Abstract
Fungi involved in lichen symbioses produce a large array of secondary metabolites that are often diagnostic in the taxonomic delimitation of lichens. The most common lichen secondary metabolites—polyketides—are synthesized by polyketide synthases, particularly by Type I PKS (TI-PKS). Here, we present a comparative genomic analysis of the TI-PKS gene content of 23 lichen-forming fungal genomes from Ascomycota, including the de novo sequenced genome of Bacidia rubella. Firstly, we identify a putative atranorin cluster in B. rubella. Secondly, we provide an overview of TI-PKS gene diversity in lichen-forming fungi, and the most comprehensive Type I PKS phylogeny of lichen-forming fungi to date, including 624 sequences. We reveal a high number of biosynthetic gene clusters and examine their domain composition in the context of previously characterized genes, confirming that PKS genes outnumber known secondary substances. Moreover, two novel groups of reducing PKSs were identified. Although many PKSs remain without functional assignments, our findings highlight that genes from lichen-forming fungi represent an untapped source of novel polyketide compounds.
1. Introduction
Fungi synthesize an extensive array of chemically and functionally diverse natural products, termed secondary metabolites, with roles in defense, self-protection and development [1,2]. Based on their properties and the core enzymes and precursors involved in their biosynthesis, four major groups of fungal secondary metabolites are distinguished: polyketides, non-ribosomal peptides (NRPS), terpenoids and tryptophan derivatives [3]. Most fungal secondary metabolites are encoded by genes located adjacent to each other (i.e., “clustered”) in the genome [2,4].
In lichen-forming fungi, polyketides are the most common class of secondary metabolites [5,6]. Although lichen polyketides can be produced by mycobionts grown axenically under appropriate conditions [7,8,9,10], they are usually formed in intact symbioses [11,12]. Many lichen polyketides are synthesized by Type I polyketide synthases (TI-PKS) [13], the closest structural and functional analogue of which is the mammalian fatty acid synthase [14]. In the minimal configuration, the domain structure of TI-PKSs always includes a ketoacyl synthase (KS), an acyltransferase (AT), and an acyl carrier protein (ACP). These domains are essential for polyketide synthesis [3]. This configuration can be supplemented with domains such as starter unit-ACP transacylase (SAT), ketoreductase (KR), dehydratase (DH), enoyl reductase (ER), methyltransferase (CMeT), and thioesterase (TE) [3].
The presence of optional domains has led to the classification of fungal PKSs into three subgroups based on their ability to perform redox reactions. The first subgroup comprises non-reducing (NR) PKSs, which lack reductive domains and mainly produce aromatic polyketides [13,15]. The second subgroup contains partially reducing (PR) PKSs, typically having a single KR or a KR and DH domain. Finally, reducing (R) PKSs contain a complete set of reductive domains, viz. KR, DH, and ER. All three subgroups can be found in lichen-forming fungi. Previous work suggests various ecological roles of polyketides in lichens ranging from light-screening and chemical weathering to allelopathic effects and herbivore defense [3,6,16,17]. More broadly, secondary metabolite profiles including polyketides are often characteristic of taxonomic groups and are thus extensively used to identify lichens.
The first PKS gene from a lichen-forming fungus was cloned and analyzed by Armaleo et al. in 2011 [18], and in recent years secondary metabolite research has benefited from genome mining approaches in bacteria, fungi, and plants, uncovering hidden diversity (e.g., [19,20,21]). The fact that genes encoding natural product biosynthetic pathways are often clustered in the genome [2] facilitates the identification of biosynthetic gene clusters (BGCs) in whole genome sequences. In many cases, the chemical structure of their products can be predicted from the biosynthetic logic of enzymes encoded in a BGC and their similarity to known counterparts [22,23,24,25]. Despite this progress, the biosynthetic genes for most secondary metabolites of lichen-forming fungi remain uncharacterized. Observations that lichen-forming fungi contain more biosynthetic genes than characterized chemical compounds also complicates clear gene assignments to particular metabolites. Consequently, previous studies have focused only on a few well-known compounds, e.g., usnic acid, grayanic acid, and atranorin (e.g., [18,25,26,27,28,29]). However, the increasing availability of lichen-forming fungal genomes provides a largely untapped resource for identifying additional biosynthetic genes [30].
Here, we investigate the diversity of BGCs in the de novo sequenced Bacidia rubella (Hoffm.) A. Massal. (Ramalinaceae) genome within a two-level comparative genomic framework. We aim to identify genes involved in the biosynthesis of atranorin, a secondary metabolite present in many lichens including B. rubella. To achieve this, we examine the secondary metabolite biosynthetic potential of B. rubella by comparing its genome to that of Ramalinaceae species: Bacidia gigantensis, Ramalina intermedia and R. peruviana. The secondary metabolites of these four closely-related species are known, and the chemical substance profiles overlap. This facilitates comparison of the presence, absence and structure of PKS genes, and enables the evaluation of BGCs for previously characterized genes, including those encoding for atranorin biosynthesis. We then extend this comparative approach to include an additional nineteen, publicly available fungal genomes from the Lecanoromycetes. Most of these genomes were obtained from pure cultures, with high genome completeness and little contamination. By employing an in-silico approach combined with phylogenetic reconstructions of previously characterized sequences, we gain insight into the putative functions of TI-PKS BGCs in lichen-forming fungi. Our comparative genomic results are congruent with previous work, indicating a high diversity of BGCs in lichen-forming fungi that extends beyond what can be observed from chemical profiles (e.g., [9,25]). This sheds new light on the potential of lichen–forming fungi to produce different secondary metabolites, with direct relevance for natural product research and production.
2. Materials and Methods
2.1. In Vitro Cultivation of the Bacidia rubella Mycobiont
The lichen-forming fungus Bacidia rubella was axenically cultivated from a specimen collected from Germany (Bavaria, Lkr. Neuburg-Schrobenhausen, Markt Rennertshofen, south-east of Bertoldsheim, Naturwaldreservat “Mooser Schütt”; mixed forest, on bark of the trunk of Fraxinus sp., ca. 1.0 m above the ground, ca. 400 m asl.; M-0307710) in April 2019. The mycobiont culture was obtained from a multispore discharge of a single apothecium of B. rubella following the method of Yoshimura et al. [31]. Briefly, young and middle-aged apothecia were detached from the thallus and soaked in sterile water for about an hour. Then, they were fixed to the top of a Petri dish lid using petroleum jelly while keeping the lid slightly open to let the apothecia dry slowly. We used Bold’s Basal Media (BBM) with doubled nitrate (25 g/L) as an initial substrate. Upon germination, the spores were transferred to a malt-yeast extract medium (Lichen medium) [32]. The mycobiont cultures were stored in a growth chamber (Wachstumsschrank Binder KBWF 720; Binder GmbH, Tuttlingen) at 16 °C and 60% relative humidity. They were subcultured every two to three months until a sufficient biomass for genomic analysis was obtained (ca. one year).
2.2. DNA Isolation and Sequencing
To obtain concentrated high-molecular-weight genomic DNA, about 1 cm2 of mycelium was taken and ground in liquid nitrogen with a pre-cooled pistil. Genomic DNA was isolated using the MagAttract HMW DNA Kit following the manufacturer’s protocol and subsequent purification steps with magnetic beads (HMW genomic DNA from fresh or frozen tissue protocol), resulting in a total yield of 1 µg. The final concentration was 9.53 ng/µL, measured with a NanoDrop 1000 spectrophotometer (Peqlab Biotechnologie, GmbH, Erlangen, Germany) and Qubit 4 Fluorometer (ThermoFisher Scientific, Waltham, MA, USA) using 1X dsDNA HS Assay Kits. DNA extraction, PCR amplification and Sanger sequencing (using nrITS primers) were performed to evaluate possible contamination and confirm the cultures’ identities. First, a paired-end library was constructed using Illumina DNA Prep (earlier known as Nextera DNA Flex Library Prep) and sequenced on NovaSeq 6000 (NovaSeq SP 150 bp Paired-end Flow Cell, Illumina) at the Biomedical Sequencing Facility (BSF, Vienna, Austria). A total concentration of 0.2 µg was used for Illumina library preparation. To supplement the Illumina data, we prepared several Oxford Nanopore libraries using the SQK-LSK109 kit and sequenced them on a MinION sequencer using R9.4.1 flow cells (altogether, four runs were conducted). The total yield of DNA was in the range of 0.12–0.31 µg.
2.3. Data Generation and Initial Read-Quality Assessment
To obtain a high-quality genome, we used a hybrid assembly approach using Illumina short-reads and Oxford Nanopore long reads. In total, we produced 17 Gbp of raw paired-end Illumina reads with 500× coverage. Raw data inspection with FastQC v0.11.7 [33] indicated high-quality reads (quality score (Q) above 34) and no excessive adapter contamination or read duplication. Basecalling of Nanopore raw signals was performed using Flappie v2.1.3 (git commit 4de542f; https://github.com/nanoporetech/flappie) into a total of 22 Gbp of raw sequences up to 94.5 Kb of read length.
2.4. Genome Completeness and Quality Assessment
The paired-end Illumina reads and unpaired nanopore reads were assembled using hybridSPAdes [34] with k-mer sizes of 33, 55, 77 and 127. To examine the assembly for potential non-target contigs, de novo assembly was subjected to BLASTX using DIAMOND [35] against a custom database comprising the protein sets of the NCBI nr database (downloaded in July 2018). The results of this DIAMOND search were used as input for blobtools v1.1.1 [36]. The final results showed the absence of foreign contigs; therefore, further filtering was unnecessary. The quality of the assembly and genome statistics were assessed using QUAST v5.0.2 [37]. The completeness of the genomes was assessed using all single-copy BUSCO genes of the ascomycota_odb9 set (part of the phylociraptor pipeline; git commit 4cfd3c4; accessed on 2 July 2021; see below).
2.5. Dataset Construction
The dataset consists of the de novo sequenced genome of B. rubella and twenty-two additional representative genomes of Lecanoromycetes, the largest radiation of lichen-forming fungi. We included in our study twenty-three genomes obtained from pure fungal cultures with the exception of B. gigantensis and R. intermedia, which were obtained by sequencing the whole thallus (Table 1). The dataset aims to identify known and possible unknown BGCs placing them together with previously characterized PKSs from other fungi and bacteria. The genomes were downloaded using phylociraptor, which automatically downloads genomes from NCBI and combines them with additionally specified genomes provided by the user [38]. We kept the original taxon names from NCBI for convenience, even though the current taxonomic status might differ.

Table 1.
Starting material, sequencing strategy and genome quality statistics of Ramalinaceae genomes analyzed in this study.
2.6. Genome Annotation
The publicly available genomes were sequenced with different sequencing technologies at different times and are thus of varying quality. To make annotations comparable, we performed ab initio gene calling and functional annotations for all of them. The downloaded genomes were analyzed using the smsi-funannotate (https://github.com/reslp/smsi-funannotate; git commit 398a144; accessed on 4 June 2021) pipeline based on funannotate v.1.8.7 [39]. First, we removed duplicated identical contigs (funannotate clean) in each assembly. To avoid long contig/scaffold names, we sorted our assembly by contig length and then renamed the fasta headers (funannotate sort). Afterwards, we made a soft repeat masking of the assembly using tantan (funannotate mask) [40]. The following steps included gene prediction using the gene-callers Augustus v3.3.2 [41], snap [42], GlimmerHMM v3.0.4 [43] and Genemark ES v4.68 [44]. For Augustus, we used Aspergillus nidulans as a pre-trained species. All the pipelines were run on the cluster of the University of Graz and our in-house Linux Server at LMU Munich. The genome statistics are summarized in Table 4 (See Section 3.2. Biosynthetic Gene Composition in Twenty-Three Annotated Fungal Genomes). The output files of the newly annotated genomes from NCBI and JGI used in this study are available on figshare (http://doi.org/10.6084/m9.figshare.19487837).
2.7. Annotation of Biosynthetic Gene Clusters
The identification, annotation and analysis of secondary metabolite BGCs in the studied fungal genome sequences were performed with antiSMASH v6.0 as a part of the smsi-funannotate pipeline and thus all BGC numbers are referred to antiSMASH prediction. We chose all BGCs from the annotation results that contained orthologous core TI-PKSs for our comparative genomic and phylogenetic analyses. The GBK output files with annotated BGCs and predicted genes were visualized on the antiSMASH webserver (fungal version; accessed: November 2021) [45].
2.8. Identification of Atranorin Biosynthetic Gene Cluster and Phylogeny
To identify atranorin candidate genes, we downloaded sequences from PKS16 of Cladonia grayi (GenBank: ADM79459; 2089 aa) and PKS23 of Stereocaulon alpinum (GenBank: QXF68953; 2500 aa). Both PKSs were previously reported as possible candidates involved in the biosynthesis of atranorin (see Section 3.1.1 Atranorin in Section 3 Results and Discussion for details). As atranorin synthesis involves an oxidation, the cluster must contain a cytochrome P450 as well as additional genes, O-methyltransferase (OMT), and transporter gene [29]. The presence of these genes was verified by our annotation results, showing OMT (PF08241), cytochrome P450 (PF00067), and transporter gene (PF00400) present in the putative atranorin BGC in B. rubella. The gene arrows plot, comparing two atranorin clusters in Bacidia rubella and Cladonia rangiferina, was drawn using the gggenes v0.4.1 R package (https://github.com/wilkox/gggenes/; accessed on 20 April 2022).
We used BLASTP v2.9.0+ [46] to detect the orthologs of those characterized sequences in all PKS sequences we identified on the twenty-three studied genomes. We filtered BLAST output to retain sequences with a minimum identity of 30% over the alignment length and a minimum query coverage of 50%, sorted for the highest bit score and lowest e-value. Additionally, we used the best blast subject hits with the highest percent similarity to the query gene. The best hits from the blast results (>30%) were selected for the PKS23 alignment. The taxon selection was confirmed with the clade from the large TI-PKS phylogenetic tree.
We aligned the 29 identified amino acid sequences using MAFFT v7.480 [47] and calculated the maximum likelihood using IQ-TREE v2.1.4 [48] with 1000 ultrafast bootstrap replicates, after selecting the best-fitting substitution model (LG+G8+F) with ModelFinder [49]. The final alignment comprised 29 sequences from 16 taxa, with 2320 amino acid sites, 2071 distinct patterns, 1756 parsimony-informative, 221 singleton sites, and 343 constant sites.
2.9. Topology Test to Confirm Atranorin Biosynthetic Genes
Our maximum-likelihood phylogeny recovered the B. rubella (Ramalinaceae) atranorin gene (bacrubpred_000804) as sister to a clade comprised of Cladonia rangiferina and S. alpinum (claranpred_005882 and QXF68953, respectively), which both belong to Cladoniaceae. Miadlikowska et al. [50] recovered Parmeliaceae as the closest relative of Cladoniaceae, with a clade of these two families being sister to Ramalinaceae. Thus, we tested if the monophyly of a clade comprising Bacidia + Cladoniaceae is significantly supported against the expected phylogenetic relationship comprising Parmeliaceae + Cladoniaceae. For this, we compared (1) an unconstrained ML tree to recover Bacidia + Cladoniaceae as monophyletic, and (2) a constrained tree with Bacidia sister to Cladoniaceae + Parmeliaceae (Figure 1).
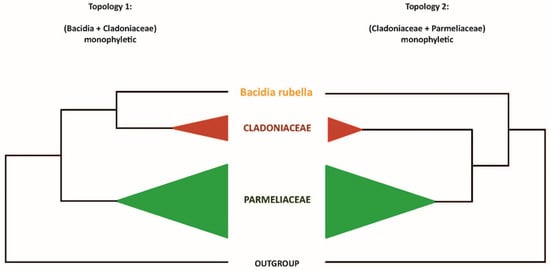
Figure 1.
Two alternative topologies used for comparison in the topology test. Topology 1, recovering Bacidia + Cladoniaceae as monophyletic; Topology 2, recovering Cladoniaceae + Parmeliaceae as monophyletic.
For the topology test, we used PKS23 sequences in the strict sense, i.e., from the species known as atranorin produces with Gyalolechia flavorubescens and Xanthoria elegans as outgroups. The constrained tree can be multifurcating and need not contain all species; therefore, finally, we shortened our tree to the taxa we wanted to test. First, we performed a constrained search for both topologies using the LG model for the amino acid dataset. Then, we concatenated both trees and implemented tree topology tests, based on the Kishino-Hasegawa test [51], Shimodaira–Hasegawa test [52], Expected Likelihood Weight [53], and approximately unbiased (AU) test [54] using IQ-TREE (-m LG -z concatenated_trees.treels -n 0 -zb 10,000 -au). We considered tree topology to be unlikely if its test p-value was <0.05 (marked with a “-” sign). The trees were visualized and examined using FigTree 1.3.1 [55].
2.10. Identification of Orthologues and Orthogroups with BUSCO and OrthoFinder
Orthofinder uses a hybrid approach based on sequence similarity estimated by a BLAST all-vs-all search with DIAMOND and subsequent reconstruction of gene trees and a species tree to identify (single copy) orthologs and paralogs. To identify orthologues of extracted PKSs, we inferred orthogroups with Orthofinder v2.5.4 [56] for (1) all predicted proteins and (2) all extracted TI-PKSs, independently. The independent runs for two datasets were conducted to check for the concordance of the final results. As the results were not contradictory, we discussed the result of TI-PKS only. The Markov Cluster (MCL) inflation parameter was set up by default (1.5). The trees utilized by Orthofinder were reconstructed using Fasttree v2.1.10 (-m msa, -A muscle, and -S diamond (default)) [57].
2.11. Type I Iterative PKS Alignment
We performed a phylogenetic analysis of all TI-PKSs identified in twenty-three studied genomes of Lecanoromycetes, to assess the relationship of non-reducing PKSs (NR-PKS) and reducing PKSs (R-PKS) (including partly reducing PKSs). First, we prepared a list of all identified TI-PKS based on our functional annotations. We retrieved the sequences based on the corresponding gene ID numbers included in the gff3 file of each genome using a custom python script (select_transcript.py: https://github.com/reslp/genomics/blob/master/select_transcripts.py; accessed on 13 October 2021). We included predicted ketoacyl synthase (KS) amino acid alignment reported by Kroken et al. [14] to enhance our dataset. The inclusion of reference PKSs enables us to compare the tree topology proposed in Kroken et al. [14] to our larger sampling of putative PKS genes.
All TI-PKS sequences were combined into a single file and aligned using MAFFT v7.480 [47]. We chose the E-INS-i alignment strategy because it performs better when aligning sequences with several conserved motifs interspersed in long, unalignable regions. We trimmed the alignment using trimAl v.1.4.rev15 [58]. Initial testing of different trimming settings (e.g., -gappyout, -strict, and -automated), showed that parameter combination (-gt 0.70 -resoverlap 0.70 -seqoverlap 60) is a good trade-off between removing ambiguously aligned sites and keeping phylogenetically informative sites of the PKS sequences. The final data matrix consisted of 624 amino acid sequences with 1049 amino acid sites, 1049 distinct patterns, and 1043 parsimony-informative sites from 61 taxa (23 studied genomes and 38 other fungal and bacterial taxa from Kroken et al. [14]). In addition, PKS23 from Stereocaulon alpinum (QXF68953) and PKS16 from Cladonia grayi (ADM79459) from NCBI were included. The alignment is available on figshare (http://doi.org/10.6084/m9.figshare.19487837).
We selected the best-fitting substitution model (LG+F+G4) according to the Akaike and Bayesian Information Criteria using the ModelFinder implemented in IQ-TREE [49] on our TI-PKS alignment. We calculated a phylogenetic tree using maximum-likelihood (ML) analysis implemented in IQ-TREE v2.1.4 [48], with 1000 ultrafast bootstrap replicates [59]. We also calculated a tree using RAxML-NG v1.0.3 [60] with parameters --all --bs-trees 100 --model LG+F+G4 --threads 16 --data-type AA. The inferred phylogenetic tree was then rooted with Bacterial Type II polyketide sequences using phyx [61].
To compare the congruence of the trees, we utilized Dendroscope v3.7.6 [62] using Tanglegram (Algorithms). The resulting tree was visualized using FigTree 1.3.1 [55] and a custom R script, with additional annotations added in Adobe Illustrator v24.0.3.
3. Results and Discussion
3.1. General Characteristics of De Novo Bacidia rubella Genome
We report the de novo assembled genome of the lichen-forming fungus Bacidia rubella obtained from an axenic fungal culture. Our hybrid approach to sequencing, using Illumina short-read with Oxford Nanopore long-read, resulted in a high-quality genome assembly of B. rubella. The final assembly had a size of 33.52 Mb in 51 scaffolds, an N50 of 1.77 Mb (Table 1), and was 98% BUSCO complete (fungi_odb9). Using multiple ab-initio gene-calling methods, 8773 genes were identified (Table 1). The similarity of standard genome metrics between B. rubella and B. gigantensis [63] suggests a high-quality B. rubella genome (Table 1).
The genome of B. rubella contains 31 BGCs (Table 2), including six non-reducing and four reducing TI-PKS sequences. This TI-PKS biosynthetic arsenal is similar to that of B. gigantensis, containing 11 identified TI-PKS genes (seven NR-PKSs and four R-PKSs, respectively; Table 2). Despite the overall similarity of TI-PKS gene numbers in B. rubella and B. gigantensis, our BLAST-based comparison of these biosynthetic clusters showed only two significant hits between the two species: one for R-PKS (bacgigpred_003963 and bacrubpred_000636), and one for NR-PKS (bacgigpred_008278 and bacrubpred_000202), with 62.8 and 63% similarity in amino-acid sequences, respectively. This result is not surprising, given that both fungal species differ in their secondary metabolite profiles. The only secondary compound of B. rubella is atranorin, which is the most widespread secondary metabolite found in the genus Bacidia s. lat. (Ekman 1996) [64]. In contrast, B. gigantensis is currently the only known Bacidia species to produce homosekikaic acid [65].
Table 2.
Overview of biosynthetic gene clusters and polyketide synthase families found in the twenty-three studied fungal genomes. The occurrence of the major secondary substance of B. rubella, atranorin, is highlighted. All sequences except the de novo sequenced B. rubella genome are from NCBI, if not otherwise specified in the first column.
The two species Ramalina intermedia and R. peruviana belong to the same family as the genus Bacidia but differ in having many more BGCs compared to B. gigantensis and B. rubella. In detail, R. intermedia contains 54 BGCs, including thirteen non-reducing, seventeen reducing and one partially reducing TI-PKS sequences. Ramalina peruviana, on the other hand, contains 43 BGCs including nine non-reducing and seven reducing genes, and one partially reducing gene (Table 2).
3.1.1. Atranorin
Atranorin is the main secondary compound known in B. rubella [64], and its biosynthetic pathway has received attention in previous studies on lichen-forming fungi [26,29]. Our phylogenetic results—which included previously identified sequences of genes involved in atranorin production from Cladonia species and Stereocaulon alpinum—revealed a putative PKS23 homolog in B. rubella. This is consistent with the phylogenetic investigation of Kim et al. [29], where PKS23 sequences were reported to group together with sequences from atranorin-producing lichen-forming fungi (see Section 3.3 Type I PKS phylogeny). The domain configuration of B. rubella PKS23 also supports our phylogenetic results, showing the same organization of putative atranorin BGC in B. rubella as has been reported for Cladonia rangiferina (Figure 2). The B. rubella PKS23 cluster contains a cytochrome P450 domain (atr2) required for oxidation, as well as an O-methyltransferase (OMT) domain (atr3) and transporter gene (atr4), which are involved in atranorin biosynthesis [29]. A BLASTp search of the OMT domain (atr3) in B. rubella PKS23 resulted in 36% protein sequence identity to Trt5 (UniProtKB accession no. Q0C8A3). This level of similarity is consistent with previous findings by Kim et al. [29] for C. rangiferina PKS23.
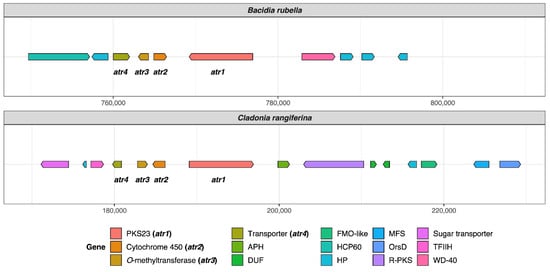
Figure 2.
Organization of putative atranorin BGCs in Bacidia rubella and Cladonia rangiferina. The genes PKS23 (atr1), Cytochrome 450 (atr2), O-methyltransferase (atr3), and transporter gene (atr4) are present on the left side of the gene arrow plot. The BLAST percent similarity between two genes is 73% for atr1, 74% for atr2, 78% for atr3, and 91.5% for atr4. APH, phosphotransferase enzyme family (PF01636); DUF, domain of unknown function; FMO-like, flavin-containing monooxygenase family (PF00743); HCP60, HSP60 chaperone family (PF00118); HP, hypothetical protein; MFS, Major Facilitator Superfamily, transporter gene (PF07690); R-PKS, reducing PKS (in this case from R-VIII group); OrsD, Orsellinic acid/F9775 biosynthesis cluster protein D family (PF12013); TFIIH, Ssl1-like protein, subunit of the transcription factor II H complex (PF04056); Sugar_tr, Sugar transporter family (PF00083); WD-40, WD40 repeat (PF00400).
The newly identified, putative PKS23 sequence of B. rubella was recovered as a sister to PKS23 sequences from C. rangiferina and S. alpinum, suggesting a sister-group relationship of Bacidia with Cladoniaceae (See Figure 3 in Section 3.3: Type I PKS Phylogeny), whereby this clade formed a sister-group to the PKS23 sequences from Parmeliaceae (schematically shown in Figure 1).
This is in contrast to the family-level taxonomy previously established, e.g., by Peršoh et al. [66] and Miadlikowska et al. [50], where Cladoniaceae is more closely related to Parmeliaceae than to Ramalinaceae. To test the monophyly of the clade comprising Bacidia + Cladoniaceae compared to a group comprised of Parmeliaceae and Cladoniaceae, we performed a phylogenetic tree topology test on the PKS23 dataset. Specifically, we compared the unconstrained ML tree recovering Bacidia + Cladoniaceae as monophyletic (Topology 1) versus the constrained tree recovering Cladoniaceae + Parmeliaceae as monophyletic and Bacidia sister to that branch (Topology 2; Figure 1).
Our results showed that the topology constraining Cladoniaceae + Parmeliaceae as monophyletic is significantly less likely (0.8%, Figure 1, Table 3). Possible explanations are that the ancestor of Bacidia may have acquired the PKS23 gene from an ancestor of Cladoniaceae via horizontal gene transfer or that the PKS23 genes were generated by convergent evolution. It is obvious that these hypotheses require additional testing with augmented sampling of PKS23 sequences from Cladoniaceae, Parmeliaceae, and Ramalinaceae. Both scenarios may also explain the scattered occurrence of atranorin in Ramalinaceae.
Table 3.
Parameters for the topology test. Topology 1, recovering Bacidia + Cladoniaceae as monophyletic; Topology 2, recovering Cladoniaceae + Parmeliaceae as monophyletic. Value for the Kishino–Hasegawa test (Kishino and Hasegawa, 1989), Shimodaira and Hasegawa, 1989), expected likelihood weights (Strimmer and Rambaut, 2002) and approximately unbiased (AU) test (Shimodaira, 2002) are given.
Even though a PKS16 gene from Cladonia grayi was first shown to be involved in grayanic acid production by Armaleo et al. [18], a homolog of this gene from C. rangiferina was later suggested to be involved in atranorin production by Elshobary et al. [26]. In our phylogeny, these genes group together with genes from several other Cladonia and lichen-forming fungi (PKS16 clade, See Figure 3 in Section 3.3: Type I PKS Phylogeny), but not all of them are known as atranorin producers (Table 2). Moreover, atranorin is a B-orcinol depside, and a corresponding biosynthetic gene would require a CMeT domain to add a methyl group to the depside ring. This is not the case for PKS16, which is thought to be involved in the synthesis of orcinol depsides [25]. Our comparison of known metabolites in lichen-forming fungi containing PKS16 does not reveal a clear pattern either (See Figure 3 in section: Type I PKS Phylogeny, Group NR-I, PKS16 clade; Table 2). As such, the biosynthetic role of PKS16 genes remains elusive.
3.1.2. Homosekikaic Acid
Homosekikaic acid is a major secondary metabolite in Bacidia gigantensis as well as in both Ramalina intermedia and R. peruviana. However, it has not been found in B. rubella. To our knowledge, there was no putative PKS reported as being involved in the biosynthesis of homosekikaic acid, and its biosynthesis has not been characterized using gene expression or heterologous expression experiments. To identify candidate genes involved in the biosynthesis of homosekikaic acid in these three species, we used BLASTp on all predicted BGC sequences from the two Bacidia and Ramalina species. We identified three BGC candidates with BLAST similarity ranging from 53 to 93%, but none of them were a TI-PKS. Instead, we recovered Type 3-PKS homologs from B. gigantensis (BGC 1.2), R. intermedia (BGC 6.1) and R. peruviana (BGC 223.1) as potential homosekikaic biosynthetic genes; however, none of B. rubella BGCs showed a high similarity to them. Furthermore, our TI-PKS phylogeny did not reveal any clade with both Ramalina species and B. gigantensis that excluded B. rubella. A possible explanation for the observed pattern is that the BGC responsible for synthesizing homosekikaic acid is present in the genome of B. rubella, but not expressed, and therefore homosekikaic acid is not produced in detectable amount. This hypothesis requires testing with gene expression experiments and analyses of substance profiles.
3.1.3. Other Biosynthetic Genes Identified In Silico in the Two Bacidia Species
Using further in silico analyses with antiSMASH, we were able to identify genes encoding enzymes to synthesize secondary metabolites such as clavaric acid (100% similarity) and squalestatin S1 (40% similarity). These terpenes have been identified in both Bacidia. Moreover, we identified a monascorubrin biosynthetic gene in B. rubella confirmed by 100% BLAST identity to the monascorubrin biosynthetic gene from Talaromyces (Penicillium) marneffei (PKS3: HM070047) [67]. Apart from monascorubrin biosynthesis, PKS3 genes were suggested to be involved in the production of the well-known toxin citrinin, as well as a yellow pigment, ankaflavin [67]. Monascorubrin and its related compounds are polyketides used as natural red colorants for food [68]. In B. rubella, monascorubrin could be responsible for the characteristic orange to the orange-brown coloration of the apothecia. However, these substances have not yet been reported from B. rubella.
In the genome of B. gigantensis, we identified genes possessing high sequence similarity with genes involved in the production of pyranonigrin E (100% similarity to BGC0001124), naphthopyrone (100% similarity to BGC0000107), and melanin (100% similarity to BGC0001265). However, in most cases, only a part of the sequences showed a high percentage of similarity; therefore, our in-silico based report is provisional. Additional detailed studies are necessary to confirm these first functional assignments.
3.2. Biosynthetic Gene Composition in Twenty-Three Annotated Fungal Genomes
The basic statistics for all twenty-three studied genomes are provided in Table 4. According to BUSCO homology searches against the fungal dataset (fungi_odb9), most genomes were highly gene complete. We included only two genomes with completeness below 90%, namely Alectoria sarmentosa (75.8%) and Graphis scripta (88.6%). The number of predicted genes was in the range of 6756 to 11,072. The lowest number of predicted genes was observed for Ramalina peruviana (most likely due to issues in the quality of the assembly given the lower sequencing depth of 2×), and the highest number was observed for Evernia prunastri (Table 4).
Table 4.
Genome basics for twenty-three studied fungal genomes.
We investigated the BGCs predicted in all twenty-three studied genomes that belong to different taxonomic groups and synthesize a plethora of secondary metabolites (Table 2; Table 4). Our results revealed a high number of BGCs, with an average of 48 clusters per genome. The smallest number was recovered in the genome of Lasallia pustulata (26 BGCs), and the highest in Evernia prunastri (86 BGCs), which agrees with the results reported by Calchera et al. based on 15 lichenized genomes [6]. In nearly half the genomes, NR-PKSs are more common than R-PKS (Table 2). This evidence is in contrast to previous results reporting that R-PKS gene numbers exceed the number of NR-PKS genes [6]. As such, a larger data set is necessary to confirm whether R-PKS or NR-PKSs are more numerous in the genomes of lichen-forming fungi. The total number of TI-PKSs identified across all studied genomes was 478. Of those, 44.35% were of the non-reducing (NR), 53.35% of reducing (R), and 2.3% of partly reducing (PR) PKSs. The highest number of PKS genes was found in E. prunastri (36 TI-PKS clusters) and C. rangiferina (34 clusters) and the lowest numbers were in B. rubella (10 TI-PKS clusters), B. gigantensis (11 clusters), and Cyanodermella asteris (10 clusters) (Table 2).
Our broad genomic sampling provides phylogenetic context for the NR-PKS genes of B. rubella and B. gigantensis. The six TI-PKS genes from B. rubella are NR-PKSs (three in Subclade I and three in Subclade II), while for B. gigantensis seven TI-PKS genes are NR-PKSs (all are in Subclade I).
The discrepancy between the large number of recovered TI-PKS sequences and the few experimentally verified secondary metabolites (Table 2) raises questions about the role these genes play in the secondary metabolism, and about the products they produce (e.g., [9,24,25,29]. However, results linking PKS genes to lichen secondary metabolites beyond in silico methods are still scarce.
3.3. Type I PKS Phylogeny
Our maximum-likelihood phylogeny of TI-PKS with a total of 624 sequences recovered from twenty-three fungal genomes and supplemented with previously published sequences is the largest analysis of biosynthetic gene content of various TI-PKS genes in lichen-forming fungi to date.
The TI-PKSs phylogenetic tree is divided into six main subgroups (Figure 3): Bacterial Type II PKS (including bacterial and mitochondrial ketoacyl-ACP-synthetases; used as outgroup), Bacterial Type I PKS (also including some fungal sequences), animal fatty acid synthase (FAS), PR-PKS, NR-PKS, and R-PKS. Lichen PKS genes are distributed across the three of these main subgroups, viz. PR-PKS, NR-PKS, and R-PKS. These groups were also recognized by Kroken et al. [14] and Calchera et al. [6], but both used the single KS domain for tree reconstruction, and the latter subsumed PR-PKS and R-PKS.
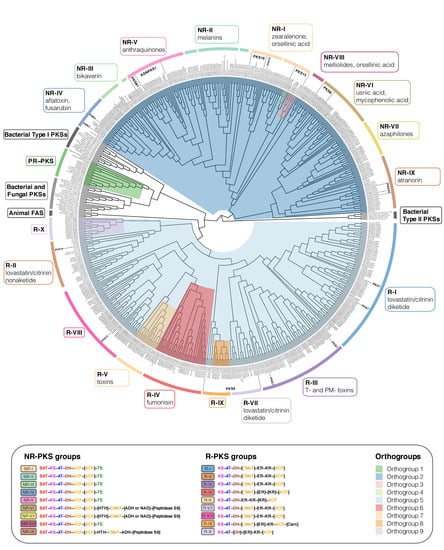
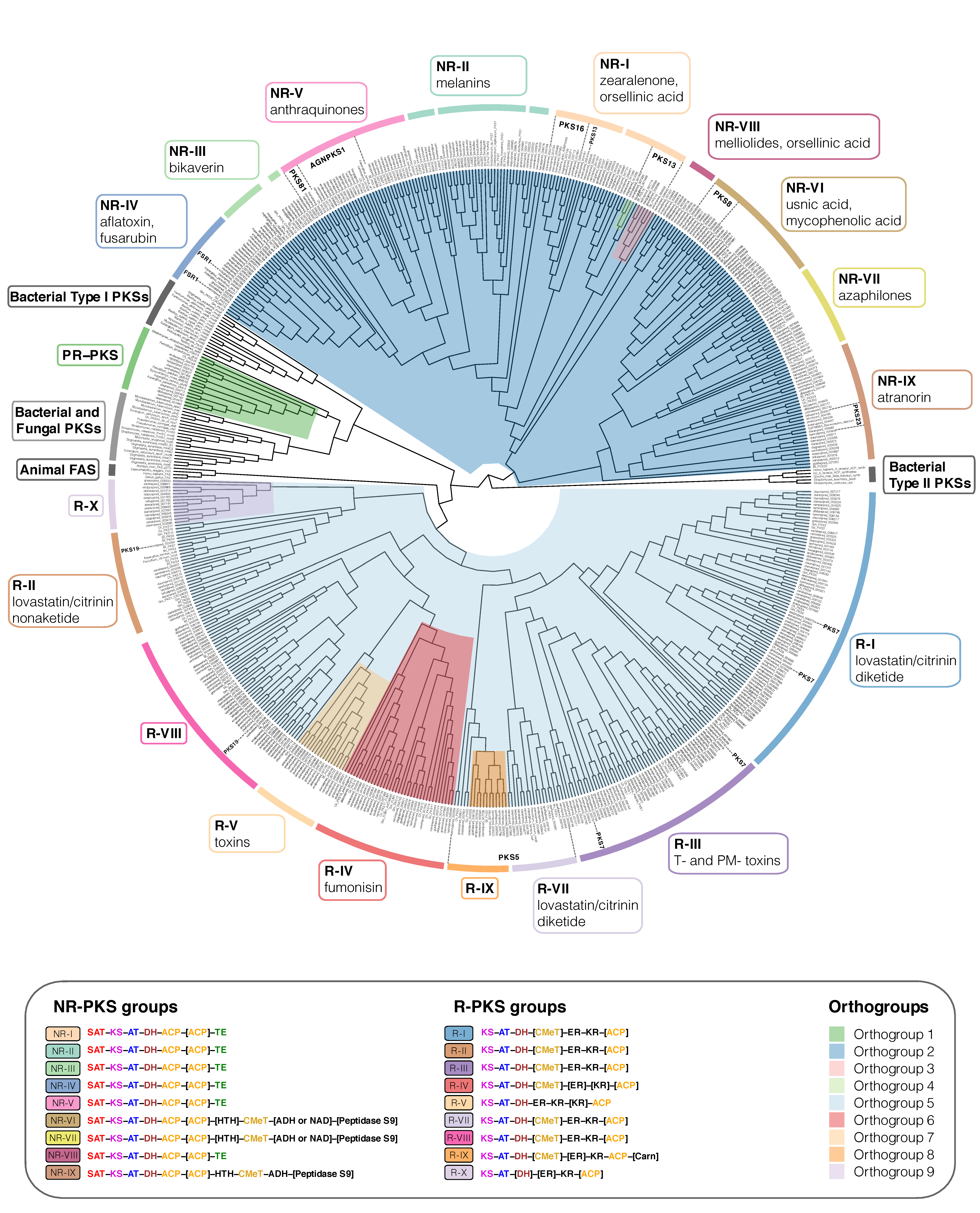
Figure 3.
Maximum-likelihood phylogeny of Type I PKS genes inferred by IQ-TREE using Type II Bacterial PKSs as outgroup. Clades containing lichen-forming fungi are highlighted and the corresponding Orthogroups (1 to 9, respectively) are indicated by different colors. PR-PKS corresponds to Orthogroup 1 (green); the nine NR-PKS groups (NR-I to NR-IX) belong to Orthogroups 2, 3 and 4 (dark blue, pink and light green, respectively); the ten R-PKS groups (R-I to R-X) belong to Orthogroups 5, 6, 7, 8, and 9 (light blue, red, peach, orange, and lilac, respectively). Characteristic secondary substances for the groupings are given in the corresponding colored boxes. Groups not containing lichen-forming fungal genes are indicated by grey boxes. For each group, the domain arrangement of PKS is highlighted with distinct colors: SAT—starter unit-ACP transacylase; KS—ketoacyl synthase; AT—acyltransferase; ACP—acyl carrier protein; KR—ketoreductase; DH—dehydratase; ER—enoyl reductase; CMeT—methyltransferase; TE—thioesterase; HTH—helix-to-helix; ADH—adhydrolase; NAD—NAD-binding; Carn—Choline/Carnitine O-acyltransferase domain.
Figure 3.
Maximum-likelihood phylogeny of Type I PKS genes inferred by IQ-TREE using Type II Bacterial PKSs as outgroup. Clades containing lichen-forming fungi are highlighted and the corresponding Orthogroups (1 to 9, respectively) are indicated by different colors. PR-PKS corresponds to Orthogroup 1 (green); the nine NR-PKS groups (NR-I to NR-IX) belong to Orthogroups 2, 3 and 4 (dark blue, pink and light green, respectively); the ten R-PKS groups (R-I to R-X) belong to Orthogroups 5, 6, 7, 8, and 9 (light blue, red, peach, orange, and lilac, respectively). Characteristic secondary substances for the groupings are given in the corresponding colored boxes. Groups not containing lichen-forming fungal genes are indicated by grey boxes. For each group, the domain arrangement of PKS is highlighted with distinct colors: SAT—starter unit-ACP transacylase; KS—ketoacyl synthase; AT—acyltransferase; ACP—acyl carrier protein; KR—ketoreductase; DH—dehydratase; ER—enoyl reductase; CMeT—methyltransferase; TE—thioesterase; HTH—helix-to-helix; ADH—adhydrolase; NAD—NAD-binding; Carn—Choline/Carnitine O-acyltransferase domain.
- TI-PKS domain content mostly corresponds to phylogeny
TI-PKS genes encode for multi-domain enzymes, with each domain executing a specific function. The order and domain content of the PKSs thus defines the class of polyketides produced by the corresponding BGC. The presence or absence of individual domains has led to the classification of fungal PKSs into three main subgroups. The first subgroup comprises NR-PKSs, which lack reductive domains. The generalized domain content of NR-PKS in fungi studied here is SAT-KS-AT-DH-ACP-[ACP]-HTH-CMeT-[TE or ADH or NAD]-[Peptidase S9] (NR-I to NR-IX; Figure 3).
The second subgroup comprises PR-PKSs containing a single KR domain or KR and DH domains. The generalized domain content in PR-PKS according to our phylogeny is KS-AT-[DH]-KR-ACP (PR-PKS; Figure 3).
Finally, the third subgroup—R-PKSs—contains a complete set of reductive domains, viz. KR, DH, and ER and thus exhibit the following domain structure: KS-AT-DH-CMeT-ER-KR-ACP-[Carn] (R-I to R-X; Figure 3). Additional domains, such as helix-to-helix (HTH), adhydrolase (ADH), NAD-binding (NAD), Peptidase S9, and Choline/Carnitine O-acyltransferase domain (Carn) were not shown in the previous studies of TI-PKS in lichen-forming fungi. We discuss them in detail below in the corresponding sections.
- The discrepancy of grouping PKS genes
Assigning PKS genes to different groups in fungi on the account of gene domain composition and their synthesized products was introduced by Kroken et al. [14] and later refined based on DH-domain pocket sizes by Ahuja et al. [69] and Liu et al. [70]. This classification was used in several studies investigating PKS gene diversity in lichen-forming fungi, e.g., [29,71]. Despite this existing classification, it remains unclear if all groups indeed form monophyletic clades and if all genes from one group synthesize the same (or at least chemically similar) substances. In our phylogeny, previously proposed groups were not always monophyletic (e.g., NR-II; Figure 3), and experimentally characterized genes from one group have been shown to produce different substances (e.g., PKS81 and AGNPKS1 in NR-V; Figure 3). Additionally, we performed an ortholog clustering using Orthofinder on all TI-PKS sequences to provide an objective way of identifying groups of sequences. This resulted in nine orthogroups (Figure 3: colored clades), named Orthogroup 1 to Orthogroup 9, respectively. Orthogroup 1 corresponds to the PR-PKSs (Figure 3). NR-PKSs are spread across Orthogroups 2, 3, and 4 but none of them correspond explicitly to any of the nine groups identified before by Pizarro et al. [71] and Kim et al. [29]. Orthogroups 5, 6, 7, 8, and 9 contain R-PKSs, where only Orthogroup 6 (R-IV) and 7 (R-V) agree with groups defined in Kroken et al. [14] and Punya et al. [72]. However, all Orthogroups identified here form well supported clades in the PKS phylogeny (Figure 3). The discrepancy between our results and previous studies needs to be investigated in subsequent studies. In our phylogeny, several groups of the NR-PKS and R-PKS showed differences in the PKS domains present corresponding to the supported clades; these groups do contain support from different sources and thus merit discussion (see below).
3.3.1. Lichen-Forming Fungi Contain Only a Few Partially Reducing PKSs in the Phylogenetic Neighbourhood to Bacterial PKSs
The PR-PKS sequences formed a well-supported clade sister to bacterial Type I PKS sequences with other fungal PKSs (PR-PKS; Figure 3). Their domain configuration is KS-AT-[DH]-KR-ACP, including only a single reductase domain (KR). In contrast to the large NR- and R-PKS groups, PR-PKS contains genes mainly from Aspergillus and Penicillium, and only a few genes from lichen-forming fungi. Genes from the studied fungal genomes have the typical PR-PKS domain composition, but in two genes from two Ramalina (ramintpred_001715 and ramperpred_002012), the DH domain was missing. PR-PKS genes form a sister clade to Bacterial Type I PKSs (Figure 2). Bacterial T1-PKSs often possess a KR domain catalyzing the first step in the reductive modification of beta-carbonyl centers in the growing polyketide chain. This domain requires NADPH to reduce the keto- to a hydroxy group [73].
The “simple” domain configuration of Bacterial and PR-PKS genes compared to NR- and R-PKSs has not escaped our attention. Our study was not designed to specifically test how PKS genes in Ascomycetes were acquired and how they diversified. However, the placement of Bacterial and NR-PKS sequences as the earliest branches in our phylogeny supports the hypothesis suggested by Kroken et al. [14] that fungal TI-PKS genes could have been acquired by an ancient horizontal gene transfer event between bacteria and fungi. Additionally, our results suggest that ancestral TI-PKS genes may have been partially reducing. Under this scenario, NR- and R-PKS evolution could have been connected to domain structure modification (such as gaining SAT in NR-PKS) and subsequent functional diversification. However, this cannot be concluded with certainty without a greatly expanded sample of genomes from other fungal groups, including genomes from early branching fungal lineages and comprehensive ancestral state reconstructions.
3.3.2. Fungal PKSs Producing Non-Reduced Polyketides
The Diversity of NR-PKS Sequences
In previous studies, NR-PKSs were divided into nine major groups based on protein sequence similarity and PKS domain content [14,29,69,71]. Similar to previous studies, we observed characteristic domain configurations for the different clades in our phylogeny. The domain structure of NR-PKSs in the studied fungi can be generalized as SAT-KS-AT-DH-ACP-[ACP]-HTH-CMeT-[TE or ADH or NAD]-[Peptidase S9]; however, some PKS genes may deviate from this structure. The observed domain content variations are not random but rather occur in two main patterns: either through the duplication of the ACP domain, or through the addition of an N-terminal TE domain. PKS genes possessing an additional ACP domain are scattered throughout different NR-PKS clades, suggesting multiple independent gains. However, the functional significance of ACP domain duplications is still unknown [14].
Most of the NR-PKS sequences belong to a large clade containing groups NR-I to NR-V and NR-VIII, with domain configuration containing a TE domain at the N-terminal end.
The irregularly present additional TE domain is involved in a thioesterase-mediated product release, which is the most common release mechanism in TI-PKS [74]. It regularly extends to a C-C Claisen cyclization domain (TE/CLC domain), e.g., in Aspergillus parasiticus PksA [75]. Although CYC domains have previously been reported from various fungi [14,69,70], we could not identify them in our analyses and thus did not indicate them in the phylogeny.
Subclade I of NR-PKSs (Including Groups NR-I to NR-V, and NR-VIII)
- Group NR-I
The paraphyletic group NR-I contains several clades with previously characterized sequences involved in the biosynthesis of aromatic compounds derived from orsellinic acid, such as grayanic acid (PKS16), physodic acid and olivetoric acid [24,25], lecanoric acid [76], xanthones, aflatoxin, and naphthoquinones [14]. In addition, the clade also comprises PKS13 sequences from several lichen-forming fungi and Gibberella zeae as well as a PKS15 sequence from Botryotinia fuckeliana with unknown functions.
- Group NR-II
Our results show that NR-II is a diverse group, comprised of several clades containing sequences from lichen-forming fungi interspersed with characterized PKS genes proposed to be involved in the biosynthesis of various substances. The earliest branching clade contains genes involved in the 6-hydroxymellein biosynthesis (NR-II; Figure 3), a key intermediate in terrein biosynthesis [77]. Terrein, produced in large quantities by Aspergillus terreus, has phytotoxic activity and has the potential to serve as a novel antibiotic [78]. Our phylogeny revealed sequences of four lichen-forming fungi (clagrapred_001463, clauncpred_002890, psefurpred_002946, and grascrpred_004317) clustering together with terA gene in A. terreus. A BLAST search of terA (GenBank: EAU38791) against these sequences revealed 58 to 67% similarity at the amino acid level, indicating a high degree of conservation despite an approximate 350-million-year split of Eurotiomycetes (Aspergillus) and Lecanoromycetes [79]. Terrein has not been reported from lichen-forming fungi; therefore, without further experiments and detailed substance characterization, terrein production in lichen-forming fungi remains hypothetical.
Another important pattern is the occurrence of characterized melanin genes and melanin precursors in different clades. Melanins are a diverse group of substances that play a role in virulence, morphogenesis or the response to environmental stress, and can be synthesized via different pathways [80]. PKSs produce two forms of melanin: either 1,8-dihydroxynaphtalene (DHN) melanin or Deoxybostrycoidein-melanin [81]. Melanins are insoluble and thus cannot be studied by standard biochemical methods [80]. In the PKS gene survey by Kroken et al. [14], all melanin synthesizing genes were grouped together in a single monophyletic clade. In our analysis, they are recovered in at least three clades containing sequences from several Lecanoromycetes and Endocarpon pusillum. Sister to NR-II is a clade containing sequences from Colletotrichum lagenarium (PKS1), Glarea sp., Nodulisporium sp., and C. heterostrophus (PKS18), which were also characterized as producers of melanin [14]. The high number of genes from lichen-forming fungi close to characterized melanin biosynthetic genes suggests an essential role of melanins in lichens.
- Groups NR-III and NR-IV
The sister groups NR-III and NR-IV contain sequences of proteins producing large polyketide chains, such as conidial yellow pigment Alb1 (NR-III) or aflatoxin/sterigmatocystin of A. nidulans (NR-IV) [14,71,82].
NR-III additionally contains characterized PKSs producing duclauxin (35–71% similarity) and naphthopyrone (100% similarity). Duclauxins are dimeric, heptacyclic fungal polyketides with various activities [83], while bis-naphthopyrones act in herbivore defense in filamentous ascomycetes [84].
Several genes recovered in the NR-IV group are potentially involved in producing different fungal pigments. The genes from Ramalina intermedia (ramintpred_005964) and Cladonia grayi (clagrapred_006320) were assigned to an FSR1 gene, which is involved in producing highly pigmented naphthoquinones fusarubins responsible for fruiting body coloration of Fusarium fujikuroi [85]. A BLAST search of the R. intermedia gene (ramintpred_005964) revealed 72.3% similarity to a PKS of Cladonia metacorallifera (GenBank: QIX11499) that is involved in the biosynthesis of the red compound cristazarin, characterized by antibacterial and antitumor activity [86]. Sequences of Bacidia rubella and several Cladonia species producing red pigments were recovered sister to the putative Ramalina intermedia FSR1 gene. Interestingly, R. intermedia, at the same time, lacks red pigments [87]. This close phylogenetic proximity raises the possibility that combinations of substances synthesized by different PKS genes could be responsible for red-colored pigments (see also discussion on monascorubrin above), particularly for the red fruiting bodies in B. rubella.
- Group NR-V
According to the previous studies, NR-V contains PKSs without the TE domain [71]; however, in the characterized PKSs in our phylogeny, the TE or R domains were present in all clades. The genes in the NR-V group are suggested to be involved in the production of different mycotoxins, such as desertorin (Aspergillus nidulans) and atrochrysone (Aspergillus fumigatus) [71,88,89].
The crown clades in the NR-V group include the annotated PKS81 orthologs from Cladonia metacorallifera, C. macilenta, Lasallia pustulata and L. hispanica as sister to a PKS19 sequence from Cochliobolus heterostrophus and two clades with characterized Agnpks1 also from several lichen-forming fungi. Agnpks1 (Atrochrysone carboxylic acid synthase) was originally described from Penicillium divaricatum and is involved in the biosynthesis of agnestins and dihydroxy-xanthone metabolites [90]. Xanthones and related benzophenones are produced by various filamentous fungi. They exhibit insecticide, antioxidant, antibacterial, anti-inflammatory, and anticancer activities [91]. Examples include desmethyl-sterigmatocystin, a key intermediate of the aflatoxin group of mycotoxins produced by Aspergillus flavus, and a norlichexanthone from the lichen-forming Lecanora straminea [90]. An additional BLAST search of norlichexanthone synthase from L. straminea (GenBank: D7PI15) against our annotated fungal sequences showed at least 37% similarity to sequences from the clade containing PKS81 and Agnpks1. The relationship between these two genes remains unclear, but given our results, PKS81 might belong to or have evolved from Agnpks1. Additional closer investigations will be necessary to show the role of PKS81 and Agnpks1 orthologs in lichen-forming fungi and their possible role in xanthone biosynthesis.
Subclade II of NR-PKSs (Including Groups NR-VI, NR-VII, and NR-IX)
The second largest clade of NR-PKSs includes the previously recovered groups NR-VI, NR-VII, and the recently defined group NR-IX [29] (Figure 3). The generalized domain arrangement in these groups is SAT-KS-AT-DH-ACP-[ACP]-[HTH]-CMeT-[ADH/NAD]-[Peptidase S9], deviating from the generalized arrangement for other NR-PKS (see above). Although previously reported from all NR-PKS groups except group NR-V [69,71], we did not observe TE domains in groups NR-VI, NR-VII, and NR-IX. Instead, we observed NAD and ADH domains—in some cases followed by a Peptidase S9 (PFAM: PF00326) domain. Peptidase S9 belongs to proteolytic enzymes that consume serine in their catalytic activity, and they are ubiquitously found in viruses, bacteria, and eukaryotes [92]. We also observed a helix-turn-helix domain (HTH) being inserted after one or two ACP domains in several groups. The HTH domain was previously found in PKSs in fungi but it has not been shown in the arrangement of the NR-PKSs of lichen-forming fungi before. Proteins with an HTH motif are, for example, involved in DNA repair, RNA metabolism, and protein–protein interaction [93], and could thus be involved in developmental or morphogenic processes.
- Group NR-VI
Characterized PKSs in this group include PKS18 and PKS19 genes from Botryotinia fuckeliana. Genes from this group were suggested to be involved in usnic acid biosynthesis [71], and according to Kim et al. [29] they belong to PKS8. Our results show that this group is divided into two clades. The first clade contains genes of known usnic acid producers such as Ramalina intermedia, R. peruviana, Cladonia metacorallifera, C. macilenta, C. uncialis, Evernia prunastri, Alectoria sarmentosa, Letharia columbiana, and Usnea hakonensis, interspersed with genes from lichen-forming fungi known not to produce usnic acid. The second clade includes genes mostly from lichen-forming fungi not producing usnic acid, including a gene from B. rubella (bacrubpred_000551). However, sequences from Cladonia macilenta, C. uncialis, and Evernia prunastri were also recovered in this clade.
- Group NR-VII
The only genes characterized in this group are PKS17 from Botryotinia fuckeliana and PKS21 from Cochliobolus heterostrophus. These genes have been proposed to produce citrinin or lovastatin [14,94,95]. The other identified genes from the lichen-forming fungi in this group had 50 to 70% BLAST similarity to genes involved in the biosynthesis of monascorubrin, citrinin, conidial yellow, azanigerone A, and stipitatic acid (see also discussion on monascorubrin above). As mentioned above, the biosynthetic gene from B. rubella (bacrubpred_007642) has a 100% identity to the monascorubrin biosynthetic gene.
- Group NR-IX
The recently recognized group NR-IX [29] contains several predicted PKS23 sequences, involved in the biosynthesis of atranorin (see sections on 3.1.1 Atranorin and 3.1.2 Homosekikaic acid above) and methylated orsellinic acid derivatives. In contrast to the groups NR-VI and NR-VII, NR-IX sequences always contain an ADH and lack a NAD domain after CMeT, while Peptidase S9 is only sporadically present. It is necessary to note that only one ACP domain was present in the PKS23, including atranorin producers. In addition, other characterized PKSs of this group are from Cochliobolus heterostrophus (PKS22 and PKS23) and Botryotinia fuckeliana (PKS16 and PKS20).
3.3.3. Fungal PKSs Producing Reduced Polyketides
The Diversity of R-PKS Sequences
R-PKS genes are involved in synthesizing various reduced, usually linear polyketides. Many are precursors of toxins that are active in animals (e.g., lovastatin, citrinin, and fumonisin) or plants (e.g., T-toxin and PM-toxin) [96,97,98,99]. We follow this classification in our discussion whenever possible, while also highlighting deviations. Compared to NR-PKSs, less is known about the possible roles of R-PKSs. In our phylogeny, R-PKSs formed a large clade sister to NR-PKSs with many well-supported subclades. Unlike what has been shown in previous studies, all studied R-PKSs here lacked a second ACP domain, and in some cases, ACP was missing. According to Kroken et al. [14], four major groups (R-I to R-IV) of R-PKS genes can be distinguished based on their overall domain structure and produced compounds. This classification was later extended to groups R-V to R-VIII by Punya et al. [72]. Over 70% of the studied fungal sequences could be assigned to groups designated by Kroken et al. [14] and Punya et al. [72] with the general domain content: KS-AT-DH-[CMeT]-ER-KR-[ACP]. Two clades recovered by Punya et al. [72] are missing from our tree, namely R-VI and R-VIII. Clade VI contained uncommon PKS-NRPS hybrids with four additional domains after ACP, viz. condensation (C), adenylation (A), thiolation (T), and reductase (R) [72]. As our study focused on the TI-PKSs, it did not include hybrid NRPS-PKS genes. The sequences from R-VIII were not present in our phylogeny, and are thus not feasible to identify with confidence.
Based on our phylogenetic results, we identified two R-PKS clades that did not match any of the R-PKS groups reported in previous studies. Sequences in those two clades had characteristic domain structures and were recovered as distinct orthogroups in our Orthofinder analysis. This leads us to propose these clades as new reducing PKS groups, R-IX and R-X, following the numbering scheme by Kroken et al. [14] and Punya et al. [72].
Groups of R-PKSs
- Group R-I
Group R-I is the largest group of R-PKSs in our tree. It contains one of the R-PKS genes synthesizing the diketide portion of lovastatin and citrinin, T-toxin, and PM-toxin [14]. Our results show that the genes of this group, recovered in a monophyletic clade by Kroken et al. [14], were nested in different clades in our analysis; therefore, R-I assignment is not complete.
- Group R-II
Group R-II forms a well-supported clade and it was previously characterized by the absence of an ER domain [14]. In our phylogeny, however, ER domain was present in all lichen-forming fungi. The group includes many previously published sequences and orthologs from Cladonia and Letharia species, Xanthoria elegans, Pseudevernia furfuracea, and Ramalina intermedia. The R. intermedia ortholog was assigned to PKS19. It was predicted to be involved in the production of heptaketides with a role in a lesion of rice leaves formation in Magnaporthe oryzae [100]. Two characterized proteins from A. terreus lovB and P. citrinum mlcA synthesize the cyclic nonaketide portion of lovastatin and citrinin.
- Group R-III
Group R-III forms a clade comprised of four sequences previously assigned to this group by Kroken et al. [14], and a sequence from Cyanodermella asteris. PKSs from this clade do not possess a CMeT domain. The experimentally characterized gene in group R-III is C. heterostrophus PKS2, which, along with PKS1, is required for the synthesis of T-toxin [14]. This group contains several sequences from Kroken et al. [14] interspersed with sequences from various lichen-forming fungi.
- Group R-IV
Genes belonging to R-IV may contain a conserved CMeT domain. Thus, their domain configuration somewhat resembles that of groups R-I and R-II. However, N-terminal domains ER, KR, and ACP can be present or absent in R-IV. The only characterized PKS in this group is G. moniliformis FUM1 (Gm_FUM1_AAD43562), which makes the linear polyketide precursor of the toxin fumonisin [98]. This group contains several sequences from Kroken et al. [14] interspersed with sequences from various lichen-forming fungi.
- Group R-V
Sequences in group R-V have similar domain composition like sequences from Orthogroup 7: KS-AT-DH-ER-KR-[KR]-ACP. However, a second KR domain is only present in Dibaeis baeomyces (dibbaepred_000831). We recovered genes from several lichen-forming fungi in this group. Again, experimentally characterized genes from Gibberella moniliformis (Gm_PKS13) and Botryotinia fuckeliana (Bf_PKS10) also recovered here indicate a possible role in toxin biosynthesis (Figure 3) [14].
- Group R-VII
This group contains several previously characterized sequences as well as sequences from R. intermedia, P. furfuracea, G. furfuracea, and C. macilenta where we could assign PKS5 annotations. Additional sequences from lichen-forming fungi also recovered here had no functional annotations; as such the role of these genes remains unclear. The characterized sequences of Aspergillus (Aspergillus_terreus_lovF) and Penicillium (Penicillium_citrinum_mlcB) indicate a role in the production of the diketide portion of lovastatin and citrinin, but they were assigned to group R-I by Kroken et al. [14].
- Novel group R-IX
A new group of reducing PKSs can be delineated based on a specific domain composition and our orthogroup assignment. We tentatively call this group R-IX following the naming scheme of Kroken et al. [14] and Punya et al. [72]. R-IX is sister to group R-VII and differs from all other groups by having a facultative Choline/Carnitine O-acyltransferase domain (Carn) following the ACP domain: KS-AT-DH-[CMeT]-ER-KR-[ACP]-[Carn] (Figure 3). The Carn domain can be found in several eukaryotic acetyltransferases and also at the C terminus of a highly reducing polyketide synthase (SdnO), where it is part of a gene cluster mediating the biosynthesis of glycoside antibiotics sordarin and hypoxysordarin [101].
- Novel group R-X
Similar to R-IX, a second group of reducing PKSs can be identified based on a specific domain composition and orthogroup assignment. This group R-X, recovered as Orthogroup 9, contains only genes from lichen-forming fungi. Together with Group R-II, they share a sister-group relationship to all other R-PKSs in our phylogeny (Figure 3). Genes from this group may not have DH, ER and ACP domains, resulting in the following domain content: KS-AT-[DH]-[ER]-KR-[ACP]. Most fungal genes recovered here are missing the DH and ER domain. However, the genes from Lasallia hispanica (lashispred_001760) and Evernia prunastri (eveprupred_001763) contain a complete domain configuration: KS-AT-DH-ER-KR-ACP.
4. Conclusions and Limitations
This study presents a comprehensive analysis of the PKS gene content of the de novo sequenced genome of B. rubella and twenty-two publicly available genomes of mainly lichen-forming fungi. Our results reveal that a BGC including PKS23 is likely involved in biosynthesis of atranorin in B. rubella. Similar to previous studies (e.g., [6,23,24,29]), we observed a large diversity of PKS genes in lichen-forming fungi as well as B. rubella, much larger than expected based on their recorded secondary metabolite profiles. This highlights the potential of lichen-forming fungi to produce a much higher number of substances than previously assumed, as mentioned by Calchera et al. [6] focusing on foliose lichens. Our in silico approach provides first insights into the possible roles of PKS genes in crustose B. rubella; however, we are aware that this comes with limitations. Only detailed studies of individual BGCs combining gene expression analyses with analytical chemistry and metabolomics will allow thorough testing of the hypotheses proposed here. The prospects for this are promising as an increasing number of studies succeeded in the heterologous expression of lichen-forming fungal genes, (e.g., [29,76,86,102,103]). Subsequent in vivo studies of metabolite profiles in lichen-forming fungi supplemented by high-resolution MS/MS spectra [104] or metabolic profiling based on stable isotope analysis [105] will contribute to our knowledge on how secondary metabolites in lichen-forming fungi are produced and what roles they play. Combined with the growing number of available lichen-forming fungal genomes, we will be able to refine our understanding and formulate novel hypotheses about the biosynthetic potential of lichens.
Author Contributions
Conceptualization, J.V.G., A.B., S.W. and P.R.; methodology, software and formal analysis, J.V.G. and P.R.; investigation, J.V.G. and A.B.; resources and funding acquisition, S.W. and A.B.; data curation, J.V.G. and P.R.; writing—original draft preparation, J.V.G.; writing—review and editing, J.V.G., A.B., S.W. and P.R.; visualization, J.V.G.; supervision, A.B., S.W. and P.R. All authors have read and agreed to the published version of the manuscript.
Funding
J.G. was supported by a BAYHOST fellowship from the Bayerische Staatsministerium für Bildung und Kultus, Wissenschaft und Kunst and an LMU travel grant (April 2021). Laboratory work and genome sequencing were funded by the LMU Munich start-up funds of S.W. and by the Staatliche Naturwissenschaftliche Sammlungen Bayerns, grant SNSB innovativ to A.B.
Data Availability Statement
The data supporting the findings of this study are available open access in figshare at http://doi.org/10.6084/m9.figshare.19487837. These include gff3 files and protein sequences from twenty-two re-annotated genomes downloaded from the NCBI. In addition, the alignment used for the PKS tree calculation is deposited. The de novo sequenced genome of Bacidia rubella is available under the NCBI BioProject accession number PRJNA821848 and on the Sequence Read Archive (SRA).
Acknowledgments
We thank Diego F. Morales-Briones (LMU Munich) for his valuable help in the figure preparation and Elizabeth Joyce (LMU Munich) for improving the English text. Andrea Brandl and Tanja Ernst (both LMU Munich) helped with the lab work. We thank the government of the administrative district of Upper Bavaria for the sampling permit.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Keller, N.P. Fungal Secondary Metabolism: Regulation, Function and Drug Discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.; Mead, M.E.; Steenwyk, J.L.; Raja, H.A.; Oberlies, N.H. Biosynthetic Gene Clusters and the Evolution of Fungal Chemodiversity. Nat. Prod. Rep. 2020, 37, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P.; Turner, G.; Bennett, J.W. Fungal Secondary Metabolism—from Biochemistry to Genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P.; Hohn, T.M. Metabolic Pathway Gene Clusters in Filamentous Fungi. Fungal Genet. Biol. 1997, 21, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huneck, S.; Yoshimura, I. Identification of Lichen Substances. In Identification of Lichen Substances; Huneck, S., Yoshimura, I., Eds.; Springer: Berlin/Heidelberg, Germany, 1996; pp. 11–123. ISBN 978-3-642-85243-5. [Google Scholar]
- Calchera, A.; Dal Grande, F.; Bode, H.B.; Schmitt, I. Biosynthetic Gene Content of the ‘Perfume Lichens’ Evernia prunastri and Pseudevernia furfuracea. Molecules 2019, 24, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culberson, C.F.; Armaleo, D. Induction of a Complete Secondary-Product Pathway in a Cultured Lichen Fungus. Exp. Mycol. 1992, 16, 52–63. [Google Scholar] [CrossRef]
- Culberson, C.F.; Culberson, W.L.; Johnson, A. Characteristic Lichen Products in Cultures of Chemotypes of the Ramalina siliquosa Complex. Mycologia 1992, 84, 705–714. [Google Scholar] [CrossRef]
- Hamada, N. The Effect of Various Culture Conditions on Depside Production by an Isolated Lichen Mycobiont. Bryologist 1989, 92, 310–313. [Google Scholar] [CrossRef]
- Stocker-Wörgötter, E.; Elix, J.A. Secondary Chemistry of Cultured Mycobionts: Formation of a Complete Chemosyndrome by the Lichen Fungus of Lobaria spathulata. Lichenologist 2002, 34, 351–359. [Google Scholar] [CrossRef]
- Hamada, N.; Tanahashi, T.; Miyagawa, H.; Miyawaki, H. Characteristics of Secondary Metabolites from Isolated Lichen Mycobionts. Symbiosis 2001, 31, 23–33. [Google Scholar]
- Molina, M.C.; Crespo, A.; Vicente, C.; Elix, J.A. Differences in the Composition of Phenolics and Fatty Acids of Cultured Mycobiont and Thallus of Physconia distorta. Plant Physiol. Biochem. 2003, 41, 175–180. [Google Scholar] [CrossRef]
- Cox, R.J. Polyketides, Proteins and Genes in Fungi: Programmed Nano-Machines Begin to Reveal Their Secrets. Org. Biomol. Chem. 2007, 5, 2010–2026. [Google Scholar] [CrossRef] [PubMed]
- Kroken, S.; Glass, N.L.; Taylor, J.W.; Yoder, O.C.; Turgeon, B.G. Phylogenomic Analysis of Type I Polyketide Synthase Genes in Pathogenic and Saprobic Ascomycetes. Proc. Natl. Acad. Sci. USA 2003, 100, 15670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, T.P.; Rudd, B.A.M.; Dawson, M.; Lazarus, C.M.; Simpson, T.J.; Cox, R.J. Design and Utility of Oligonucleotide Gene Probes for Fungal Polyketide Synthases. Chem. Biol. 2001, 8, 157–178. [Google Scholar] [CrossRef] [Green Version]
- Lawrey, J.D. Biological Role of Lichen Substances. Bryologist 1986, 89, 111–122. [Google Scholar] [CrossRef]
- Rundel, P.W. The Ecological Role of Secondary Lichen Substances. Biochem. Syst. Ecol. 1978, 6, 157–170. [Google Scholar] [CrossRef]
- Armaleo, D.; Sun, X.; Culberson, C. Insights from the First Putative Biosynthetic Gene Cluster for a Lichen Depside and Depsidone. Mycologia 2011, 103, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Mullins, A.J.; Murray, J.A.H.; Bull, M.J.; Jenner, M.; Jones, C.; Webster, G.; Green, A.E.; Neill, D.R.; Connor, T.R.; Parkhill, J.; et al. Genome Mining Identifies Cepacin as a Plant-Protective Metabolite of the Biopesticidal Bacterium Burkholderia ambifaria. Nat. Microbiol. 2019, 4, 996–1005. [Google Scholar] [CrossRef]
- Nielsen, J.C.; Grijseels, S.; Prigent, S.; Ji, B.; Dainat, J.; Nielsen, K.F.; Frisvad, J.C.; Workman, M.; Nielsen, J. Global Analysis of Biosynthetic Gene Clusters Reveals Vast Potential of Secondary Metabolite Production in Penicillium Species. Nat. Microbiol. 2017, 2, 17044. [Google Scholar] [CrossRef]
- Lichman, B.R.; Godden, G.T.; Buell, C.R. Gene and Genome Duplications in the Evolution of Chemodiversity: Perspectives from Studies of Lamiaceae. Physiol. Metab. 2020, 55, 74–83. [Google Scholar] [CrossRef]
- Medema, M.H.; Fischbach, M.A. Computational Approaches to Natural Product Discovery. Nat. Chem. Biol. 2015, 11, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A Computational Framework to Explore Large-Scale Biosynthetic Diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Armaleo, D.; Dal Grande, F.; Schmitt, I. Depside and Depsidone Synthesis in Lichenized Fungi Comes into Focus through a Genome-Wide Comparison of the Olivetoric Acid and Physodic Acid Chemotypes of Pseudevernia furfuracea. Biomolecules 2021, 11, 1445. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Calchera, A.; Merges, D.; Otte, J.; Schmitt, I.; Grande, F.D. A Candidate Gene Cluster for the Bioactive Natural Product Gyrophoric Acid in Lichen-Forming Fungi. bioRxiv 2022. [Google Scholar] [CrossRef]
- Elshobary, M.E.; Becker, M.G.; Kalichuk, J.L.; Chan, A.C.; Belmonte, M.F.; Piercey-Normore, M.D. Tissue-Specific Localization of Polyketide Synthase and Other Associated Genes in the Lichen, Cladonia rangiferina, Using Laser Microdissection. Phytochemistry 2018, 156, 142–150. [Google Scholar] [CrossRef]
- Abdel-Hameed, M.; Bertrand, R.L.; Piercey-Normore, M.D.; Sorensen, J.L. Identification of 6-Hydroxymellein Synthase and Accessory Genes in the Lichen Cladonia uncialis. J. Nat. Prod. 2016, 79, 1645–1650. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Hameed, M.; Bertrand, R.L.; Piercey-Normore, M.D.; Sorensen, J.L. Putative Identification of the Usnic Acid Biosynthetic Gene Cluster by de novo Whole-Genome Sequencing of a Lichen-Forming Fungus. Fungal Biol. 2016, 120, 306–316. [Google Scholar] [CrossRef]
- Kim, W.; Liu, R.; Woo, S.; Bin Kang, K.; Park, H.; Yu, Y.H.; Ha, H.-H.; Oh, S.-Y.; Yang, J.H.; Kim, H.; et al. Linking a Gene Cluster to Atranorin, a Major Cortical Substance of Lichens, through Genetic Dereplication and Heterologous Expression. mBio 2021, 12, e01111-21. [Google Scholar] [CrossRef]
- Boustie, J.; Tomasi, S.; Grube, M. Bioactive Lichen Metabolites: Alpine Habitats as an Untapped Source. Phytochem. Rev. 2011, 10, 287–307. [Google Scholar] [CrossRef]
- Yoshimura, I.; Yamamoto, Y.; Nakano, T.; Finnie, J. Isolation and Culture of Lichen Photobionts and Mycobionts. In Protocols in Lichenology: Culturing, Biochemistry, Ecophysiology and Use in Biomonitoring; Kranner, I.C., Beckett, R.P., Varma, A.K., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; pp. 3–33. ISBN 978-3-642-56359-1. [Google Scholar]
- Honegger, R.; Zippler, U.; Gansner, H.; Scherrer, S. Mating Systems in the Genus Xanthoria (Lichen-Forming Ascomycetes). Mycol. Res. 2004, 108, 480–488. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 April 2022).
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. HybridSPAdes: An Algorithm for Hybrid Assembly of Short and Long Reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Laetsch, D.; Blaxter, M. BlobTools: Interrogation of Genome Assemblies [Version 1; Peer Review: 2 Approved with Reservations]. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Resl, P.; Hahn, C. Phylociraptor—Rapid Phylogenomic Tree Calculator. 2021. Available online: https://github.com/reslp/phylociraptor (accessed on 20 April 2022).
- Palmer, J.M.; Stajich, J. Funannotate: Eukaryotic Genome Annotation; Zenodo. Available online: https://zenodo.org/record/4054262#.YmeT83ZBxPY (accessed on 15 April 2022).
- Frith, M.C. A New Repeat-Masking Method Enables Specific Detection of Homologous Sequences. Nucleic Acids Res. 2011, 39, e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene Prediction in Eukaryotes with a Generalized Hidden Markov Model That Uses Hints from External Sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Korf, I. Gene Finding in Novel Genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two Open Source Ab Initio Eukaryotic Gene-Finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene Prediction in Novel Fungal Genomes Using an Ab Initio Algorithm with Unsupervised Training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving Cluster Detection and Comparison Capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Miadlikowska, J.; Kauff, F.; Högnabba, F.; Oliver, J.C.; Molnár, K.; Fraker, E.; Gaya, E.; Hafellner, J.; Hofstetter, V.; Gueidan, C.; et al. A Multigene Phylogenetic Synthesis for the Class Lecanoromycetes (Ascomycota): 1307 Fungi Representing 1139 Infrageneric Taxa, 317 Genera and 66 Families. Mol. Phylogenet. Evol. 2014, 79, 132–168. [Google Scholar] [CrossRef] [Green Version]
- Kishino, H.; Hasegawa, M. Evaluation of the Maximum Likelihood Estimate of the Evolutionary Tree Topologies from DNA Sequence Data, and the Branching Order in Hominoidea. J. Mol. Evol. 1989, 29, 170–179. [Google Scholar] [CrossRef]
- Shimodaira, H.; Hasegawa, M. Multiple Comparisons of Log-Likelihoods with Applications to Phylogenetic Inference. Mol. Biol. Evol. 1999, 16, 1114. [Google Scholar] [CrossRef] [Green Version]
- Strimmer, K.; Rambaut, A. Inferring Confidence Sets of Possibly Misspecified Gene Trees. Proc. R. Soc. Lond. B Biol. Sci. 2002, 269, 137–142. [Google Scholar] [CrossRef]
- Shimodaira, H. An Approximately Unbiased Test of Phylogenetic Tree Selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree: Tree Figure Drawing Tool. 2009. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 21 February 2022).
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.W.; Walker, J.F.; Smith, S.A. Phyx: Phylogenetic Tools for Unix. Bioinformatics 2017, 33, 1886–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An Interactive Tool for Rooted Phylogenetic Trees and Networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.L.; Jones, S.J.M.; McMullin, R.T.; Rokas, A. Draft Genome Sequence of the Lichenized Fungus Bacidia gigantensis. Microbiol. Resour. Announc. 2021, 10, e00686-21. [Google Scholar] [CrossRef]
- Ekman, S. The Corticolous and Lignicolous Species of Bacidia and Bacidina in North America. Opera Bot. 1996, 127, 1–148. [Google Scholar]
- McMullin, R.T.; McCune, B.; Lendemer, J.C. Bacidia gigantensis (Ramalinaceae), a New Species with Homosekikaic Acid from the North Shore of Lake Superior in Ontario, Canada. Bryologist 2020, 123, 215–224. [Google Scholar] [CrossRef]
- Peršoh, D.; Beck, A.; Rambold, G. The Distribution of Ascus Types and Photobiontal Selection in Lecanoromycetes (Ascomycota) against the Background of a Revised SSU NrDNA Phylogeny. Mycol. Prog. 2006, 3, 103–121. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lam, C.-W.; Tam, E.W.T.; Lee, K.-C.; Yung, K.K.Y.; Leung, C.K.F.; Sze, K.-H.; Lau, S.K.P.; Yuen, K.-Y. The Biosynthetic Pathway for a Thousand-Year-Old Natural Food Colorant and Citrinin in Penicillium marneffei. Sci. Rep. 2014, 4, 6728. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, R.; Liu, Q.; He, Y.; He, K.; Ding, X.; Kang, L.; Guo, X.; Xie, N.; Zhou, Y.; et al. Orange, Red, Yellow: Biosynthesis of Azaphilone Pigments in Monascus Fungi. Chem. Sci. 2017, 8, 4917–4925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, M.; Chiang, Y.-M.; Chang, S.-L.; Praseuth, M.B.; Entwistle, R.; Sanchez, J.F.; Lo, H.-C.; Yeh, H.-H.; Oakley, B.R.; Wang, C.C.C. Illuminating the Diversity of Aromatic Polyketide Synthases in Aspergillus nidulans. J. Am. Chem. Soc. 2012, 134, 8212–8221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Zhang, Z.; Shao, C.-L.; Wang, J.-L.; Bai, H.; Wang, C.-Y. Bioinformatical Analysis of the Sequences, Structures and Functions of Fungal Polyketide Synthase Product Template Domains. Sci. Rep. 2015, 5, 10463. [Google Scholar] [CrossRef] [Green Version]
- Pizarro, D.; Divakar, P.K.; Grewe, F.; Crespo, A.; Dal Grande, F.; Lumbsch, H.T. Genome-Wide Analysis of Biosynthetic Gene Cluster Reveals Correlated Gene Loss with Absence of Usnic Acid in Lichen-Forming Fungi. Genome Biol. Evol. 2020, 12, 1858–1868. [Google Scholar] [CrossRef]
- Punya, J.; Swangmaneecharern, P.; Pinsupa, S.; Nitistaporn, P.; Phonghanpot, S.; Kunathigan, V.; Cheevadhanarak, S.; Tanticharoen, M.; Amnuaykanjanasin, A. Phylogeny of Type I Polyketide Synthases (PKSs) in Fungal Entomopathogens and Expression Analysis of PKS Genes in Beauveria bassiana BCC 2660. Fungal Biol. 2015, 119, 538–550. [Google Scholar] [CrossRef]
- Bonnett, S.A.; Whicher, J.R.; Papireddy, K.; Florova, G.; Smith, J.L.; Reynolds, K.A. Structural and Stereochemical Analysis of a Modular Polyketide Synthase Ketoreductase Domain Required for the Generation of a Cis-Alkene. Chem. Biol. 2013, 20, 772–783. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Lou, L. PKS and NRPS Release Mechanisms. Nat. Prod. Rep. 2010, 27, 255–278. [Google Scholar] [CrossRef]
- Korman, T.P.; Crawford, J.M.; Labonte, J.W.; Newman, A.G.; Wong, J.; Townsend, C.A.; Tsai, S.-C. Structure and Function of an Iterative Polyketide Synthase Thioesterase Domain Catalyzing Claisen Cyclization in Aflatoxin Biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6246–6251. [Google Scholar] [CrossRef] [Green Version]
- Kealey, J.T.; Craig, J.P.; Barr, P.J. Identification of a Lichen Depside Polyketide Synthase Gene by Heterologous Expression in Saccharomyces cerevisiae. Metab. Eng. Commun. 2021, 13, e00172. [Google Scholar] [CrossRef]
- Zaehle, C.; Gressler, M.; Shelest, E.; Geib, E.; Hertweck, C.; Brock, M. Terrein biosynthesis in Aspergillus terreus and its impact on phytotoxicity. Chem. Biol. 2014, 21, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asfour, H.Z.; Awan, Z.A.; Bagalagel, A.A.; Elfaky, M.A.; Abdelhameed, R.F.A.; Elhady, S.S. Large-Scale Production of Bioactive Terrein by Aspergillus terreus Strain S020 Isolated from the Saudi Coast of the Red Sea. Biomolecules 2019, 9, 480. [Google Scholar] [CrossRef] [Green Version]
- Beimforde, C.; Feldberg, K.; Nylinder, S.; Rikkinen, J.; Tuovila, H.; Dörfelt, H.; Gube, M.; Jackson, D.J.; Reitner, J.; Seyfullah, L.J.; et al. Estimating the Phanerozoic History of the Ascomycota Lineages: Combining Fossil and Molecular Data. Mol. Phylogenet. Evol. 2014, 78, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Eisenman, H.C.; Casadevall, A. Synthesis and Assembly of Fungal Melanin. Appl. Microbiol. Biotechnol. 2012, 93, 931–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, A.V.; Franco, M.E.E.; Yanil Lopez, S.M.; Troncozo, M.I.; Saparrat, M.C.N.; Balatti, P.A. Melanins in Fungi: Types, Localization and Putative Biological Roles. Physiol. Mol. Plant Pathol. 2017, 99, 2–6. [Google Scholar] [CrossRef]
- Yu, J.H.; Leonard, T.J. Sterigmatocystin Biosynthesis in Aspergillus nidulans Requires a Novel Type I Polyketide Synthase. J. Bacteriol. 1995, 177, 4792–4800. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.-S.; Zhang, T.; Garcia-Borràs, M.; Hung, Y.-S.; Billingsley, J.M.; Houk, K.N.; Hu, Y.; Tang, Y. Biosynthesis of Heptacyclic Duclauxins Requires Extensive Redox Modifications of the Phenalenone Aromatic Polyketide. J. Am. Chem. Soc. 2018, 140, 6991–6997. [Google Scholar] [CrossRef]
- Xu, Y.; Vinas, M.; Alsarrag, A.; Su, L.; Pfohl, K.; Rohlfs, M.; Schäfer, W.; Chen, W.; Karlovsky, P. Bis-Naphthopyrone Pigments Protect Filamentous Ascomycetes from a Wide Range of Predators. Nat. Commun. 2019, 10, 3579. [Google Scholar] [CrossRef] [Green Version]
- Studt, L.; Wiemann, P.; Kleigrewe, K.; Humpf, H.-U.; Tudzynski, B. Tudzynski Bettina Biosynthesis of Fusarubins Accounts for Pigmentation of Fusarium fujikuroi Perithecia. Appl. Environ. Microbiol. 2012, 78, 4468–4480. [Google Scholar] [CrossRef] [Green Version]
- Jeong, M.-H.; Park, C.-H.; Kim, J.A.; Choi, E.D.; Kim, S.; Hur, J.-S.; Park, S.-Y. Production and Activity of Cristazarin in the Lichen-Forming Fungus Cladonia metacorallifera. J. Fungi 2021, 7, 601. [Google Scholar] [CrossRef]
- Bowler, P.A.; Rundel, P.W. The Ramalina intermedia Complex in North America. Bryologist 1974, 77, 617–623. [Google Scholar] [CrossRef]
- Galagan, J.E.; Calvo, S.E.; Cuomo, C.; Ma, L.-J.; Wortman, J.R.; Batzoglou, S.; Lee, S.-I.; Baştürkmen, M.; Spevak, C.C.; Clutterbuck, J.; et al. Sequencing of Aspergillus nidulans and Comparative Analysis with A. fumigatus and A. oryzae. Nature 2005, 438, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.Y.; Hou, Y.; Chen, Y.; Oh, J.-H.; Lee, I.; Bugni, T.S.; Keller, N.P. Genome-Based Cluster Deletion Reveals an Endocrocin Biosynthetic Pathway in Aspergillus fumigatus. Appl. Environ. Microbiol. 2012, 78, 4117–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szwalbe, A.J.; Williams, K.; Song, Z.; de Mattos-Shipley, K.; Vincent, J.L.; Bailey, A.M.; Willis, C.L.; Cox, R.J.; Simpson, T.J. Characterisation of the Biosynthetic Pathway to Agnestins A and B Reveals the Reductive Route to Chrysophanol in Fungi. Chem. Sci. 2019, 10, 233–238. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.-A.; Su, B.-N.; Keller, W.J.; Mehta, R.G.; Kinghorn, A.D. Antioxidant Xanthones from the Pericarp of Garcinia mangostana (Mangosteen). J. Agric. Food Chem. 2006, 54, 2077–2082. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J. Families of Serine Peptidases. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1994; Volume 244, pp. 19–61. ISBN 0076-6879. [Google Scholar]
- Roy, S.; Kundu, T.K. V-Chemical Principles of DNA Sequence Recognition and Gene Regulation. In Chemical Biology of the Genome; Roy, S., Kundu, T.K., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 171–223. ISBN 978-0-12-817644-3. [Google Scholar]
- Chiang, Y.-M.; Szewczyk, E.; Davidson, A.D.; Keller, N.; Oakley, B.R.; Wang, C.C.C. A Gene Cluster Containing Two Fungal Polyketide Synthases Encodes the Biosynthetic Pathway for a Polyketide, Asperfuranone, in Aspergillus nidulans. J. Am. Chem. Soc. 2009, 131, 2965–2970. [Google Scholar] [CrossRef] [Green Version]
- Gallo, A.; Ferrara, M.; Perrone, G. Phylogenetic Study of Polyketide Synthases and Nonribosomal Peptide Synthetases Involved in the Biosynthesis of Mycotoxins. Toxins 2013, 5, 717–742. [Google Scholar] [CrossRef] [Green Version]
- Hendrickson, L.; Davis, C.R.; Roach, C.; Nguyen, D.K.; Aldrich, T.; McAda, P.C.; Reeves, C.D. Lovastatin Biosynthesis in Aspergillus terreus: Characterization of Blocked Mutants, Enzyme Activities and a Multifunctional Polyketide Synthase Gene. Chem. Biol. 1999, 6, 429–439. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Cox, R.J. The Molecular Steps of Citrinin Biosynthesis in Fungi. Chem. Sci. 2016, 7, 2119–2127. [Google Scholar] [CrossRef] [Green Version]
- Proctor, R.H.; Brown, D.W.; Plattner, R.D.; Desjardins, A.E. Co-Expression of 15 Contiguous Genes Delineates a Fumonisin Biosynthetic Gene Cluster in Gibberella moniliformis. Fungal Genet. Biol. 2003, 38, 237–249. [Google Scholar] [CrossRef]
- Yang, G.; Rose, M.S.; Turgeon, B.G.; Yoder, O.C. A Polyketide Synthase Is Required for Fungal Virulence and Production of the Polyketide T-Toxin. Plant Cell 1996, 8, 2139–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, S.; Grötsch, T.; Foster, A.J.; Schüffler, A.; Rieger, P.H.; Sandjo, L.P.; Liermann, J.C.; Opatz, T.; Thines, E. Unravelling the Biosynthesis of Pyriculol in the Rice Blast Fungus Magnaporthe oryzae. Microbiology 2017, 163, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Kudo, F.; Matsuura, Y.; Hayashi, T.; Fukushima, M.; Eguchi, T. Genome Mining of the Sordarin Biosynthetic Gene Cluster from Sordaria araneosa Cain ATCC 36386: Characterization of Cycloaraneosene Synthase and GDP-6-Deoxyaltrose Transferase. J. Antibiot. 2016, 69, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Sinnemann, S.J.; Andrésson, Ó.S.; Brown, D.W.; Miao, V.P.W. Cloning and Heterologous Expression of Solorina crocea PyrG. Curr. Genet. 2000, 37, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, R.L.; Sorensen, J.L. Transcriptional Heterologous Expression of Two Type III PKS from the Lichen Cladonia uncialis. Mycol. Prog. 2019, 18, 1437–1447. [Google Scholar] [CrossRef]
- Olivier-Jimenez, D.; Chollet-Krugler, M.; Rondeau, D.; Beniddir, M.A.; Ferron, S.; Delhaye, T.; Allard, P.-M.; Wolfender, J.-L.; Sipman, H.J.M.; Lücking, R.; et al. A Database of High-Resolution MS/MS Spectra for Lichen Metabolites. Sci. Data 2019, 6, 294. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, V.; Geisberger, T.; Huber, C.; Beck, A.; Eisenreich, W. A Facile in Vivo Procedure to Analyze Metabolic Pathways in Intact Lichens. New Phytol. 2019, 224, 1657–1667. [Google Scholar] [CrossRef]
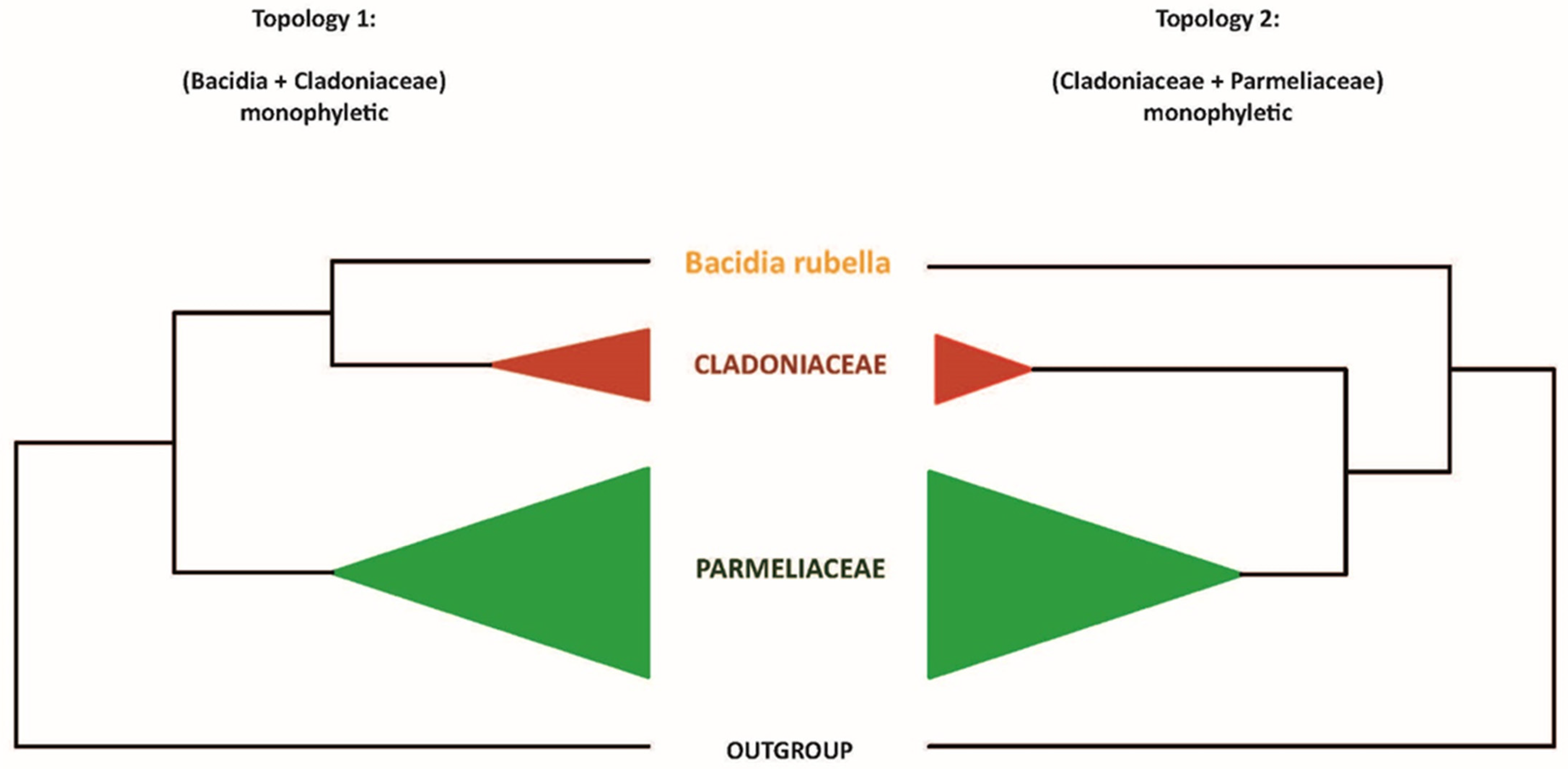
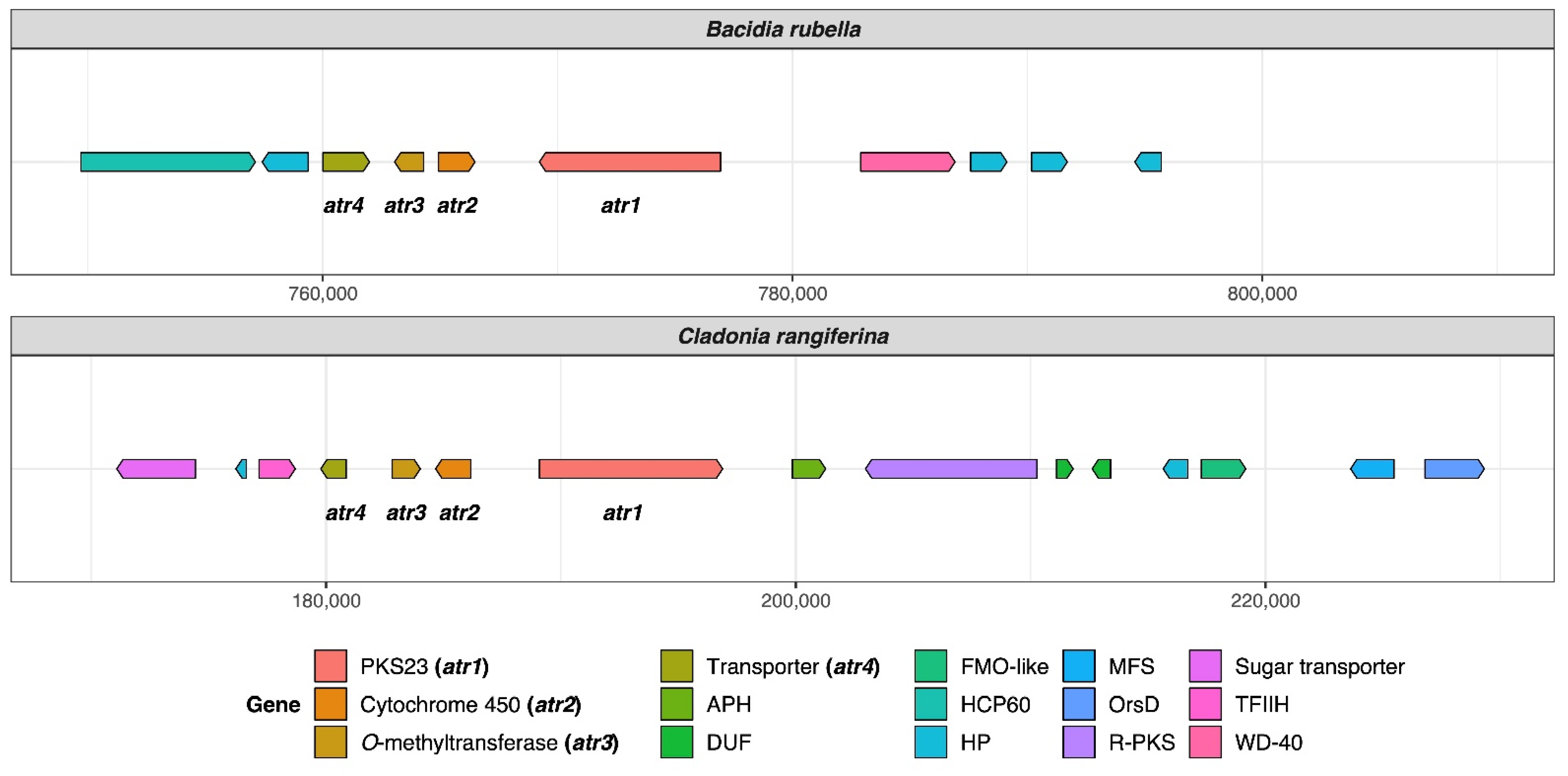
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).