green fluorescent protein. Microbiology 1999, 145 Pt 3, 729–734. [Google Scholar] [CrossRef] [Green Version]
- Fleissner, A.; Dersch, P. Expression and export: Recombinant protein production systems for Aspergillus. Appl. Microbiol. Biotechnol. 2010, 87, 1255–1270. [Google Scholar] [CrossRef] [PubMed]
- Meyer, V.; Wanka, F.; van Gent, J.; Arentshorst, M.; van den Hondel, C.A.; Ram, A.F. Fungal gene expression on demand: An inducible, tunable, and metabolism-independent expression system for Aspergillus niger. Appl. Environ. Microbiol. 2011, 77, 2975–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanka, F.; Cairns, T.; Boecker, S.; Berens, C.; Happel, A.; Zheng, X.; Sun, J.; Krappmann, S.; Meyer, V. Tet-on, or Tet-off, that is the question: Advanced conditional gene expression in Aspergillus. Fungal. Genet. Biol. 2016, 89, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, A.H.; Dersch, P. A novel expression system for intracellular production and purification of recombinant affinity-tagged proteins in Aspergillus niger. Appl. Microbiol. Biotechnol. 2010, 86, 659–670. [Google Scholar] [CrossRef]
- Blumhoff, M.; Steiger, M.G.; Marx, H.; Mattanovich, D.; Sauer, M. Six novel constitutive promoters for metabolic engineering of Aspergillus niger. Appl. Microbiol. Biotechnol. 2013, 97, 259–267. [Google Scholar] [CrossRef]
- Punt, P.J.; Dingemanse, M.A.; Kuyvenhoven, A.; Soede, R.D.; Pouwels, P.H.; van den Hondel, C.A. Functional elements in the promoter region of the Aspergillus nidulans gpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene 1990, 93, 101–109. [Google Scholar] [CrossRef]
- el-Enshasy, H.; Hellmuth, K.; Rinas, U. GpdA-promoter-controlled production of glucose oxidase by recombinant Aspergillus niger using nonglucose carbon sources. Appl. Biochem. Biotechnol. 2001, 90, 57–66. [Google Scholar] [CrossRef]
- Zhang, H.; Yan, J.N.; Zhang, H.; Liu, T.Q.; Xu, Y.; Zhang, Y.Y.; Li, J. Effect of gpd box copy numbers in the gpdA promoter of Aspergillus nidulans on its transcription efficiency in Aspergillus niger. FEMS Microbiol. Lett. 2018, 365, fny154. [Google Scholar] [CrossRef]
- Alazi, E.; Knetsch, T.; Di Falco, M.; Reid, I.D.; Arentshorst, M.; Visser, J.; Tsang, A.; Ram, A.F.J. Inducer-independent production of pectinases in Aspergillus niger by overexpression of the D-galacturonic acid-responsive transcription factor gaaR. Appl. Microbiol. Biotechnol. 2018, 102, 2723–2736. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, G.; Diano, A.; Nielsen, J. Recombinant bacterial hemoglobin alters metabolism of Aspergillus niger. Metab. Eng. 2009, 11, 8–12. [Google Scholar] [CrossRef]
- Dhillon, N.; Shelansky, R.; Townshend, B.; Jain, M.; Boeger, H.; Endy, D.; Kamakaka, R. Permutational analysis of Saccharomyces cerevisiae regulatory elements. Synth. Biol. 2020, 5, ysaa007. [Google Scholar] [CrossRef]
- Redden, H.; Alper, H.S. The development and characterization of synthetic minimal yeast promoters. Nat. Commun. 2015, 6, 7810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubertozzi, D.; Keasling, J.D. Marker and promoter effects on heterologous expression in Aspergillus nidulans. Appl. Microbiol. Biotechnol. 2006, 72, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Díaz, J.; Chávez, R.; Larrondo, L.F.; Eyzaguirre, J.; Bull, P. Functional analysis of the endoxylanase B (xynB) promoter from Penicillium purpurogenum. Curr. Genet. 2008, 54, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Ouyang, L.; Qi, J.; Chu, J. Exploration and characterization of hypoxia-inducible endogenous promoters in Aspergillus niger. Appl. Microbiol. Biotechnol. 2021, 105, 5529–5539. [Google Scholar] [CrossRef] [PubMed]
- Rantasalo, A.; Landowski, C.P.; Kuivanen, J.; Korppoo, A.; Reuter, L.; Koivistoinen, O.; Valkonen, M.; Penttila, M.; Jantti, J.; Mojzita, D. A universal gene expression system for fungi. Nucleic. Acids Res. 2018, 46, e111. [Google Scholar] [CrossRef]
- Wang, G.; Jia, W.; Chen, N.; Zhang, K.; Wang, L.; Lv, P.; He, R.; Wang, M.; Zhang, D. A GFP-fusion coupling FACS platform for advancing the metabolic engineering of filamentous fungi. Biotechnol. Biofuels 2018, 11, 232. [Google Scholar] [CrossRef]
- Huang, X.; Lu, X.; Li, J.J. Cloning, characterization and application of a glyceraldehyde-3-phosphate dehydrogenase promoter from Aspergillus terreus. J. Ind. Microbiol. Biotechnol. 2014, 41, 585–592. [Google Scholar] [CrossRef]
- Schape, P.; Kwon, M.J.; Baumann, B.; Gutschmann, B.; Jung, S.; Lenz, S.; Nitsche, B.; Paege, N.; Schutze, T.; Cairns, T.C.; et al. Updating genome annotation for the microbial cell factory Aspergillus niger using gene co-expression networks. Nucleic. Acids Res. 2019, 47, 559–569. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.H.; Zheng, X.M.; Cairns, T.C.; Zhang, Z.D.; Wang, D.P.; Zheng, P.; Sun, J.B. Disruption or reduced expression of the orotidine-5’-decarboxylase gene pyrG increases citric acid production: A new discovery during recyclable genome editing in Aspergillus niger. Microb. Cell Fact. 2020, 19, 76. [Google Scholar] [CrossRef]
- Carvalho, N.D.; Arentshorst, M.; Jin Kwon, M.; Meyer, V.; Ram, A.F. Expanding the ku70 toolbox for filamentous fungi: Establishment of complementation vectors and recipient strains for advanced gene analyses. Appl. Microbiol. Biotechnol. 2010, 87, 1463–1473. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Shen, B.; Zhang, C.; Huang, X.; Zhang, Y. sgRNAcas9: A software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS ONE 2014, 9, e100448. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zheng, P.; Sun, J.; Kun, Z.; Ma, Y. Heterologous and endogenous U6 snRNA promoters enable CRISPR/Cas9 mediated genome editing in Aspergillus niger. Fungal. Biol. Biotechnol. 2018, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Yu, J.; Cairns, T.C.; Zhang, L.; Zhang, Z.; Zhang, Q.; Zheng, P.; Sun, J.; Ma, Y. Comprehensive improvement of sample preparation methodologies facilitates dynamic metabolomics of Aspergillus niger. Biotechnol. J. 2019, 14, e1800315. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Cairns, T.C.; Ni, X.; Zhang, L.; Zhai, H.; Meyer, V.; Zheng, P.; Sun, J. Comprehensively dissecting the hub regulation of PkaC on high-productivity and pellet macromorphology in citric acid producing Aspergillus niger. Microb. Biotechnol. 2022, e14020. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic. Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Storms, R.; Zheng, Y.; Li, H.; Sillaots, S.; Martinez-Perez, A.; Tsang, A. Plasmid vectors for protein production, gene expression and molecular manipulations in Aspergillus niger. Plasmid 2005, 53, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Dave, K.; Punekar, N.S. Utility of Aspergillus niger citrate synthase promoter for heterologous expression. J. Biotechnol. 2011, 155, 173–177. [Google Scholar] [CrossRef]
- Meyer, V.; Arentshorst, M.; El-Ghezal, A.; Drews, A.C.; Kooistra, R.; van den Hondel, C.A.; Ram, A.F. Highly efficient gene targeting in the Aspergillus niger kusA mutant. J. Biotechnol. 2007, 128, 770–775. [Google Scholar] [CrossRef]
- Aleksandrov, R.; Hristova, R.; Stoynov, S.; Gospodinov, A. The chromatin response to double-strand dna breaks and their repair. Cells 2020, 9, 1853. [Google Scholar] [CrossRef] [PubMed]
- Mathis, H.; Margeot, A.; Bouix, M. Optimization of flow cytometry parameters for high-throughput screening of spores of the filamentous fungus Trichoderma reesei. J. Biotechnol. 2020, 321, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Steiger, M.G. Flow Cytometry for Filamentous Fungi. Methods Mol. Biol 2021, 2234, 147–155. [Google Scholar]
- Yang, Y.J.; Liu, Y.; Liu, D.D.; Guo, W.Z.; Wang, L.X.; Wang, X.J.; Lv, H.X.; Yang, Y.; Liu, Q.; Tian, C.G. Development of a flow cytometry-based plating-free system for strain engineering in industrial fungi. Appl. Microbiol. Biotechnol. 2022, 106, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Beneyton, T.; Wijaya, I.P.; Postros, P.; Najah, M.; Leblond, P.; Couvent, A.; Mayot, E.; Griffiths, A.D.; Drevelle, A. High-throughput screening of filamentous fungi using nanoliter-range droplet-based microfluidics. Sci. Rep. 2016, 6, 27223. [Google Scholar] [CrossRef]
- Lu, X.; Sun, J.; Nimtz, M.; Wissing, J.; Zeng, A.P.; Rinas, U. The intra- and extracellular proteome of Aspergillus niger growing on defined medium with xylose or maltose as carbon substrate. Microb. Cell Fact. 2010, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Laothanachareon, T.; Bruinsma, L.; Nijsse, B.; Schonewille, T.; Suarez-Diez, M.; Tamayo-Ramos, J.A.; Martins Dos Santos, V.A.P.; Schaap, P.J. Global transcriptional response of Aspergillus niger to blocked active citrate export through deletion of the exporter gene. J. Fungi 2021, 7, 409. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Motif name | Sequence (5′ to 3′) 1 | Location |
|---|---|---|
| Motif I | CCGAacaaCTGGcAaaAcaTtCtCGAGAT | −631, −603 |
| Motif II | ATTGGtCcgtacGGcAgC | −560, −543 |
| Motif III (gpdA-box) | CCAaATATCGTGagTCTCCTGCTTTGCCCGGTGTATGAAACCGGAAAgG | −521, −473 |
| Motif IV | GCGGCGCAagcCGGGAA | −453, −437 |
| Motif V | GCggCaTCTGTgcctCCtCCaGGaG | −381, −361 |
| Motif VI | TGAcctGgTCGTTGCGTCAGTC | −293, −272 |
| Gene ID | Name | Function | Foldchange | ||
|---|---|---|---|---|---|
| XMD6.1 vs. D353.8 | XMD7.1 vs. D353.8 | XMD7.1 vs. XMD6.1 | |||
| An17g01710 | cexA | Citrate exporter | 2.79 | 6.12 | 2.19 |
| An12g06930 | amyA | Amylase | 2.03 | 3.76 | 1.85 |
| An03g06550 | glaA | Glucoamylase | 2.83 | 6.49 | 2.29 |
| An02g03540 | mstC | Low affinity glucose transporter | 1.25 | 1.58 | 1.26 |
| An05g01290 | mstG | High affinity glucose transporter | 1.25 | 1.39 | 1.11 |
| An15g03940 | mstH | High affinity glucose transporter | 2.73 | 5.75 | 2.11 |
| An04g06910 | amyR | Transcription factor | 2.35 | 2.59 | 1.10 |
| An08g02260 | pgkA | Phosphoglycerate kinase | 1.32 | 1.37 | 1.04 |
| An11g02550 | ppcA | Phosphoenolpyruvate carboxykinase | 2.57 | 4.30 | 1.67 |
| An07g02100 | pfkB | 6-phosphofructo-2-kinase | 1.46 | 1.90 | 1.31 |
| An02g12140 | gsdA | Glucose-6-phosphate dehydrogenase | 0.88 | 0.71 | 0.80 |
| An02g02930 | rpiB | Ribose-5-phosphate isomerase | 1.35 | 1.68 | 1.24 |
| An11g11230 | ctpA | Citrate/malate carrier protein | 1.57 | 1.26 | 0.80 |
| An08g10530 | acoA | cis-aconitase | 0.66 | 0.52 | 0.79 |
| An11g00510 | acsA | ATP-citrate lyase | 0.45 | 0.46 | 1.02 |
| An11g00530 | acsB | ATP-citrate lyase | 0.51 | 0.50 | 0.98 |
| An10g00820 | oahA | Oxaloacetate acetylhydrolase | 0.19 | 0.62 | 3.23 |
| An15g00070 | mdhAc | Cytoplasmic malate dehydrogenase | 1.26 | 1.44 | 1.14 |
| An11g04810 | aox1 | Alternative oxidase | 1.37 | 2.14 | 1.56 |
| An01g09270 | iclA | Isocitrate lyase | 1.11 | 5.23 | 4.72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Zheng, X.; Wang, Y.; Zhang, L.; Wang, L.; Lei, Y.; Zhang, T.; Zheng, P.; Sun, J. Evaluation of Aspergillus niger Six Constitutive Strong Promoters by Fluorescent-Auxotrophic Selection Coupled with Flow Cytometry: A Case for Citric Acid Production. J. Fungi 2022, 8, 568. https://doi.org/10.3390/jof8060568
Lu Y, Zheng X, Wang Y, Zhang L, Wang L, Lei Y, Zhang T, Zheng P, Sun J. Evaluation of Aspergillus niger Six Constitutive Strong Promoters by Fluorescent-Auxotrophic Selection Coupled with Flow Cytometry: A Case for Citric Acid Production. Journal of Fungi. 2022; 8(6):568. https://doi.org/10.3390/jof8060568
Chicago/Turabian StyleLu, Yudan, Xiaomei Zheng, Yu Wang, Lihui Zhang, Lixian Wang, Yu Lei, Tongcun Zhang, Ping Zheng, and Jibin Sun. 2022. "Evaluation of Aspergillus niger Six Constitutive Strong Promoters by Fluorescent-Auxotrophic Selection Coupled with Flow Cytometry: A Case for Citric Acid Production" Journal of Fungi 8, no. 6: 568. https://doi.org/10.3390/jof8060568
APA StyleLu, Y., Zheng, X., Wang, Y., Zhang, L., Wang, L., Lei, Y., Zhang, T., Zheng, P., & Sun, J. (2022). Evaluation of Aspergillus niger Six Constitutive Strong Promoters by Fluorescent-Auxotrophic Selection Coupled with Flow Cytometry: A Case for Citric Acid Production. Journal of Fungi, 8(6), 568. https://doi.org/10.3390/jof8060568