
Transcriptional Profiles Elucidate Differential Host Responses to Infection with Cryptococcus neoformans and Cryptococcus gattii

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Inoculum Preparation
2.2. C. gattii and C. neoformans Infection
2.3. RNA Preparation
2.4. Statistical Analysis
2.5. Pathway Analysis
2.6. Classifier Development
2.7. Validation of Findings in Human PBMCs
3. Results
3.1. Mice Infected with C. neoformans Develop More Severe Fungal Burden
3.2. Peripheral White Blood Cell Differentials of Mice Infected with Both C. gattii and C. neoformans Differ from Control Mice
3.3. Mice Infected with Cryptococcus Exhibit a Powerful Transcriptomic Response to Infection with Many Broadly Conserved Components Regardless of Fungal Species
3.4. Differences in the Responses to C. gattii and C. neoformans Are Reflected in the Host Transcriptome
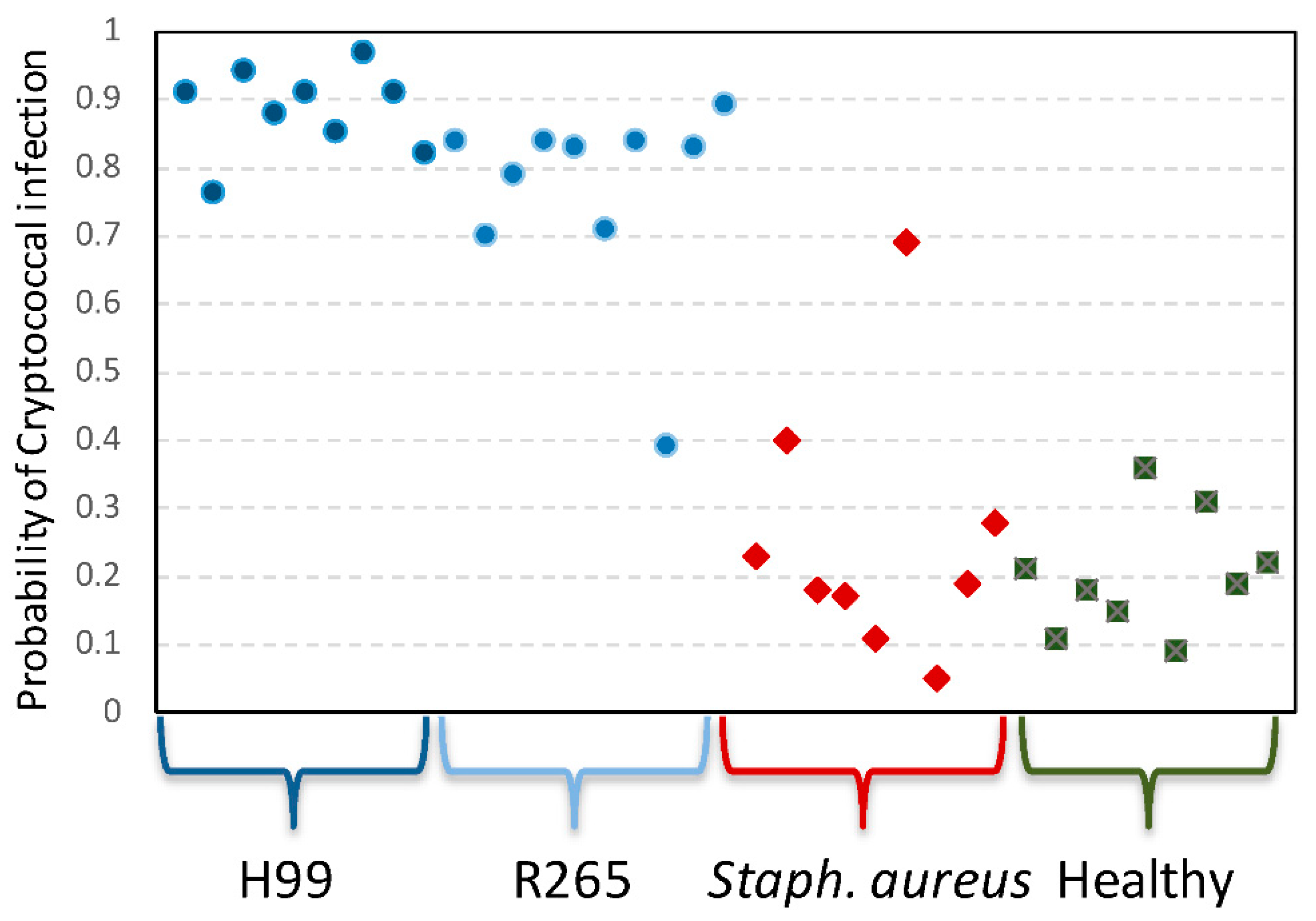
3.5. A Transcriptomic Classifier Accurately Identifies Cryptococcal Infection
3.6. Translation to Human Applications
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desalermos, A.; Kourkoumpetis, T.K.; Mylonakis, E. Update on the epidemiology and management of cryptococcal meningitis. Expert Opin. Pharmacother. 2012, 13, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Rajasingham, R.; Smith, R.M.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.M.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global burden of disease of HIV-associated cryptococcal meningitis: An updated analysis. Lancet Infect. Dis. 2017, 17, 873–881. [Google Scholar] [CrossRef]
- Lewis, J.L.; Rabinovich, S. The wide spectrum of cryptococcal infections. Am. J. Med. 1972, 53, 315–322. [Google Scholar] [CrossRef]
- Galanis, E.; Macdougall, L.; Kidd, S.; Morshed, M. Epidemiology of cryptococcus gattii, British Columbia, Canada, 1999–2007. Emerg. Infect. Dis. 2010, 16, 251–257. [Google Scholar] [CrossRef]
- Gibson, J.F.; Johnston, S.A. Immunity to cryptococcus neoformans and C. gattii during cryptococcosis. Fungal Genet. Biol. FG B 2015, 78, 76–86. [Google Scholar] [CrossRef]
- Dromer, F.; Perronne, C.; Barge, J.; Vilde, J.L.; Yeni, P. Role of IgG and complement component C5 in the initial course of experimental cryptococcosis. Clin. Exp. Immunol. 1989, 78, 412–417. [Google Scholar]
- Zhang, Y.; Wang, F.; Tompkins, K.C.; McNamara, A.; Jain, A.V.; Moore, B.B.; Toews, G.B.; Huffnagle, G.B.; Olszewski, M.A. Robust Th1 and Th17 immunity supports pulmonary clearance but cannot prevent systemic dissemination of highly virulent cryptococcus neoformans H99. Am. J. Pathol. 2009, 175, 2489–2500. [Google Scholar] [CrossRef]
- Flaczyk, A.; Duerr, C.U.; Shourian, M.; Lafferty, E.I.; Fritz, J.H.; Qureshi, S.T. IL-33 signaling regulates innate and adaptive immunity to Cryptococcus neoformans. J. Immunol. 2013, 191, 2503–2513. [Google Scholar] [CrossRef]
- Muller, U.; Stenzel, W.; Kohler, G.; Werner, C.; Polte, T.; Hansen, G.; Schutze, N.; Straubinger, R.K.; Blessing, M.; McKenzie, A.N.; et al. IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with cryptococcus neoformans. J. Immunol. 2007, 179, 5367–5377. [Google Scholar] [CrossRef]
- Jain, A.V.; Zhang, Y.; Fields, W.B.; McNamara, D.A.; Choe, M.Y.; Chen, G.H.; Erb-Downward, J.; Osterholzer, J.J.; Toews, G.B.; Huffnagle, G.B.; et al. Th2 but not Th1 immune bias results in altered lung functions in a murine model of pulmonary cryptococcus neoformans infection. Infect. Immun. 2009, 77, 5389–5399. [Google Scholar] [CrossRef]
- Wozniak, K.L.; Hardison, S.E.; Kolls, J.K.; Wormley, F.L. Role of IL-17A on resolution of pulmonary C. neoformans infection. PLoS ONE 2011, 6, e17204. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, J.N.; Casazza, J.P.; Stone, H.H.; Meintjes, G.; Lawn, S.D.; Levitz, S.M.; Harrison, T.S.; Koup, R.A. The phenotype of the cryptococcus-specific CD4+ memory T-cell response is associated with disease severity and outcome in HIV-associated cryptococcal meningitis. J. Infect. Dis. 2013, 207, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Voelz, K.; Lammas, D.A.; May, R.C. Cytokine signaling regulates the outcome of intracellular macrophage parasitism by cryptococcus neoformans. Infect. Immun. 2009, 77, 3450–3457. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Olszewski, M.A.; Tsang, T.M.; McDonald, R.A.; Toews, G.B.; Huffnagle, G.B. Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with cryptococcus neoformans. Infect. Immun. 2011, 79, 1915–1926. [Google Scholar] [CrossRef]
- Hardison, S.E.; Ravi, S.; Wozniak, K.L.; Young, M.L.; Olszewski, M.A.; Wormley, F.L., Jr. Pulmonary infection with an interferon-gamma-producing cryptococcus neoformans strain results in classical macrophage activation and protection. Am. J. Pathol. 2010, 176, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Islam, A.; Li, S.S.; Oykhman, P.; Timm-McCann, M.; Huston, S.M.; Stack, D.; Xiang, R.F.; Kelly, M.M.; Mody, C.H. An acidic microenvironment increases NK cell killing of cryptococcus neoformans and cryptococcus gattii by enhancing perforin degranulation. PLoS Pathog. 2013, 9, e1003439. [Google Scholar] [CrossRef]
- Kawakami, K.; Kinjo, Y.; Uezu, K.; Yara, S.; Miyagi, K.; Koguchi, Y.; Nakayama, T.; Taniguchi, M.; Saito, A. Monocyte chemoattractant protein-1-dependent increase of V alpha 14 NKT cells in lungs and their roles in Th1 response and host defense in cryptococcal infection. J. Immunol. 2001, 167, 6525–6532. [Google Scholar] [CrossRef]
- Kawakami, K.; Kinjo, Y.; Yara, S.; Koguchi, Y.; Uezu, K.; Nakayama, T.; Taniguchi, M.; Saito, A. Activation of Valpha14(+) natural killer T cells by alpha-galactosylceramide results in development of Th1 response and local host resistance in mice infected with cryptococcus neoformans. Infect. Immun. 2001, 69, 213–220. [Google Scholar] [CrossRef]
- Saha, D.C.; Xess, I.; Biswas, A.; Bhowmik, D.M.; Padma, M.V. Detection of cryptococcus by conventional, serological and molecular methods. J. Med. Microbiol. 2009, 58, 1098–1105. [Google Scholar] [CrossRef]
- Jarvis, J.N.; Percival, A.; Bauman, S.; Pelfrey, J.; Meintjes, G.; Williams, G.N.; Longley, N.; Harrison, T.S.; Kozel, T.R. Evaluation of a novel point-of-care cryptococcal antigen test on serum, plasma, and urine from patients with HIV-associated cryptococcal meningitis. Clin. Infect.Dis. 2011, 53, 1019–1023. [Google Scholar] [CrossRef]
- Boulware, D.R.; Rolfes, M.A.; Rajasingham, R.; von Hohenberg, M.; Qin, Z.; Taseera, K.; Schutz, C.; Kwizera, R.; Butler, E.K.; Meintjes, G.; et al. Multisite validation of cryptococcal antigen lateral flow assay and quantification by laser thermal contrast. Emerg. Infect. Dis. 2014, 20, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Ramilo, O.; Allman, W.; Chung, W.; Mejias, A.; Ardura, M.; Glaser, C.; Wittkowski, K.M.; Piqueras, B.; Banchereau, J.; Palucka, A.K.; et al. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood 2007, 109, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Zaas, A.K.; Chen, M.; Varkey, J.; Veldman, T.; Hero, A.O., 3rd; Lucas, J.; Huang, Y.; Turner, R.; Gilbert, A.; Lambkin-Williams, R.; et al. Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe 2009, 6, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Mejias, A.; Dimo, B.; Suarez, N.M.; Garcia, C.; Suarez-Arrabal, M.C.; Jartti, T.; Blankenship, D.; Jordan-Villegas, A.; Ardura, M.I.; Xu, Z.; et al. Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS Med. 2013, 10, e1001549. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.W.; McClain, M.T.; Chen, M.; Zaas, A.K.; Nicholson, B.P.; Varkey, J.; Veldman, T.; Kingsmore, S.F.; Huang, Y.; Lambkin-Williams, R.; et al. A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLoS ONE 2013, 8, e52198. [Google Scholar] [CrossRef]
- Berry, M.P.; Graham, C.M.; McNab, F.W.; Xu, Z.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Banchereau, R.; Skinner, J.; Wilkinson, R.J.; et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010, 466, 973–977. [Google Scholar] [CrossRef]
- Zak, D.E.; Penn-Nicholson, A.; Scriba, T.J.; Thompson, E.; Suliman, S.; Amon, L.M.; Mahomed, H.; Erasmus, M.; Whatney, W.; Hussey, G.D.; et al. A blood RNA signature for tuberculosis disease risk: A prospective cohort study. Lancet 2016, 387, 2312–2322. [Google Scholar] [CrossRef]
- Holcomb, Z.E.; Tsalik, E.L.; Woods, C.W.; McClain, M.T. Host-based peripheral blood gene expression analysis for diagnosis of infectious diseases. J. Clin. Microbiol. 2017, 55, 360–368. [Google Scholar] [CrossRef]
- Zaas, A.K.; Aziz, H.; Lucas, J.; Perfect, J.R.; Ginsburg, G.S. Blood gene expression signatures predict invasive candidiasis. Sci. Transl. Med. 2010, 2, 21ra17. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; Available online: http://www.R-project.org (accessed on 5 April 2022).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Ngamskulrungroj, P.; Chang, Y.; Sionov, E.; Kwon-Chung, K.J. The primary target organ of cryptococcus gattii is different from that of cryptococcus neoformans in a murine model. mBio 2012, 3, e00103-12. [Google Scholar] [CrossRef]
- Cormier, S.A.; Yuan, S.; Crosby, J.R.; Protheroe, C.A.; Dimina, D.M.; Hines, E.M.; Lee, N.A.; Lee, J.J. T(H)2-mediated pulmonary inflammation leads to the differential expression of ribonuclease genes by alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2002, 27, 678–687. [Google Scholar] [CrossRef]
- Ivanov, I.; Kuhn, H.; Heydeck, D. Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene 2015, 573, 1–32. [Google Scholar] [CrossRef]
- Nair, M.G.; Cochrane, D.W.; Allen, J.E. Macrophages in chronic type 2 inflammation have a novel phenotype characterized by the abundant expression of Ym1 and Fizz1 that can be partly replicated in vitro. Immunol. Lett. 2003, 85, 173–180. [Google Scholar] [CrossRef]
- Nair, M.G.; Du, Y.; Perrigoue, J.G.; Zaph, C.; Taylor, J.J.; Goldschmidt, M.; Swain, G.P.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.; et al. Alternatively activated macrophage-derived RELM-{alpha} is a negative regulator of type 2 inflammation in the lung. J. Exp. Med. 2009, 206, 937–952. [Google Scholar] [CrossRef] [PubMed]
- Novince, C.M.; Koh, A.J.; Michalski, M.N.; Marchesan, J.T.; Wang, J.; Jung, Y.; Berry, J.E.; Eber, M.R.; Rosol, T.J.; Taichman, R.S.; et al. Proteoglycan 4, a novel immunomodulatory factor, regulates parathyroid hormone actions on hematopoietic cells. Am. J. Pathol. 2011, 179, 2431–2442. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.A.; Yee, M.; Buczynski, B.W.; Vitiello, P.F.; Keng, P.C.; Welle, S.L.; Finkelstein, J.N.; Dean, D.A.; Lawrence, B.P. Neonatal oxygen increases sensitivity to influenza A virus infection in adult mice by suppressing epithelial expression of Ear1. Am. J. Pathol. 2012, 181, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Schroder, W.A.; Le, T.T.; Major, L.; Street, S.; Gardner, J.; Lambley, E.; Markey, K.; MacDonald, K.P.; Fish, R.J.; Thomas, R.; et al. A physiological function of inflammation-associated SerpinB2 is regulation of adaptive immunity. J. Immunol. 2010, 184, 2663–2670. [Google Scholar] [CrossRef]
- Yamada, K.J.; Barker, T.; Dyer, K.D.; Rice, T.A.; Percopo, C.M.; Garcia-Crespo, K.E.; Cho, S.; Lee, J.J.; Druey, K.M.; Rosenberg, H.F. Eosinophil-associated ribonuclease 11 is a macrophage chemoattractant. J. Biol. Chem. 2015, 290, 8863–8875. [Google Scholar] [CrossRef]
- Diamond, R.D.; May, J.E.; Kane, M.A.; Frank, M.M.; Bennett, J.E. The role of the classical and alternate complement pathways in host defenses against cryptococcus neoformans infection. J. Immunol. 1974, 112, 2260–2270. [Google Scholar]
- Mershon, K.L.; Vasuthasawat, A.; Lawson, G.W.; Morrison, S.L.; Beenhouwer, D.O. Role of complement in protection against cryptococcus gattii infection. Infect. Immun. 2009, 77, 1061–1070. [Google Scholar] [CrossRef]
- Mills, C.D. M1 and M2 macrophages: Oracles of health and disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef]
- Hidore, M.R.; Nabavi, N.; Sonleitner, F.; Murphy, J.W. Murine natural killer cells are fungicidal to cryptococcus neoformans. Infect. Immun. 1991, 59, 1747–1754. [Google Scholar] [CrossRef]
- Ma, L.L.; Wang, C.L.; Neely, G.G.; Epelman, S.; Krensky, A.M.; Mody, C.H. NK cells use perforin rather than granulysin for anticryptococcal activity. J. Immunol. 2004, 173, 3357–3365. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, J.C.; Ma, L.L.; Marr, K.J.; Jones, G.J.; Mody, C.H. Perforin-dependent cryptococcal microbicidal activity in NK cells requires PI3K-dependent ERK1/2 signaling. J. Immunol. 2007, 178, 6456–6464. [Google Scholar] [CrossRef] [PubMed]
- Marr, K.J.; Jones, G.J.; Zheng, C.; Huston, S.M.; Timm-McCann, M.; Islam, A.; Berenger, B.M.; Ma, L.L.; Wiseman, J.C.; Mody, C.H. Cryptococcus neoformans directly stimulates perforin production and rearms NK cells for enhanced anticryptococcal microbicidal activity. Infect. Immun. 2009, 77, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Steinbrink, J.M.; Myers, R.A.; Hua, K.; Johnson, M.D.; Seidelman, J.L.; Tsalik, E.L.; Henao, R.; Ginsburg, G.S.; Woods, C.W.; Alexander, B.D.; et al. The host transcriptional response to Candidemia is dominated by neutrophil activation and heme biosynthesis and supports novel diagnostic approaches. Genome Med. 2021, 13, 108. [Google Scholar] [CrossRef] [PubMed]
- Diamond, R.D.; May, J.E.; Kane, M.; Frank, M.M.; Bennett, J.E. The role of late complement components and the alternate complement pathway in experimental cryptococcosis. Proc. Soc. Exp. Biol. Med. 1973, 144, 312–315. [Google Scholar] [CrossRef]
- Shapiro, S.; Beenhouwer, D.O.; Feldmesser, M.; Taborda, C.; Carroll, M.C.; Casadevall, A.; Scharff, M.D. Immunoglobulin G monoclonal antibodies to cryptococcus neoformans protect mice deficient in complement component C3. Infect. Immun. 2002, 70, 2598–2604. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Sun, T.; Du, W.; Li, C.; Suo, C.; Meng, Y.; Liang, Q.; Lan, T.; Zhong, M.; et al. Unveil the transcriptional landscape at the cryptococcus-host axis in mice and nonhuman primates. PLoS Negl. Trop. Dis. 2019, 13, e0007566. [Google Scholar] [CrossRef]
- Farrer, R.A.; Ford, C.B.; Rhodes, J.; Delorey, T.; May, R.C.; Fisher, M.C.; Cloutman-Green, E.; Balloux, F.; Cuomo, C.A. Transcriptional heterogeneity of cryptococcus gattii VGII compared with non-VGII Lineages underpins key pathogenicity pathways. mSphere 2018, 3, e00445-18. [Google Scholar] [CrossRef]
- Movahed, E.; Munusamy, K.; Tan, G.M.; Looi, C.Y.; Tay, S.T.; Wong, W.F. Genome-wide transcription study of cryptococcus neoformans H99 clinical strain versus environmental strains. PLoS ONE 2015, 10, e0137457. [Google Scholar] [CrossRef]
- Altfeld, M.; Addo, M.M.; Kreuzer, K.A.; Rockstroh, J.K.; Dumoulin, F.L.; Schliefer, K.; Leifeld, L.; Sauerbruch, T.; Spengler, U. T(H)1 to T(H)2 shift of cytokines in peripheral blood of HIV-infected patients is detectable by reverse transcriptase polymerase chain reaction but not by enzyme-linked immunosorbent assay under nonstimulated conditions. J. Acquir. Immune Defic. Syndr. 2000, 23, 287–294. [Google Scholar] [CrossRef]
- Wright, L.C.; Santangelo, R.M.; Ganendren, R.; Payne, J.; Djordjevic, J.T.; Sorrell, T.C. Cryptococcal lipid metabolism: Phospholipase B1 is implicated in transcellular metabolism of macrophage-derived lipids. Eukaryot. Cell 2007, 6, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Huston, S.M.; Li, S.S.; Stack, D.; Timm-McCann, M.; Jones, G.J.; Islam, A.; Berenger, B.M.; Xiang, R.F.; Colarusso, P.; Mody, C.H. Cryptococcus gattii is killed by dendritic cells, but evades adaptive immunity by failing to induce dendritic cell maturation. J. Immunol. 2013, 191, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yan, H.; Zhang, L.; Kong, W.; Sun, Y.; Zhang, W.; Chen, Y.; Deng, A. Cryptococcus neoformans infection and immune cell regulation in human monocytes. Cell. Physiol. Biochem. 2015, 37, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Tsalik, E.L.; Henao, R.; Montgomery, J.L.; Nawrocki, J.W.; Aydin, M.; Lydon, E.C.; Ko, E.R.; Petzold, E.; Nicholson, B.P.; Cairns, C.B.; et al. Discriminating bacterial and viral infection using a rapid host gene expression test. Crit. Care Med. 2021, 49, 1651–1663. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.T.; Nicholson, B.P.; Park, L.P.; Liu, T.Y.; Hero, A.O., 3rd; Tsalik, E.L.; Zaas, A.K.; Veldman, T.; Hudson, L.L.; Lambkin-Williams, R.; et al. A genomic signature of influenza infection shows potential for presymptomatic detection, guiding early therapy, and monitoring clinical responses. Open Forum Infect. Dis. 2016, 3, ofw007. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leukocyte Subtype | H99 | R265 | Controls | p-Values (** = Statistically Significant) |
|---|---|---|---|---|
| Neutrophils | 10.5% | 11.3% | 19.2% | H99 vs. R265 (p = 0.8786) H99 vs. Controls (p = 0.0055) ** R265 vs. Controls (p = 0.0014) ** |
| Lymphocytes | 67.4% | 65.1% | 75.5% | H99 vs. R265 (p = 0.2552) H99 vs. Controls (p = 0.0636) R265 vs. Controls (p = 0.0025) ** |
| Atypical Lymphocytes | 10.1% | 15.4% | 1.0% | H99 vs. R265 (p = 0.1033) H99 vs. Controls (p = 0.0119) ** R265 vs. Controls (p = 0.0001) ** |
| Monocytes | 3.7% | 1.1% | 2.4% | H99 v. R265 (p = 0.0296) ** H99 v. Control (p = 0.0116) ** R265 v. Control (p = 0.0004) ** |
| Eosinophils | 8.3% | 7.1% | 1.9% | H99 vs. R265 (p = 0.3803) H99 vs. Controls (p = 0.0003) ** R265 vs. Controls (p = 0.0004) ** |
| Gene | Fold-Change (Infected vs. Control) | Function |
|---|---|---|
| Rnase2a | +14.15 | Produced in response to TH2 cytokine stimulation and serves as a macrophage chemoattractant [39,46]. |
| Retnla | +6.85 | Produced by macrophages in response to IL-4 and TH2-mediated inflammation [41]. Inhibits TH2-mediated immunity and inflammation [42]. |
| Serpinb2 | +5.77 | Expressed by myeloid antigen-presenting cells and plays a role in suppression of the TH1 immune response [45]. |
| C1qb | +5.37 | Component of the classical pathway of complement activation. |
| Chil3 | +5.35 | Produced by macrophages in response to IL-4 and TH2-mediated inflammation [41]. |
| Alox15 | +5.26 | Involved in arachidonic acid metabolism [40] |
| C1qa | +4.76 | Component of the classical pathway of complement activation. |
| Prg4 | +4.65 | Produces a glycoprotein for articular joint protection and PTH-responsive hematopoiesis and megakaryopoiesis [43]. |
| C1qc | +4.48 | Component of the classical pathway of complement activation. |
| Ear1 | +4.33 | Produces a host defense protein secreted by eosinophils, macrophages, and neutrophils [44]. |
| GO Term | Bonferroni-Corrected p-Value |
|---|---|
| Immune response (GO:0006955) | 0.00002 |
| Acute inflammatory response (GO:0002526) | 0.00389 |
| Complement activation (GO:0006956) | 0.01135 |
| Activation of plasma proteins involved in acute inflammatory response (GO:0002541) | 0.01135 |
| Innate immune response (GO:0045087) | 0.01166 |
| Immune effector process (GO:0002252) | 0.02201 |
| Defense response (GO:0006952) | 0.03399 |
| Humoral immune response (GO:0006959) | 0.03808 |
| Immunoglobulin mediated immune response (GO:0016064) | 0.05967 |
| B cell mediated immunity (GO:0019724) | 0.06529 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holcomb, Z.E.; Steinbrink, J.M.; Zaas, A.K.; Betancourt, M.; Tenor, J.L.; Toffaletti, D.L.; Alspaugh, J.A.; Perfect, J.R.; McClain, M.T. Transcriptional Profiles Elucidate Differential Host Responses to Infection with Cryptococcus neoformans and Cryptococcus gattii. J. Fungi 2022, 8, 430. https://doi.org/10.3390/jof8050430
Holcomb ZE, Steinbrink JM, Zaas AK, Betancourt M, Tenor JL, Toffaletti DL, Alspaugh JA, Perfect JR, McClain MT. Transcriptional Profiles Elucidate Differential Host Responses to Infection with Cryptococcus neoformans and Cryptococcus gattii. Journal of Fungi. 2022; 8(5):430. https://doi.org/10.3390/jof8050430
Chicago/Turabian StyleHolcomb, Zachary E., Julie M. Steinbrink, Aimee K. Zaas, Marisol Betancourt, Jennifer L. Tenor, Dena L. Toffaletti, J. Andrew Alspaugh, John R. Perfect, and Micah T. McClain. 2022. "Transcriptional Profiles Elucidate Differential Host Responses to Infection with Cryptococcus neoformans and Cryptococcus gattii" Journal of Fungi 8, no. 5: 430. https://doi.org/10.3390/jof8050430
APA StyleHolcomb, Z. E., Steinbrink, J. M., Zaas, A. K., Betancourt, M., Tenor, J. L., Toffaletti, D. L., Alspaugh, J. A., Perfect, J. R., & McClain, M. T. (2022). Transcriptional Profiles Elucidate Differential Host Responses to Infection with Cryptococcus neoformans and Cryptococcus gattii. Journal of Fungi, 8(5), 430. https://doi.org/10.3390/jof8050430