
Phylogenomics and Comparative Genomics Highlight Specific Genetic Features in Ganoderma Species

 ,
,  , , , , ,
, , , , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Strain and Culture Conditions
2.2. DNA Extractions
2.3. Genome Sequencing and Assembly
2.4. Genome Annotation and Quality Check
2.5. Synteny Analyses
2.6. Phylogenetic Analyses
2.7. Phylogenomic Analyses
2.8. Annotation of Transposable Elements
2.9. Protein Functionnal Annotation
3. Results
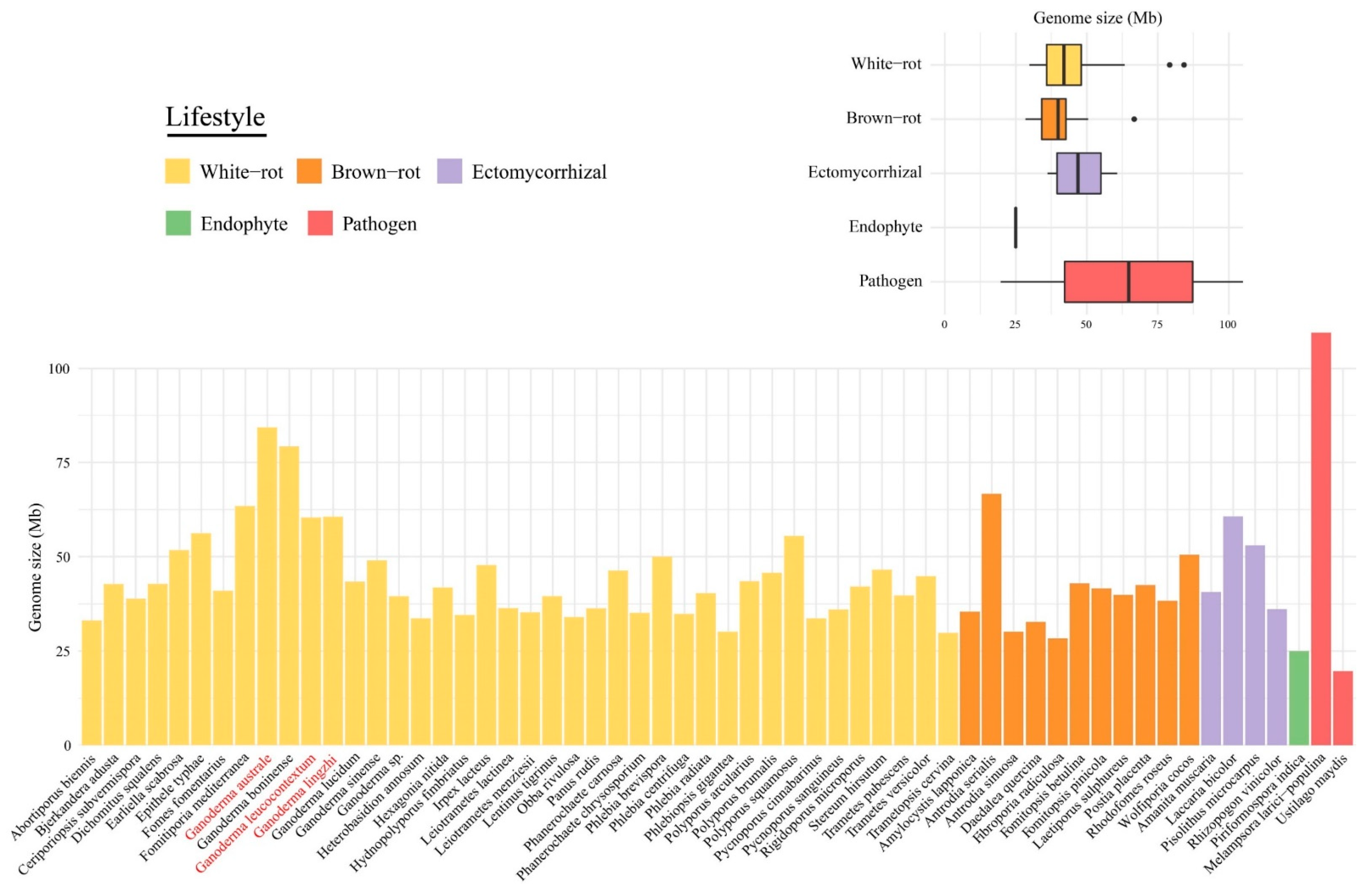
3.1. Main Genome Features
3.2. Macrosynteny between Ganoderma Genomes
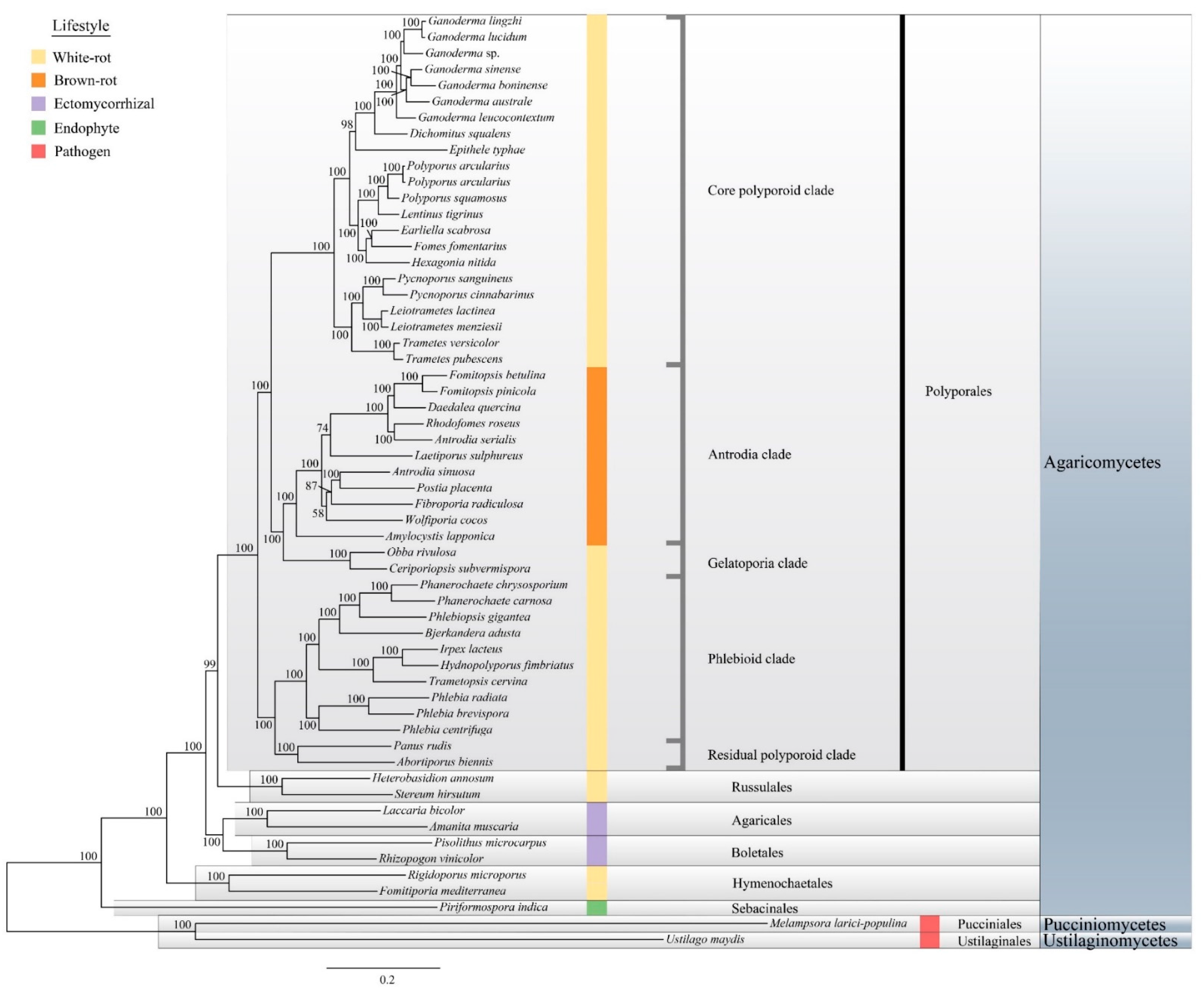
3.3. Phylogenetic Analyses of Ganoderma Strains
3.4. Phylogenomic Analyses of the Ganoderma Species and Related Polyporales
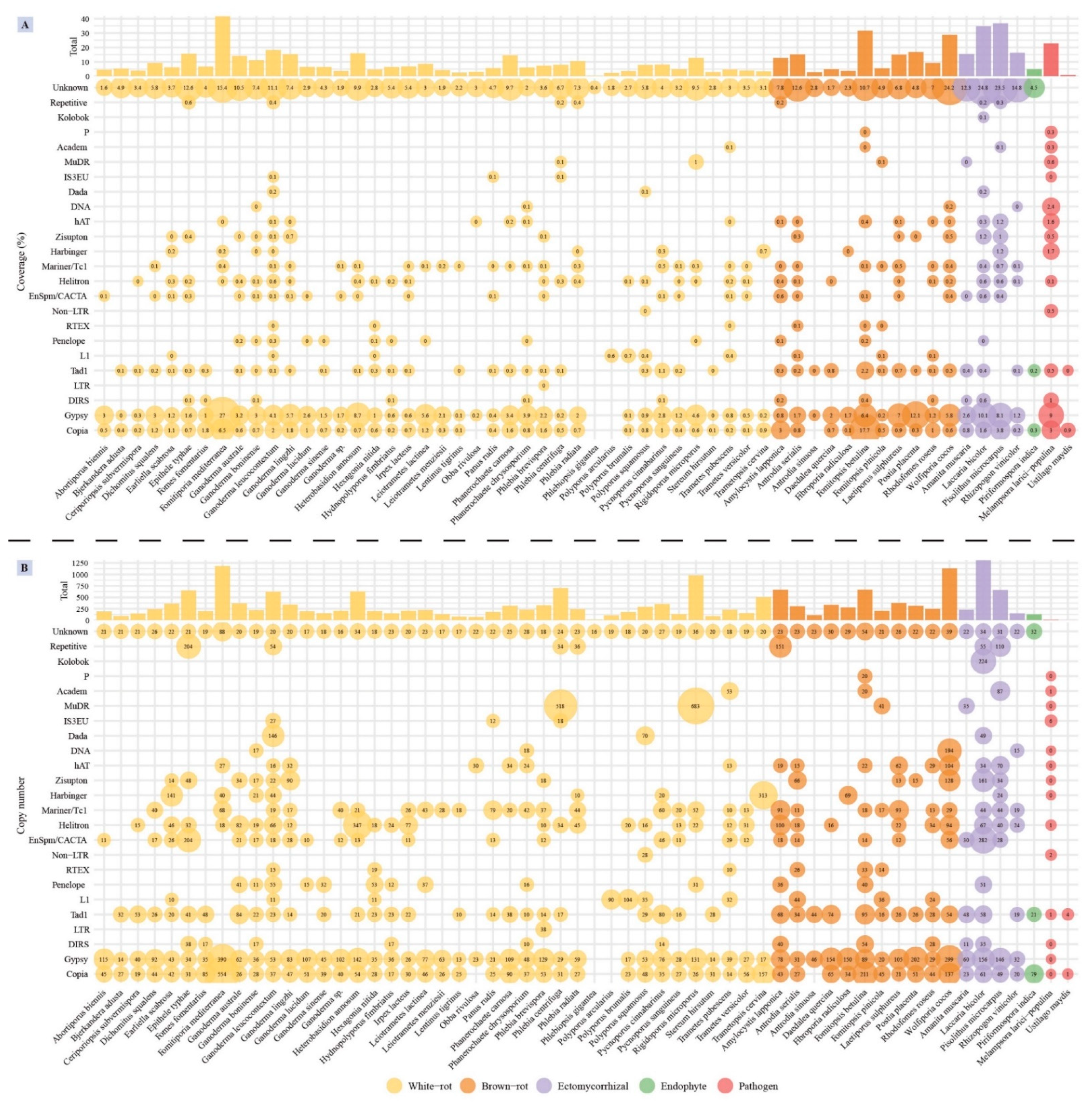
3.5. Transposable Element Identification
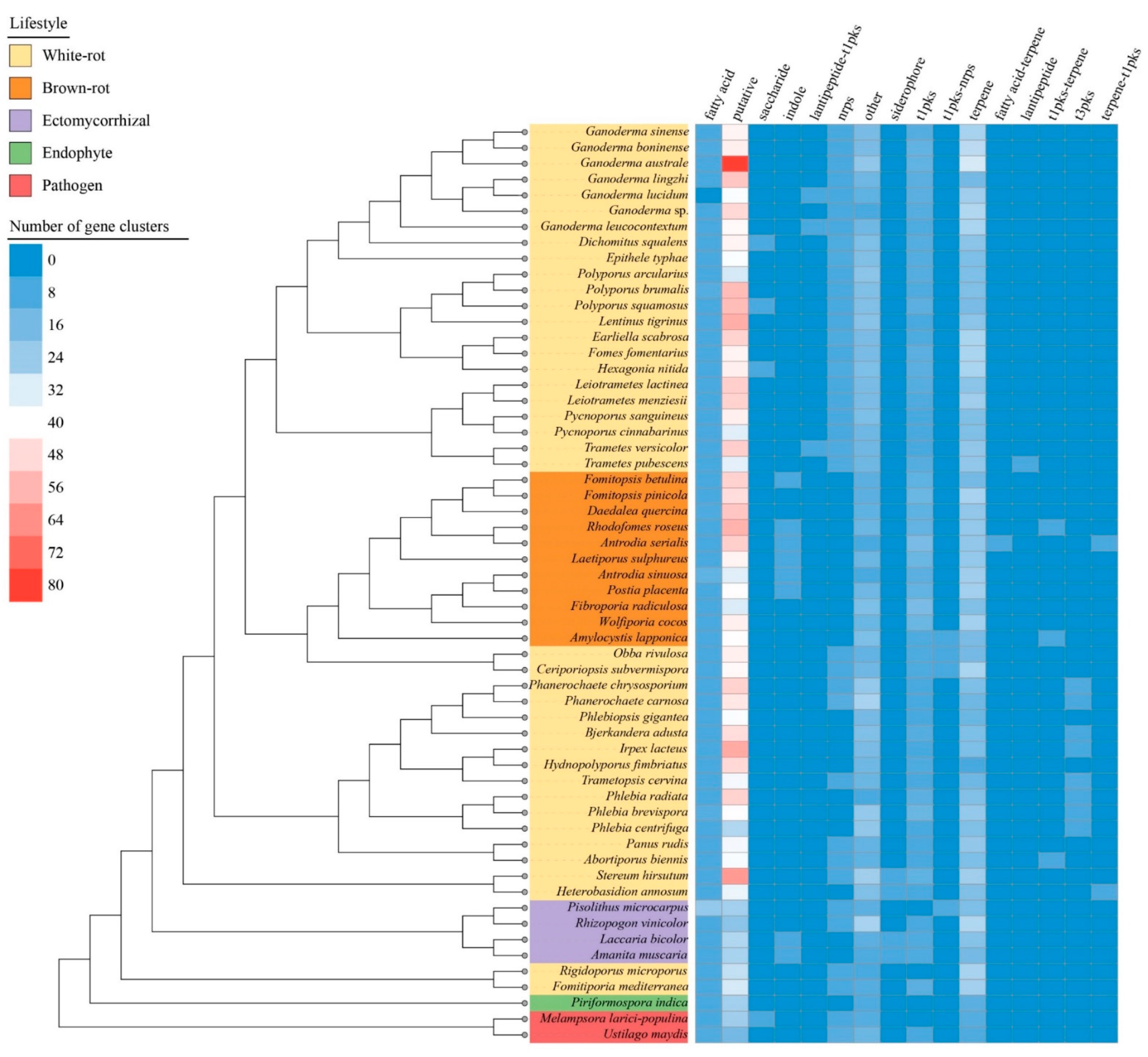
3.6. Biosynthetic Gene Clusters
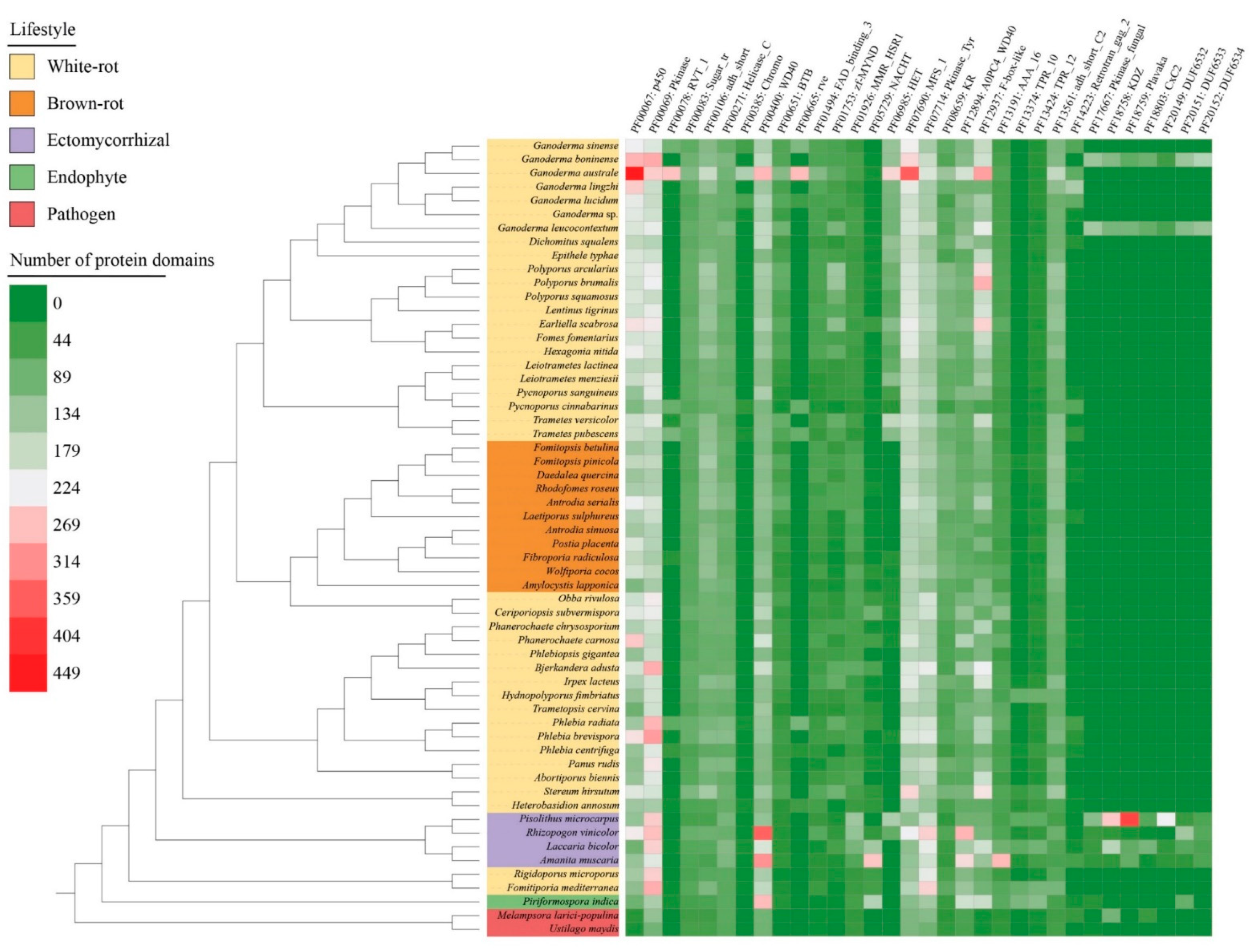
3.7. Pfam Protein Domains Found in the Genomes
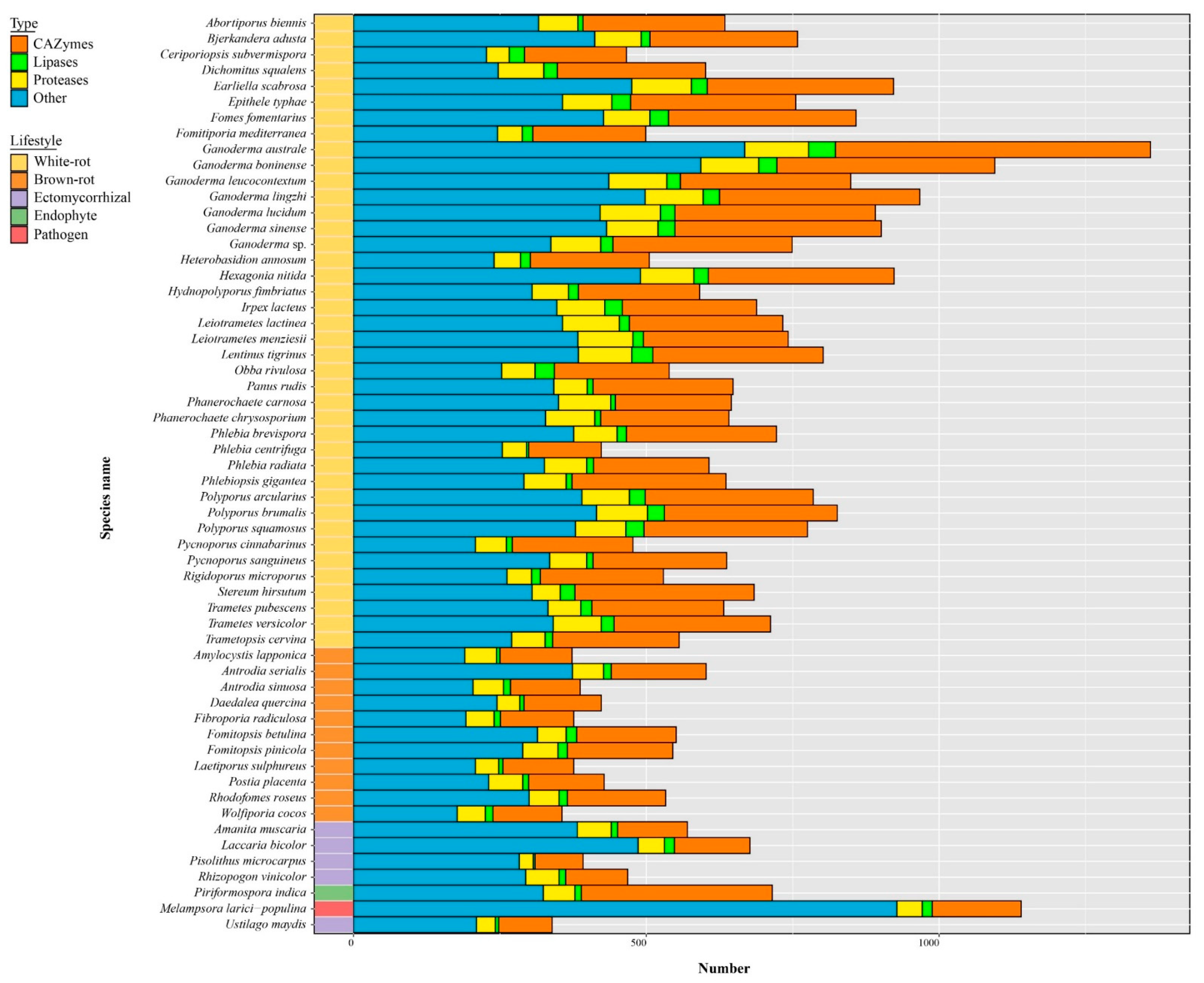
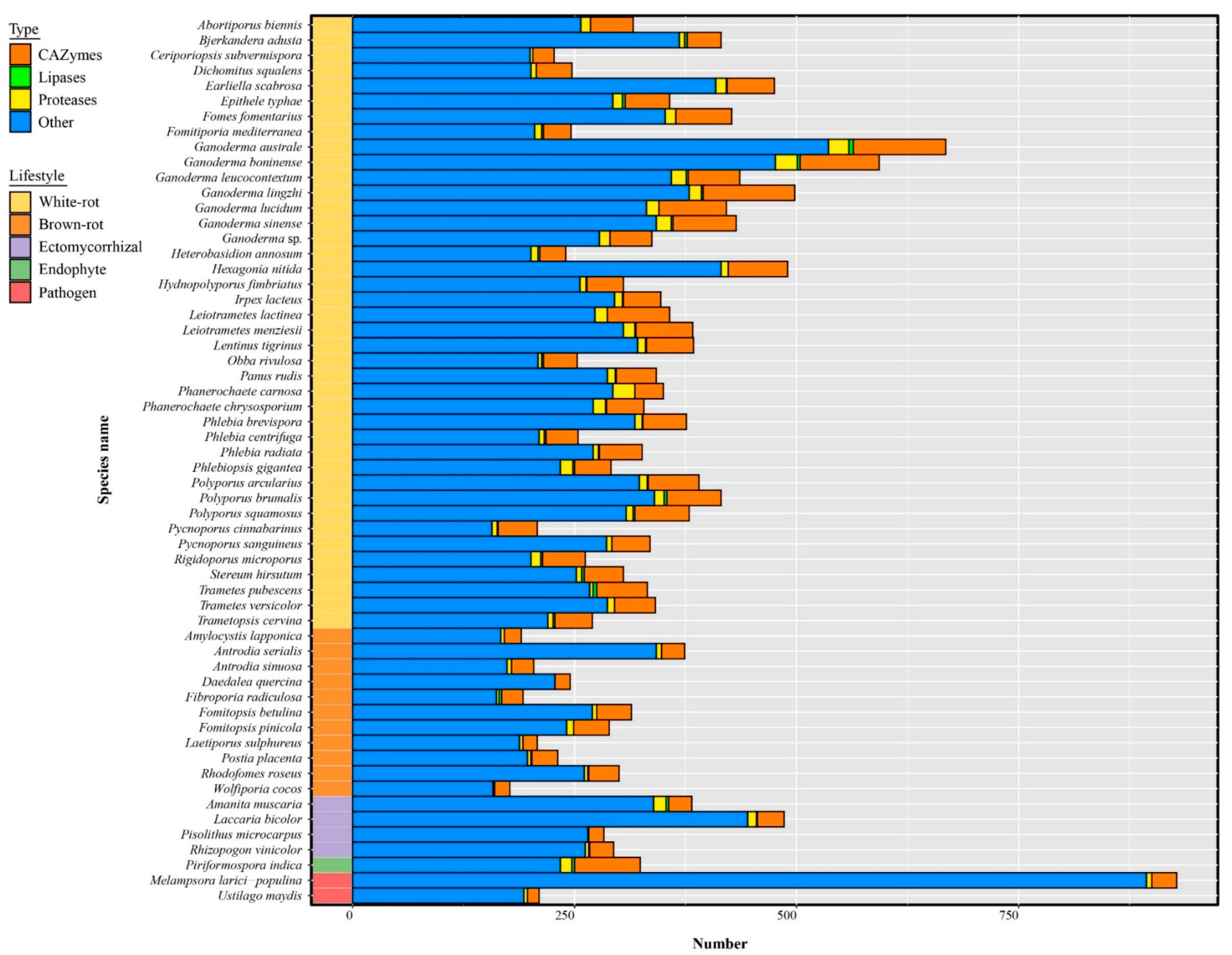
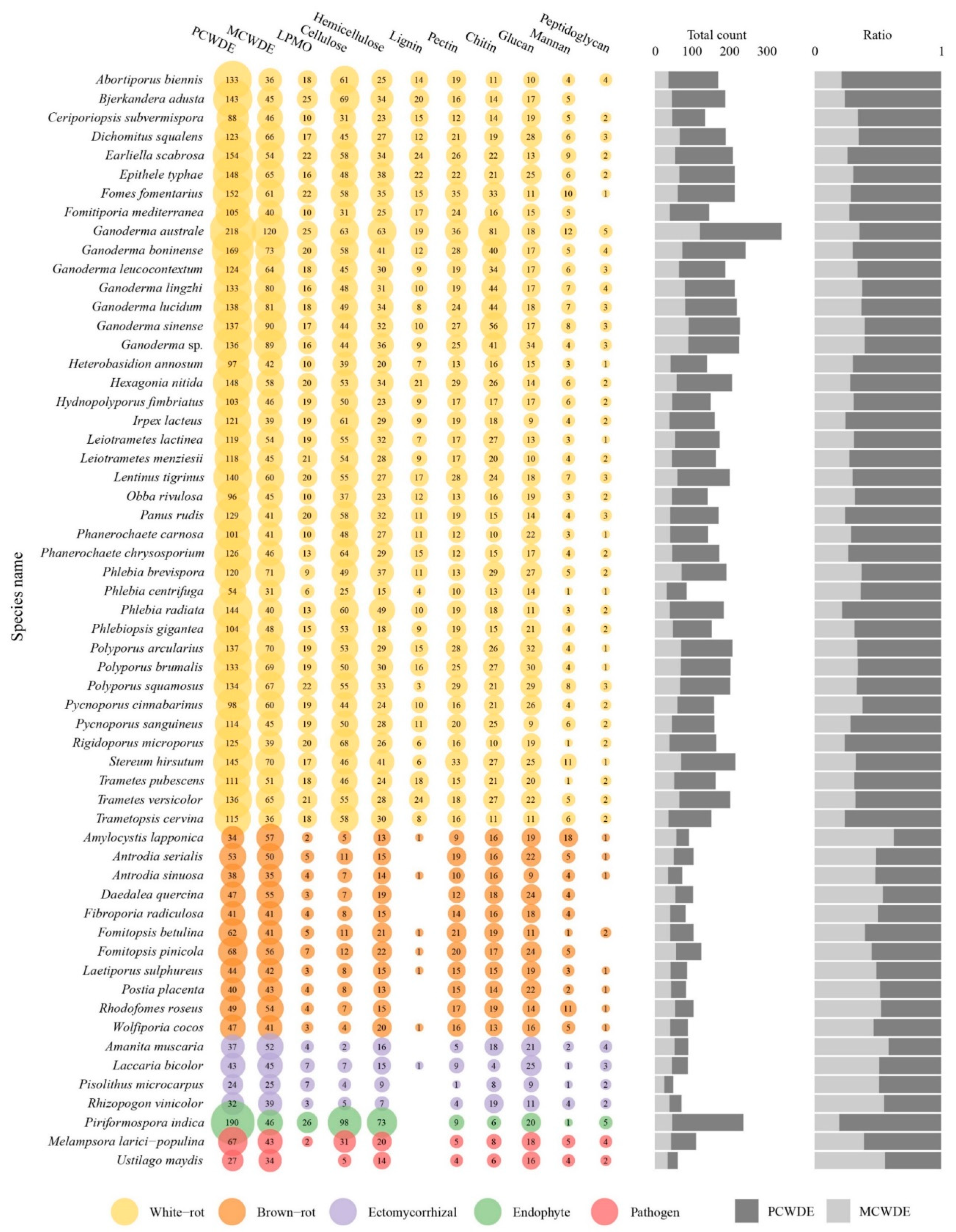
3.8. The Predicted Secretome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rodríguez-Couto, S. Industrial an environmental application of white-rot fungi. Mycosphere 2017, 8, 456–466. [Google Scholar] [CrossRef]
- Pilotti, C.A. Stem rots of oil palm caused by Ganoderma boninense: Pathogen biology and epidemiology. Mycopathologia 2005, 159, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Seman, I.B. R&D on Biology, Detection and Management of Ganoderma Disease in Oil Palm. Ph.D. Thesis, Universiti Putra Malaysia, Selangor, Malaysia, 2018. [Google Scholar]
- Si, J.; Wu, Y.; Ma, H.F.; Cao, Y.J.; Sun, Y.F.; Cui, B.K. Selection of a pH- and temperature-stable laccase from Ganoderma australe and its application for bioremediation of textile dyes. J. Environ. Manag. 2021, 299, 113619. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Deng, W.; Shen, M.; Yan, G.; Zhao, W.; Yang, Y. A laccase Gl-LAC-4 purified from white-rot fungus Ganoderma lucidum had a strong ability to degrade and detoxify the alkylphenol pollutants 4-n-octylphenol and 2-phenylphenol. J. Hazard. Mater. 2021, 408, 124775. [Google Scholar] [CrossRef] [PubMed]
- Boh, B.; Berovic, M.; Zhang, J.S.; Lin, Z.B. Ganoderma lucidum and its pharmaceutically active compounds. Biotechnol. Annu. Rev. 2007, 13, 265–301. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wu, S.H.; Dai, Y.C. Species clarification of the prize medicinal Ganoderma mushroom ‘Lingzhi’. Fungal Divers. 2012, 56, 49–62. [Google Scholar] [CrossRef]
- Zhang, Y.R.; Jiang, Y.F.; Zhang, M.; Zhang, L.J. Ganoderma sinense polysaccharide: An adjunctive drug used for cancer treatment. Prog. Mol. Biol. Transl. Sci. 2019, 163, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.M.; Yue, G.G.L.; Li, P.; Wong, E.C.W.; Lee, J.K.M.; Kennelly, E.J.; Lau, C.B.S. Screening and analysis of potential anti-tumor components from the stipe of Ganoderma sinense using high-performance liquid chromatography/time-of-flight mass spectrometry with multivariate statistical tool. J. Chromatogr. A 2017, 1487, 162–167. [Google Scholar] [CrossRef]
- Chen, S.L.; Xu, J.; Liu, C.; Zhu, Y.J.; Nelson, D.R.; Zhou, S.G.; Li, C.F.; Wang, L.Z.; Guo, X.; Sun, Y.Z.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef] [PubMed]
- Binder, M.; Justo, A.; Riley, R.; Salamov, A.; Lopez-Giraldez, F.; Sjökvist, E.; Copeland, A.; Foster, B.; Sun, H.; Larsson, E.; et al. Phylogenetic and phylogenomic overview of the Polyporales. Mycologia 2013, 105, 1350–1373. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; Xu, J.; Sun, C.; Zhou, S.G.; Xu, H.B.; Nelson, D.R.; Qian, J.; Song, J.Y.; Luo, H.M.; Xiang, L.; et al. Chromosome-level genome map provides insights into diverse defense mechanisms in the medicinal fungus Ganoderma sinense. Sci. Rep. 2015, 5, 11087. [Google Scholar] [CrossRef] [PubMed]
- Utomo, C.; Tanjung, Z.A.; Aditama, R.; Buana, R.F.N.; Pratomo, A.D.M.; Tryono, R.; Liwang, T. Draft genome sequence of the phytopathogenic fungus Ganoderma boninense, the causal agent of basal stem rot disease on oil palm. Genome Announc. 2018, 6, e00122-18. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.J.; Lee, E.H.; Yoon, Y.; Chua, B.; Son, A. Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. J. Appl. Microbiol. 2016, 120, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Fulton, T.M.; Chunwongse, J.; Tanksley, S.D. Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol. Biol. Report. 1995, 13, 207–209. [Google Scholar] [CrossRef]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; Riley, R.; Salamov, A.; Zhao, X.L.; Korzeniewski, F.; et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014, 42, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.; Bushnell, B.; Grigoriev, I.V. Fungal genomics: Sequencing and annotation. In Fungi; Martin, F., Ed.; Elsevier Academic Press: Cambridge, UK, 2014; pp. 1–52. [Google Scholar]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.S. DECIPHER: Harnessing local sequence context to improve protein multiple sequence alignment. BMC Bioinform. 2015, 16, 322. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.G.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Hage, H.; Miyauchi, S.; Virágh, M.; Drula, E.; Min, B.; Chaduli, D.; Navarro, D.; Favel, A.; Norest, M.; Lesage-Meessen, L.; et al. Gene family expansions and transcriptome signatures uncover fungal adaptations to wood decay. Environ. Microbiol. 2021, 23, 5716–5732. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A tool for phylogenetic analyses and post analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fueyoa, E.; Ruiz-Dueñasa, F.J.; Ferreirab, P.; Floudas, D.; Hibbett, D.S.; Canessa, P.; Larrondo, L.F.; James, T.Y.; Seelenfreund, D.; Lobos, S.; et al. Comparative genomics of Ceriporiopsis subvermispora and Phanerochaete chrysosporium provide insight into selective ligninolysis. Proc. Natl. Acad. Sci. USA 2011, 109, 5458–5463. [Google Scholar] [CrossRef] [PubMed]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martínez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Olson, Å.; Aerts, A.; Asiegbu, F.; Belbahri, L.; Bouzid, O.; Broberg, A.; Canbäck, B.; Coutinho, P.M.; Cullen, D.; Dalman, K.; et al. Insight into trade-off between wood decay and parasitism from the genome of a fungal forest pathogen. New Phytol. 2012, 194, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.J.; Xu, Z.Y.; Knudson, A.; Carlson, A.; Chen, N.Y.; Kovaka, S.; LaButti, K.; Lipzen, A.; Pennachio, C.; Riley, R.; et al. Genomics and development of Lentinus tigrinus: A white-rot wood-decaying mushroom with dimorphic fruiting bodies. Genome Biol. Evol. 2018, 10, 3250–3261. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, O.; Riley, R.; Barry, K.; Cullen, D.; de Vries, R.P.; Hainaut, M.; Hatakka, A.; Henrissat, B.; Hildén, K.; Kuo, R.; et al. Draft genome sequence of the white-rot fungus Obba rivulosa 3A-2. Genome Announc. 2016, 4, e00976-16. [Google Scholar] [CrossRef]
- Suzuki, H.; MacDonald, J.; Syed, K.; Salamov, A.; Hori, C.; Aerts, A.; Henrissat, B.; Wiebenga, A.D.; VanKuyk, P.A.; Barry, K.; et al. Comparative genomics of the white-rot fungi, Phanerochaete carnosa and P. chrysosporium, to elucidate the genetic basis of the distinct wood types they colonize. BMC Genom. 2012, 13, 444. [Google Scholar] [CrossRef] [PubMed]
- Ohm, R.A.; Riley, R.; Salamov, A.; Min, B.; Choi, I.; Grigoriev, I.V. Genomics of wood-degrading fungi. Fungal Genet. Biol. 2014, 72, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, M.R.; Peng, M.; Granchi, Z.; Chin-A-Woeng, T.; Hegi, R.; Pelt, S.I.V.; Ahrendt, S.; Riley, R.; Hainaut, M.; Henrissat, B.; et al. Draft genome sequence of the basidiomycete white-rot fungus Phlebia centrifuga. Genome Announc. 2018, 6, e01414-17. [Google Scholar] [CrossRef]
- Kuuskeri, J.; Häkkinen, M.; Laine, P.; Smolander, O.; Tamene, F.; Miettinen, S.; Nousiainen, P.; Kemell, M.; Auvinen, P.; Lundell, T. Time-scale dynamics of proteome and transcriptome of the white-rot fungus Phlebia radiata: Growth on spruce wood and decay effect on lignocellulose. Biotechnol. Biofuels 2016, 9, 192. [Google Scholar] [CrossRef]
- Hori, C.; Ishida, T.; Igarashi, K.; Samejima, M.; Suzuki, H.; Master, E.; Ferreira, P.; Ruiz-Dueñas, F.J.; Held, B.; Canessa, P.; et al. Analysis of the Phlebiopsis gigantea genome, transcriptome and secretome provides insight into its pioneer colonization strategies of wood. PLoS Genet. 2014, 10, e1004759. [Google Scholar] [CrossRef]
- Varga, T.; Krizsán, K.; Földi, C.; Dima, B.; Sánchez-García, M.; Sánchez-Ramírez, S.; Szöllősi, G.J.; Szarkándi, J.G.; Papp, V.; Albert, L.; et al. Megaphylogeny resolves global patterns of mushroom evolution. Nat. Ecol. Evol. 2019, 3, 668–678. [Google Scholar] [CrossRef]
- Miyauchi, S.; Rancon, A.; Drula, E.; Hage, H.; Chaduli, D.; Favel, A.; Grisel, S.; Henrissat, B.; Herpoël-Gimbert, I.; Ruiz-Dueñas, F.J.; et al. Integrative visual omics of the white-rot fungus Polyporus brumalis exposes the biotechnological potential of its oxidative enzymes for delignifying raw plant biomass. Biotechnol. Biofuels 2018, 11, 201. [Google Scholar] [CrossRef]
- Levasseur, A.; Lomascolo, A.; Chabrol, O.; Ruiz-Dueñas, F.J.; Boukhris-Uzan, E.; Piumi, F.; Kües, U.; Ram, A.F.J.; Murat, C.; Haon, M.; et al. The genome of the white-rot fungus Pycnoporus cinnabarinus: A basidiomycete model with a versatile arsenal for lignocellulosic biomass breakdown. BMC Genom. 2014, 15, 486. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, S.; Hage, H.; Drula, E.; Lesage-Meessen, L.; Berrin, J.G.; Navarro, D.; Favel, A.; Chaduli, D.; Grisel, S.; Haon, M.; et al. Conserved white-rot enzymatic mechanism for wood decay in the Basidiomycota genus Pycnoporus. DNA Res. 2020, 27, dsaa011. [Google Scholar] [CrossRef]
- Oghenekaro, A.O.; Kovalchuk, A.; Raffaello, T.; Camarero, S.; Gressler, M.; Henrissat, B.; Lee, J.; Liu, M.; Martínez, A.T.; Miettinen, O.; et al. Genome sequencing of Rigidoporus microporus provides insights on genes important for wood decay, latex tolerance and interspecific fungal interactions. Sci. Rep. 2020, 10, 5250. [Google Scholar] [CrossRef] [PubMed]
- Granchi, Z.; Peng, M.; Chi-A-Woeng, T.; de Vries, R.P.; Hildén, K.; Mäkelä, M.R. Genome sequence of the basidiomycete white-rot fungus Trametes pubescens FBCC735. Genome Announc. 2017, 5, e01643-16. [Google Scholar] [CrossRef]
- Nagy, L.G.; Riley, R.; Tritt, A.; Adam, C.; Daum, C.; Floudas, D.; Sun, H.; Yadav, J.S.; Pangilinan, J.; Larsson, K.H.; et al. Comparative genomics of early-diverging mushroom-forming fungi provides insights into the origins of lignocellulose decay capabilities. Mol. Biol. Evol. 2016, 33, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.D.; Perkins, A.D.; Sonstegard, T.S.; Schroeder, S.G.; Burgess, S.C.; Diehla, S.V. Short-read sequencing for genomic analysis of the brown rot fungus Fibroporia radiculosa. Appl. Environ. Microbiol. 2012, 78, 2272–2281. [Google Scholar] [CrossRef] [PubMed]
- Gaskell, J.; Kersten, P.; Larrondo, L.F.; Canessa, P.; Martinez, D.; Hibbett, D.; Schmoll, M.; Kubicek, C.P.; Martinez, A.T.; Yadav, I.; et al. Draft genome sequence of a monokaryotic model brown-rot fungus Postia (Rhodonia) placenta SB12. Genom. Data 2017, 14, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Kohler, A.; Kuo, A.; Nagy, L.; Morin, E.; Barry, K.; Buscot, F.; Canbäck, B.; Choi, C.; Cichocki, N.; Clum, A.; et al. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat. Genet. 2015, 47, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Aerts, A.; Ahrén, D.; Brun, A.; Danchin, E.G.; Duchaussoy, F.; Gibon, J.; Kohler, A.; Lindquist, E.; Pereda, V.; et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 2008, 452, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Mujic, A.B.; Kuo, A.; Tritt, A.; Lipzen, A.; Chen, C.; Johnson, J.; Sharma, A.; Barry, K.; Grigoriev, I.V.; Spatafora, J.W. Comparative genomics of the ectomycorrhizal sister species Rhizopogon vinicolor and Rhizopogon vesiculosus (Basidiomycota: Boletales) reveals a divergence of the mating type B Locus. Genes Genomes Genet. 2017, 7, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Zuccaro, A.; Lahrmann, U.; Güldener, U.; Langen, G.; Pfiffi, S.; Biedenkopf, D.; Wong, P.; Samans, B.; Grimm, C.; Basiewicz, M.; et al. Endophytic life strategies decoded by genome and transcriptome analyses of the mutualistic root symbiont Piriformospora indica. PLoS Pathog. 2011, 7, e1002290. [Google Scholar] [CrossRef]
- Duplessisa, S.; Cuomob, C.A.; Lin, Y.C.; Aerts, A.; Tisserant, E.; Veneault-Fourrey, C.; Joly, D.L.; Hacquard, S.; Amselem, J.; Cantarel, B.L.; et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. USA 2011, 108, 9166–9171. [Google Scholar] [CrossRef]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O.; et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable, and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2019, 37, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Payen, T.; Murat, C.; Martin, F. Reconstructing the evolutionary history of gypsy retrotransposons in the Périgord black truffle (Vittad.). Mycorrhiza 2016, 26, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, 351–358. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Blin, K.; Wolf, T.; Chevrette, M.G.; Lu, X.W.; Schwalen, C.J.; Kautsar, S.A.; Duran, H.G.S.; de Los Santos, E.L.C.; Kim, H.U.K.; Nave, M.; et al. antiSMASH 4.0—Improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017, 45, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, C.; Morin, E.; Martin, F.M.; Veneault-Fourrey, C. Comparative analysis of secretomes from ectomycorrhizal fungi with an emphasis on small-secreted proteins. Front. Microbiol. 2015, 6, 1278. [Google Scholar] [CrossRef]
- Justo, A.; Miettinen, O.; Floudas, D.; Ortiz-Santana, B.; Sjökvist, E.; Lindner, D.; Nakasone, K.; Niemelä, T.; Larsson, K.H.; Ryvarden, L.; et al. A revised family-level classification of the Polyporales (Basidiomycota). Fungal Biol. 2017, 121, 798–824. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Chen, J.J.; Ji, X.H.; Vlasák, J.; Dai, Y.C. Phylogeny and diversity of the morphologically similar polypore genera Rigidoporus, Physisporinus, Oxyporus, and Leucophellinus. Mycologia 2017, 109, 749–765. [Google Scholar] [CrossRef] [PubMed]
- Mendes, D.B.; Da Sliva, F.F.; Guarda, P.M.; Almeida, A.F.; de Oliveira, D.P.; Morais, P.B.; Guarda, E.A. Lipolytic enzymes with hydrolytic and esterification activities produced by filamentous fungi isolated from decomposition leaves in an aquatic environment. Enzym. Res. 2019, 8182425. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, C.M.D.; Nanson, J.D.; Patterson, E.I.; Forwood, J.K. Structure, function, and regulation of thioesterases. Prog. Lipid Res. 2020, 79, 101036. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Hu, S.; Peng, B.; Li, Z.H.; Yuan, X.H.; Xiao, S.J.; Fu, Y.P. Genome of Ganoderma species provides insights into the evolution, conifers substrate utilization, and terpene synthesis for Ganoderma tsugae. Front. Microbiol. 2021, 12, 724451. [Google Scholar] [CrossRef] [PubMed]
- Baby, S.; Johnson, A.J.; Govindan, B. Secondary metabolites from Ganoderma. Phytochemistry 2015, 114, 66–101. [Google Scholar] [CrossRef]
- Pattanayak, S.; Das, S.; Biswal, G. Ganoderma: The wild mushroom with wonderful health benefits. J. Pharmacogn. Phytochem. 2020, 9, 313–316. [Google Scholar] [CrossRef]
- Liu, Y.C.; Huang, L.H.; Hu, H.P.; Cai, M.J.; Liang, X.W.; Li, X.M.; Zhang, Z.; Xie, Y.Z.; Xiao, C.; Chen, S.D.; et al. Whole-genome assembly of Ganoderma leucocontextum (Ganodermataceae, Fungi) discovered from the Tibetan Plateau of China. G3 Genes Genomes Genet. 2021, 11, jkab337. [Google Scholar] [CrossRef] [PubMed]
- Schnell, N.; Entian, K.D.; Schneider, U.; Götz, F.; Zähner, H.; Kellner, R.; Jung, G. Prepeptide sequence of epidermin, a ribosomally synthesized antibiotic with four sulphide-rings. Nature 1988, 333, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Benson, K.F.; Stamets, P.; Davis, R.; Nally, R.; Taylor, A.; Slater, S.; Jensen, G.S. The mycelium of the Trametes versicolor (Turkey tail) mushroom and its fermented substrate each show potent and complementary immune activating properties in vitro. BMC Complement. Altern. Med. 2019, 19, 342. [Google Scholar] [CrossRef]
- Gao, X.; Qi, J.Y.; Ho, C.T.; Li, B.; Mu, J.J.; Zhang, Y.T.; Hu, H.P.; Mo, W.P.; Chen, Z.Z.; Xie, Y.Z. Structural characterization and immunomodulatory activity of a water-soluble polysaccharide from Ganoderma leucocontextum fruiting bodies. Carbohydr. Polym. 2020, 249, 116874. [Google Scholar] [CrossRef] [PubMed]
- Im, K.H.; Nguyen, T.K.; Choi, J.; Lee, T.S. In vitro antioxidant, anti-diabetes, anti-dementia, and inflammation inhibitory effect of Trametes pubescens fruiting body extracts. Molecules 2016, 21, 639. [Google Scholar] [CrossRef] [PubMed]
- Baldrian, P.; Valášková, V. Degradation of cellulose by basidiomycetous fungi. FEMS Microbiol. Rev. 2008, 32, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, A.; Bonnardel, F.; Dai, Y.C.; Imberty, A.; Martin, F.M.; Lisacek, F. A comprehensive phylogenetic and bioinformatics survey of lectins in the fungal kingdom. J. Fungi 2021, 7, 453. [Google Scholar] [CrossRef] [PubMed]
- Hage, H.; Rosso, M.N. Evolution of fungal carbohydrate-active enzyme portfolios and adaptation to plant cell-wall polymers. J. Fungi 2021, 7, 185. [Google Scholar] [CrossRef] [PubMed]
- Syed, K.; Nelson, D.R.; Riley, R.; Yadav, J.S. Genomewide annotation and comparative genomics of cytochrome P450 monooxygenases (P450s) in the polypore species Bjerkandera adusta, Ganoderma sp. and Phlebia brevispora. Mycologia 2013, 105, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Debets, F.; Yang, X.; Griffiths, A.J. Vegetative incompatibility in Neurospora: Its effect on horizontal transfer of mitochondrial plasmids and senescence in natural populations. Curr. Genet. 1994, 26, 113–119. [Google Scholar] [CrossRef]
- Nuss, D. Hypovirulence: Mycoviruses at the fungal–plant interface. Nat. Rev. Microbiol. 2005, 3, 632–642. [Google Scholar] [CrossRef]
- Madej, M.G. Comparative sequence–function analysis of the major facilitator superfamily: The “Mix-and-Match”. Methods Enzymol. 2015, 557, 521–549. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, Q.H.; Zhang, L.L.; Wang, Q.F.; Liang, L.; Liao, B.S. Genome-wide characterization and comparative analysis of MYB transcription factors in Ganoderma species. Genes Genomes Genet. 2020, 10, 2653–2660. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Name | Strain | Reference | Lifestyle | Order | Clade |
|---|---|---|---|---|---|
| Abortiporus biennis | CIRM-BRFM1778 | [24] | White-rot | Polyporales | residual polyporoid clade |
| Bjerkandera adusta | HHB-12826-SP | [11] | White-rot | Polyporales | phlebioid clade |
| Ceriporiopsis subvermispora | B | [26] | White-rot | Polyporales | gelatoporia clade |
| Dichomitus squalens | LYAD-421 SS1 | [27] | White-rot | Polyporales | core polyporoid clade |
| Earliella scabrosa | CIRM-BRFM 1817 | [24] | White-rot | Polyporales | core polyporoid clade |
| Epithele typhae | CBS 203.58 | [24] | White-rot | Polyporales | core polyporoid clade |
| Fomes fomentarius | CIRM-BRFM 1821 | [24] | White-rot | Polyporales | core polyporoid clade |
| Fomitiporia mediterranea | MF3/22 #7 | [27] | White-rot | Hymenochaetales | - |
| Ganoderma australe | Cui 17254 | this study | White-rot | Polyporales | core polyporoid clade |
| Ganoderma boninense | G3 | [13] | White-rot | Polyporales | core polyporoid clade |
| Ganoderma leucocontextum | Dai 12418 | this study | White-rot | Polyporales | core polyporoid clade |
| Ganoderma lingzhi | Cui 9166 | this study | White-rot | Polyporales | core polyporoid clade |
| Ganoderma lucidum | G.260125-1 | [10] | White-rot | Polyporales | core polyporoid clade |
| Ganoderma sinense | ZZ0214-1 | [12] | White-rot | Polyporales | core polyporoid clade |
| Ganoderma sp. | 10597 SS1 | [11] | White-rot | Polyporales | core polyporoid clade |
| Heterobasidion annosum | TC32-1 | [28] | White-rot | Russulales | - |
| Hexagonia nitida | CIRM-BRFM 1802 | [24] | White-rot | Polyporales | core polyporoid clade |
| Hydnopolyporus fimbriatus | CBS384.51 | [24] | White-rot | Polyporales | phlebioid clade |
| Irpex lacteus | CCBAS Fr. 238 617/93 | [24] | White-rot | Polyporales | phlebioid clade |
| Leiotrametes lactinea | CIRM-BRFM 1664 | [24] | White-rot | Polyporales | core polyporoid clade |
| Leiotrametes menziesii | CIRM-BRFM 1781 | [24] | White-rot | Polyporales | core polyporoid clade |
| Lentinus tigrinus | ALCF2SS1-7 | [29] | White-rot | Polyporales | core polyporoid clade |
| Obba rivulosa | 3A-2 | [30] | White-rot | Polyporales | gelatoporia clade |
| Panus rudis | PR-1116 ss-1 | [24] | White-rot | Polyporales | residual polyporoid clade |
| Phanerochaete carnosa | HHB-10118-Sp | [31] | White-rot | Polyporales | phlebioid clade |
| Phanerochaete chrysosporium | RP-78 | [32] | White-rot | Polyporales | phlebioid clade |
| Phlebia brevispora | HHB-7030 SS6 | [11] | White-rot | Polyporales | phlebioid clade |
| Phlebia centrifuga | FBCC195 | [33] | White-rot | Polyporales | phlebioid clade |
| Phlebia radiata | FBCC0043-79 | [34] | White-rot | Polyporales | phlebioid clade |
| Phlebiopsis gigantea | 5-6 | [35] | White-rot | Polyporales | phlebioid clade |
| Polyporus arcularius | HBB13444 | [36] | White-rot | Polyporales | core polyporoid clade |
| Polyporus brumalis | BRFM 1820 | [37] | White-rot | Polyporales | core polyporoid clade |
| Polyporus squamosus | CCBS 676 | [24] | White-rot | Polyporales | core polyporoid clade |
| Pycnoporus cinnabarinus | BRFM 137 | [38] | White-rot | Polyporales | core polyporoid clade |
| Pycnoporus sanguineus | BRFM 1264 | [39] | White-rot | Polyporales | core polyporoid clade |
| Rigidoporus microporus | ED310 | [40] | White-rot | Polyporales | - |
| Stereum hirsutum | FP-91666 SS1 | [27] | White-rot | Russulales | - |
| Trametes pubescens | FBCC735 | [41] | White-rot | Polyporales | core polyporoid clade |
| Trametes versicolor | FP-101664 SS1 | [27] | White-rot | Polyporales | core polyporoid clade |
| Trametopsis cervina | CIRM-BRFM 1824 | [24] | White-rot | Polyporales | core polyporoid clade |
| Amylocystis lapponica | SKaAmylap13 | [24] | Brown-rot | Polyporales | antrodia clade |
| Antrodia serialis | Sig1Antser10 | [24] | Brown-rot | Polyporales | antrodia clade |
| Antrodia sinuosa | LB1 | PRJNA196036 | Brown-rot | Polyporales | antrodia clade |
| Daedalea quercina | L15889 ss-12 | [42] | Brown-rot | Polyporales | antrodia clade |
| Fibroporia radiculosa | TFFH 294 | [43] | Brown-rot | Polyporales | antrodia clade |
| Fomitopsis betulina | CIRM-BRFM 1772 | [24] | Brown-rot | Polyporales | antrodia clade |
| Fomitopsis pinicola | FP-58527 SS1 | [27] | Brown-rot | Polyporales | antrodia clade |
| Laetiporus sulphureus | 93-53 | [42] | Brown-rot | Polyporales | antrodia clade |
| Postia placenta | MAD-698-R-SB12 | [44] | Brown-rot | Polyporales | antrodia clade |
| Rhodofomes roseus | CIRM-BRFM 1785 | [24] | Brown-rot | Polyporales | antrodia clade |
| Wolfiporia cocos | MD-104 SS10 | [27] | Brown-rot | Polyporales | antrodia clade |
| Amanita muscaria | Koide | [45] | Ectomycorrhizal | Agaricales | - |
| Laccaria bicolor | S238N-H82 | [46] | Ectomycorrhizal | Agaricales | - |
| Pisolithus microcarpus | 441 | [45] | Ectomycorrhizal | Boletales | - |
| Rhizopogon vinicolor | AM-OR11-026 | [47] | Ectomycorrhizal | Boletales | - |
| Piriformospora indica | DSM 11827 | [48] | Endophyte | Sebacinales | - |
| Melampsora larici-populina | 98AG31 | [49] | Pathogen | Pucciniales | - |
| Ustilago maydis | 521 | [50] | Pathogen | Ustilaginales | - |
| Genome Feature | G. australe | G. leucocontextum | G. lingzhi |
|---|---|---|---|
| Genome size (Mb) | 84.27 | 60.34 | 60.56 |
| Number of scaffolds | 93 | 843 | 342 |
| Scaffold L50 (bp) | 1,745,385 | 205,166 | 402,014 |
| Scaffold N50 | 17 | 66 | 37 |
| Longest scaffold (bp) | 4,455,856 | 1,715,371 | 2,154,085 |
| Shortest scaffold (bp) | 33,016 | 1008 | 980 |
| GC content (%) | 55.48 | 55.95 | 55.88 |
| Protein-coding genes | 20,460 | 16,952 | 15,007 |
| Average gene length (bp) | 1582 | 1846 | 1605 |
| Complete BUSCOs (%) | 84.6 | 99.8 | 73.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.-F.; Lebreton, A.; Xing, J.-H.; Fang, Y.-X.; Si, J.; Morin, E.; Miyauchi, S.; Drula, E.; Ahrendt, S.; Cobaugh, K.; et al. Phylogenomics and Comparative Genomics Highlight Specific Genetic Features in Ganoderma Species. J. Fungi 2022, 8, 311. https://doi.org/10.3390/jof8030311
Sun Y-F, Lebreton A, Xing J-H, Fang Y-X, Si J, Morin E, Miyauchi S, Drula E, Ahrendt S, Cobaugh K, et al. Phylogenomics and Comparative Genomics Highlight Specific Genetic Features in Ganoderma Species. Journal of Fungi. 2022; 8(3):311. https://doi.org/10.3390/jof8030311
Chicago/Turabian StyleSun, Yi-Fei, Annie Lebreton, Jia-Hui Xing, Yu-Xuan Fang, Jing Si, Emmanuelle Morin, Shingo Miyauchi, Elodie Drula, Steven Ahrendt, Kelly Cobaugh, and et al. 2022. "Phylogenomics and Comparative Genomics Highlight Specific Genetic Features in Ganoderma Species" Journal of Fungi 8, no. 3: 311. https://doi.org/10.3390/jof8030311
APA StyleSun, Y.-F., Lebreton, A., Xing, J.-H., Fang, Y.-X., Si, J., Morin, E., Miyauchi, S., Drula, E., Ahrendt, S., Cobaugh, K., Lipzen, A., Koriabine, M., Riley, R., Kohler, A., Barry, K., Henrissat, B., Grigoriev, I. V., Martin, F. M., & Cui, B.-K. (2022). Phylogenomics and Comparative Genomics Highlight Specific Genetic Features in Ganoderma Species. Journal of Fungi, 8(3), 311. https://doi.org/10.3390/jof8030311