
Transcriptional Network in Colletotrichum gloeosporioides Mutants Lacking Msb2 or Msb2 and Sho1


Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Strains and Culture Conditions
2.2. Analyses of Vegetative Growth and Conidiation
2.3. RNA Extraction
2.4. Preparation of Transcriptome Sequencing Libraries
2.5. Transcriptome Analysis and Sequence Assembly
2.6. Analysis of Differential Gene Expression
2.7. qRT-PCR
2.8. Statistical Analysis
3. Results
3.1. ΔCgMsb2 and ΔCgMsb2Sho1 Exhibit Defects in Vegetative Growth and Conidiation
3.2. RNA Sequencing (RNA-seq) Profiles and Identification of Differentially Expressed Genes (DEGs)
3.3. DEGs among the Transcriptomes
3.4. GO Analysis of DEGs
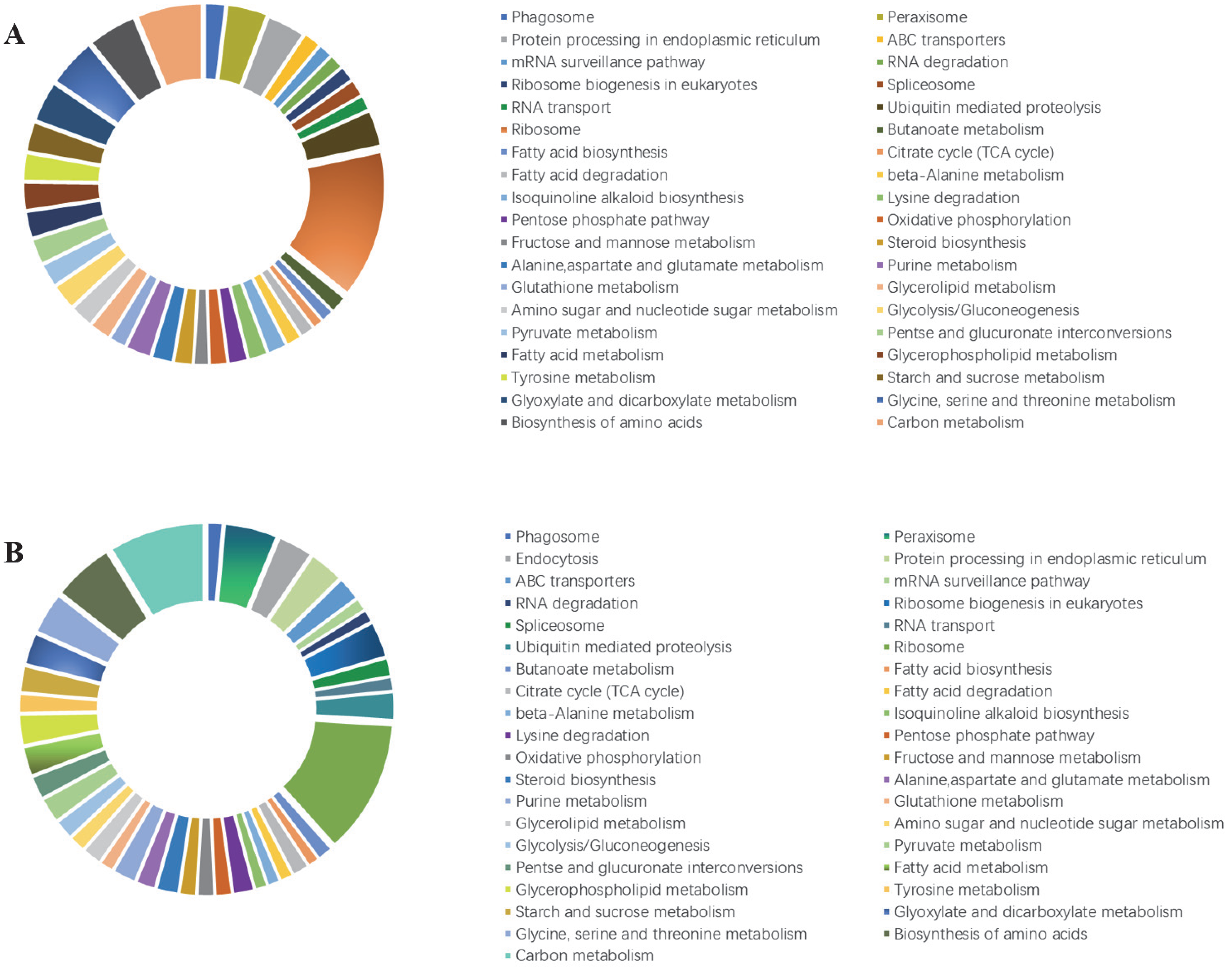
3.5. Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis of DEGs
3.6. Hydrolase-Related DEGs
3.7. Differential Expression of Candidate ABC Transporter Genes
3.8. DEGs Involved in the MAPK Signaling Pathway
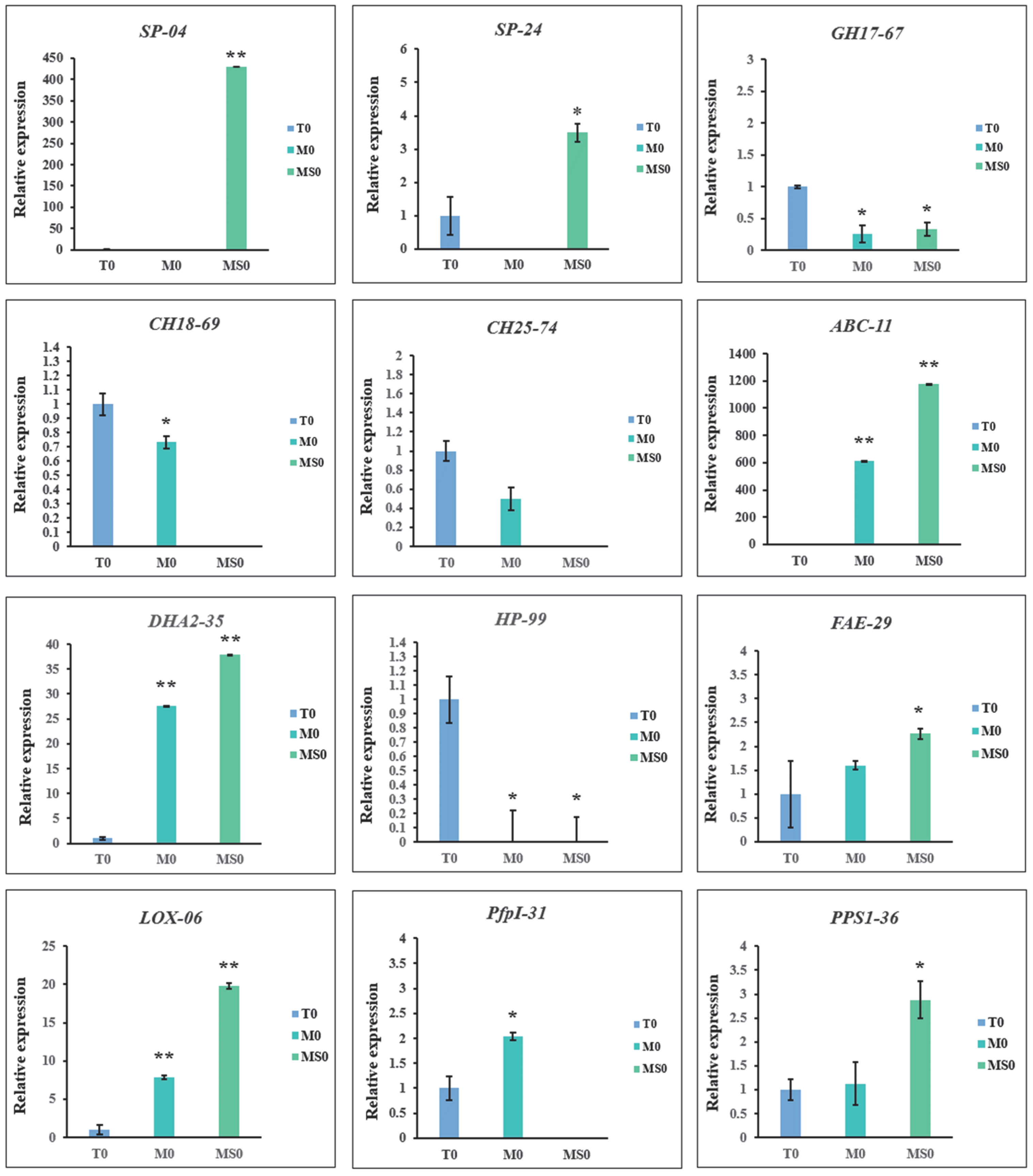
3.9. Verification of Gene Expression by qRT-PCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mansfield, J.; Genin, S.; Magori, S.; Citovsky, V.; Sriariyanum, M.; Ronald, P.; Dow, M.; Verdier, V.; Beer, S.V.; Machado, M.A. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Flaishman, M.A.; Hwang, C.S.; Kolattukudy, P.E. Involvement of protein phosphorylation in the induction of appressorium formation in Colletotrichum gloeosporioides by its host surface wax and ethylene. Physiol. Mol. Plant Pathol. 1995, 47, 103–117. [Google Scholar] [CrossRef]
- Crous, P.W.; Gams, W.; Stalpers, J.A.; Robert, V.; Stegehuis, G. MycoBank: An online initiative to launch mycology into the 21st century. Stud. Mycol. 2004, 50, 19–22. [Google Scholar]
- Droby, S. Biological control of postharvest diseases of fruits and vegetables: Difficulties and challenges. Phytopathol. Pol. 2006, 39, 105–117. [Google Scholar]
- Gomes, S.; Prieto, P.; Martins-Lopes, P.; Carvalho, T.; Martin, A.; Guedes-Pinto, H. Development of Colletotrichum acutatum on Tolerant and Susceptible Olea europaea L. cultivars: A Microscopic Analysis. Mycopathologia 2009, 168, 203. [Google Scholar] [CrossRef][Green Version]
- Money, N.P. Mechanics of Invasive Fungal Growth and the Significance of Turgor in Plant Infection. Dev. Plant Pathol. 1998, 107, 5663–5669. [Google Scholar]
- Pereira, I.S.; Abreu, M.; Alves, E.; Ferreira, J.B. Histopathological studies of the interaction Colletotrichum gloeosporioides—Coffee trees. Bragantia 2009, 68, 117–123. [Google Scholar] [CrossRef]
- Mendoza-Mendoza, A.; Berndt, P.; Djamei, A.; Weise, C.; Linne, U.; Marahiel, M.; Vraneš, M.; Kämper, J.; Kahmann, R. Physical-chemical plant-derived signals induce differentiation in Ustilago maydis. Mol. Microbiol. 2009, 71, 895–911. [Google Scholar] [CrossRef]
- Xiao, J.Z.; Watanabe, T.; Sekido, S.; Choi, W.B.; Kamakura, T.; Yamaguchi, I. An Anti-hydrotactic Response and Solid Surface Recognition of Germ Tubes of the Rice Blast Fungus, Magnaporthe gvisea. Biosci. Biotechnol. Biochem. 1997, 61, 1225–1229. [Google Scholar] [CrossRef][Green Version]
- Xu, H.; Mendgen, K. Targeted cell wall degradation at the penetration site of cowpea rust basidiosporelings. Mol. Plant-Microbe Interact. 1997, 10, 87–94. [Google Scholar] [CrossRef][Green Version]
- Abramovitch, R.B.; Yang, G.; Kronstad, J.W. The ukb1 gene encodes a putative protein kinase required for bud site selection and pathogenicity in Ustilago maydis. Fungal Genet. Biol. 2002, 37, 98–108. [Google Scholar] [CrossRef]
- Oh, Y.; Donofrio, N.; Pan, H.; Coughlan, S.; Dean, R.A. Transcriptome analysis reveals new insight into appressorium formation and function in the rice blast fungus Magnaporthe oryzae. Genome Biol. 2008, 9, R85. [Google Scholar] [CrossRef] [PubMed]
- Both, M.; Eckert, S.E.; Csukai, M.; Müller, E.; Dimopoulos, G.; Spanu, P.D. Transcript Profiles of Blumeria graminis Development During Infection Reveal a Cluster of Genes That Are Potential Virulence Determinants. Mol. Plant Microbe. Interact. 2005, 18, 125–133. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leroch, M.; Kleber, A.; Silva, E.; Coenen, T.; Koppenhofer, D.; Shmaryahu, A.; Valenzuela, P.; Hahn, M. Transcriptome Profiling of Botrytis cinerea Conidial Germination Reveals Upregulation of Infection-Related Genes during the Prepenetration Stage. Eukaryot. Cell 2013, 12, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Osés-Ruiz, M.; Cruz-Mireles, N.; Martin-Urdiroz, M.; Soanes, D.M.; Eseola, A.B.; Tang, B.; Derbyshire, P.; Nielsen, M.; Cheema, J.; Were, V. Appressorium-mediated plant infection by Magnaporthe oryzae is regulated by a Pmk1-dependent hierarchical transcriptional network. Nat. Microbiol. 2021, 6, 1383–1397. [Google Scholar] [CrossRef] [PubMed]
- Dagdas, Y.F.; Yoshino, K.; Dagdas, G.; Ryder, L.S.; Bielska, E.; Steinberg, G.; Talbot, N.J. Septin-Mediated Plant Cell Invasion by the Rice Blast Fungus, Magnaporthe oryzae. Science 2012, 336, 1590–1595. [Google Scholar] [CrossRef]
- Ryder, L.S.; Dagdas, Y.F.; Mentlak, T.A.; Kershaw, M.J.; Thornton, C.R. NADPH oxidases regulate septin-mediated cytoskeletal remodeling during plant infection by the rice blast fungus. Proc. Acad. Natl. Sci. USA 2013, 110, 3179–3184. [Google Scholar] [CrossRef]
- Ryder, L.S.; Dagdas, Y.F.; Kershaw, M.J.; Venkataraman, C.; Madzvamuse, A.; Yan, X.; Cruz-Mireles, N.; Soanes, D.M.; Oses-Ruiz, M.; Styles, V. A sensor kinase controls turgor-driven plant infection by the rice blast fungus. Nature 2019, 574, 423–427. [Google Scholar] [CrossRef]
- Liu, W.; Zhou, X.; Li, G.; Li, L.; Kong, L.; Wang, C.; Zhang, H.; Xu, J.R.; Howlett, B.J. Multiple Plant Surface Signals are Sensed by Different Mechanisms in the Rice Blast Fungus for Appressorium Formation. PLoS Pathog. 2011, 7, e1001261. [Google Scholar] [CrossRef]
- Keshet, Y.; Seger, R. The MAP Kinase Signaling Cascades: A System of Hundreds of Components Regulates a Diverse Array of Physiological Functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar]
- Wang, X.; Tournier, C. Regulation of cellular functions by the ERK5 signalling pathway. Cell. Signal. 2006, 18, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Lanver, D.; Mendoza-Mendoza, A.; Brachmann, A.; Kahmann, R. Sho1 and Msb2-related proteins regulate appressorium development in the smut fungus Ustilago maydis. Plant Cell 2010, 22, 2085–2101. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xue, Z.; Liu, H.; Xu, J.R.; Sheppard, D.C. Mitogen-activated protein kinase signaling in plant pathogenic fungi. PLoS Pathog. 2018, 14, e1006875. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, D.; Tian, C. Mucin Msb2 cooperates with the transmembrane protein Sho1 in various plant surface signal sensing and pathogenic processes in the poplar anthracnose fungus Colletotrichum gloeosporioides. Mol. Plant Pathol. 2021, 22, 1553–1573. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Dewey, C.N.; Li, B. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Young, M.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 1165–1188. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, A.; Driessen, A.J. Phylogenetic analysis of fungal ABC transporters. BMC Genom. 2010, 11, 177. [Google Scholar] [CrossRef] [PubMed]
- Kema, G.; Van, D.; Mendes, O.; Verstappen, E.; Lankhorst, R.K.; Sandbrink, H.; Ate, V.; Zwiers, L.H.; Csukai, M.; Waalwijk, C. Large-scale gene discovery in the septoria tritici blotch fungus Mycosphaerella graminicola with a focus on in planta expression. Mol. Plant-Microbe Interact. 2008, 21, 1249. [Google Scholar] [CrossRef] [PubMed]
- Turrà, D.; Segorbe, D.; Di Pietro, A. Protein Kinases in Plant-Pathogenic Fungi: Conserved Regulators of Infection. Annu. Rev. Phytopathol. 2014, 52, 267–288. [Google Scholar] [CrossRef] [PubMed]
- Levitz, S.M.; Madhani, H.D. Innate Recognition of Fungal Cell Walls. PLoS Pathog. 2010, 6, e1000758. [Google Scholar] [CrossRef]
- Levitz, S.M.; Specht, C.A. The molecular basis for the immunogenicity of Cryptococcus neoformans mannoproteins. FEMS Yeast Res. 2006, 6, 513–524. [Google Scholar] [CrossRef]
- Levitz, S.M.; Huang, H.; Ostroff, G.R.; Specht, C.A. Exploiting fungal cell wall components in vaccines. Semin. Immunopathol. 2015, 37, 199–207. [Google Scholar] [CrossRef]
- Jørgensen, H.; Vibe-Pedersen, J.; Larsen, J.; Felby, C. Liquefaction of lignocellulose at high-solids concentrations. Biotechnol. Bioeng. 2007, 96, 862–870. [Google Scholar] [CrossRef]
- Vanfossen, A.L.; Ozdemir, I.; Zelin, S.L.; Kelly, R.M. Glycoside hydrolase inventory drives plant polysaccharide deconstruction by the extremely thermophilic bacterium Caldicellulosiruptor saccharolyticus. Biotechnol. Bioeng. 2011, 108, 1559–1569. [Google Scholar] [CrossRef]
- Rodríguez-López, E.S.; González-Prieto, J.M.; Mayek-Pérez, N. La Infección de Colletotrichum gloeosporioides (Penz.) Penz. y Sacc. en Aguacatero (Persea americana Mill.): Aspectos Bioquímicos y Genéticos. Rev. Mex. Fitopatol. 2009, 27, 53–63. [Google Scholar]
- Van Vu, B.; Itoh, K.; Nguyen, Q.B.; Tosa, Y.; Nakayashiki, H. Cellulases belonging to glycoside hydrolase families 6 and 7 contribute to the virulence of Magnaporthe oryzae. Mol. Plant-Microbe Interact. 2012, 25, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012, 44, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Ab, A.; Am, B.; Jp, B.; Pf, B.; Rpa, C. ABCG: A new fold of ABC exporters and a whole new bag of riddles!—ScienceDirect. Adv. Protein Chem. Struct. Biol. 2021, 123, 163–191. [Google Scholar]
- Sun, C.B.; Suresh, A.; Naqvi, D.N.I. A Multidrug Resistance Transporter in Magnaporthe Is Required for Host Penetration and for Survival during Oxidative Stress. Plant Cell 2006, 18, 3686–3705. [Google Scholar] [CrossRef]
- Kim, Y.; Park, S.-Y.; Kim, D.; Choi, J.; Lee, Y.-H.; Lee, J.-H.; Choi, W. Genome-scale analysis of ABC transporter genes and characterization of the ABCC type transporter genes in Magnaporthe oryzae. Genomics 2013, 101, 354–361. [Google Scholar] [CrossRef]
- Kovalchuk, A.; Lee, Y.-H.; Asiegbu, F.O. Diversity and evolution of ABC proteins in basidiomycetes. Mycologia 2013, 105, 1456–1470. [Google Scholar] [CrossRef]
- Zhou, Z.; Wu, J.; Wang, M.; Zhang, J. ABC protein CgABCF2 is required for asexual and sexual development, appressorial formation and plant infection in Colletotrichum gloeosporioides. Microb. Pathog. 2017, 110, 85–92. [Google Scholar] [CrossRef]
- Ghada, A.A.; Reno, T.; Katharina, D.; Petr, K.; Deising, H.B.; Wirsel, S.; Yu, J.H. Identification of ABC Transporter Genes of Fusarium graminearum with Roles in Azole Tolerance and/or Virulence. PLoS ONE 2013, 8, e79042. [Google Scholar]
- Wang, X.; Lu, D.; Tian, C. Mitogen-activated protein kinase cascade CgSte50-Ste11-Ste7-Mk1 regulates infection-related morphogenesis in the poplar anthracnose fungus Colletotrichum gloeosporioides. Microbiol. Res. 2021, 248, 126748. [Google Scholar] [CrossRef]
- Leroch, M.; Mueller, N.; Hinsenkamp, I.; Hahn, M. The signalling mucin M sb2 regulates surface sensing and host penetration via BMP1 MAP kinase signalling in Botrytis cinerea. Mol. Plant Pathol. 2015, 16, 787–798. [Google Scholar] [CrossRef]
- Kaneko, T.; Li, L.; Li, S. The SH3 domain—A family of versatile peptide-and protein-recognition module. Front Biosci. 2008, 13, 4938–4952. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Data (bp) | Clean Reads | Q30 (%) | Total Mapping (%) | Uniquely Mapping (%) |
|---|---|---|---|---|---|
| T0 | 6,286,653,483 | 21,019,807 | 94.81% | 91.01% | 90.66% |
| M0 | 8,905,212,135 | 29,816,717 | 94.01% | 95.79% | 95.54% |
| MS0 | 9,194,480,861 | 30,799,568 | 94.74% | 95.99% | 95.74% |
| Condition | ID | Fold Change | p Value | Annotation |
|---|---|---|---|---|
| T0-M0-up | EVM0005631 | 807.4227 | 1.46 × 10−61 | Glyoxylate reductase |
| EVM0008231 | 277.6544 | 0.0000608 | DJ-1/PfpI family, transcription factor | |
| EVM0015745 | 204.4752 | 1.14 × 10−10 | Glycosyl hydrolase family 31 | |
| EVM0012935 | 133.5726 | 2.74 × 10−9 | MFS transporter | |
| EVM0008406 | 106.5425 | 3.04 × 10−27 | Linoleate 9S-lipoxygenase 1 | |
| EVM0009311 | 18.0056 | 0.001193 | BEA3, ABC transporter | |
| EVM0003035 | 12.4763 | 0.000566 | pmd1, ABC transporter | |
| EVM0006436 | 11.2868 | 0.000732 | PPS1, Protein phosphatase | |
| EVM0000077 | 9.1756 | 0.002521 | FUM19, ABC transporter | |
| EVM0007629 | 8.7358 | 5.10 × 10−14 | Feruloyl esterase | |
| T0-M0-down | EVM0008257 | 316.9458 | 3.09 × 10−19 | Pectate trisaccharide-lyase |
| EVM0002108 | 62.7619 | 1.17 × 10−20 | Pectate lyase | |
| EVM0010374 | 55.0161 | 7.21 × 10−9 | Polysaccharide deacetylase | |
| EVM0010120 | 50.3543 | 1.40 × 10−6 | Glycosyl hydrolase family 12 | |
| EVM0006919 | 31.4844 | 0.000891085 | erg-4, methyltransferase | |
| EVM0009169 | 15.2328 | 0.001032984 | Glycosyl hydrolase family 18 | |
| EVM0013863 | 14.7652 | 1.39 × 10−6 | Cutinase | |
| EVM0006990 | 13.1765 | 9.29 × 10−10 | Scytalone dehydratase | |
| EVM0010167 | 7.7034 | 1.84 × 10−5 | Glycosyl hydrolase family 17 | |
| EVM0000353 | 7.1256 | 7.81 × 10−8 | Guanine nucleotide-binding protein | |
| T0-MS0-up | EVM0002782 | 680.7201 | 5.15 × 10−63 | Carboxylesterase |
| EVM0005631 | 577.5573 | 4.67 × 10−170 | Glyoxylate reductase | |
| EVM0005555 | 156.8511 | 2.32 × 10−12 | Nitrogen assimilation transcription factor | |
| EVM0015745 | 90.8438 | 4.78 × 10−14 | Glycosyl hydrolase family 31 | |
| EVM0009311 | 19.3072 | 3.04 × 10−10 | BEA3, ABC transporter | |
| EVM0008406 | 14.4585 | 3.47 × 10−34 | Linoleate 9S-lipoxygenase 1 | |
| EVM0006436 | 4.8744 | 0.00386738 | PPS1, protein phosphatase | |
| EVM0000077 | 7.3816 | 0.000153315 | FUM19, ABC transporter | |
| EVM0005624 | 4.9106 | 7.34 × 10−3 | E3 ubiquitin-protein ligase, sporulation | |
| EVM0009704 | 4.1047 | 5.11 × 10−5 | conidium formation | |
| T0-MS0-down | EVM0001785 | 834.5262 | 1.79 × 10−10 | Pectate lyase |
| EVM0002699 | 588.0388 | 5.66 × 10−85 | Hypothetical protein | |
| EVM0008890 | 321.3349 | 3.59 × 10−33 | YheN, polysaccharide deacetylase | |
| EVM0002162 | 275.5874 | 1.33 × 10−37 | isp4, transporter protein | |
| EVM0006990 | 70.4276 | 1.47 × 10−15 | Scytalone dehydratase | |
| EVM0006183 | 15.7665 | 1.67 × 10−18 | cAMP-dependent protein kinase | |
| EVM0010120 | 13.0258 | 8.16 × 10−9 | Glycosyl hydrolase family 12 | |
| EVM0000353 | 8.8958 | 2.96 × 10−25 | Guanine nucleotide-binding protein | |
| EVM0013863 | 8.0850 | 0.000000818 | Cutinase | |
| EVM0001921 | 6.6476 | 0.005086478 | Inositol 5-phosphatase | |
| EVM0010374 | 4.8228 | 0.007172842 | Polysaccharide deacetylase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, N.; Meng, F.; Tian, C. Transcriptional Network in Colletotrichum gloeosporioides Mutants Lacking Msb2 or Msb2 and Sho1. J. Fungi 2022, 8, 207. https://doi.org/10.3390/jof8020207
Liu N, Meng F, Tian C. Transcriptional Network in Colletotrichum gloeosporioides Mutants Lacking Msb2 or Msb2 and Sho1. Journal of Fungi. 2022; 8(2):207. https://doi.org/10.3390/jof8020207
Chicago/Turabian StyleLiu, Na, Fanli Meng, and Chengming Tian. 2022. "Transcriptional Network in Colletotrichum gloeosporioides Mutants Lacking Msb2 or Msb2 and Sho1" Journal of Fungi 8, no. 2: 207. https://doi.org/10.3390/jof8020207
APA StyleLiu, N., Meng, F., & Tian, C. (2022). Transcriptional Network in Colletotrichum gloeosporioides Mutants Lacking Msb2 or Msb2 and Sho1. Journal of Fungi, 8(2), 207. https://doi.org/10.3390/jof8020207