
Whole-Genome Analysis of Starmerella bacillaris CC-PT4 against MRSA, a Non-Saccharomyces Yeast Isolated from Grape


Abstract
1. Introduction
2. Materials and Methods
2.1. Yeast Strain and Growth Conditions
2.2. Genome Sequencing and Assembly
2.3. Genome Annotation and Analyses
2.4. Nucleotide Sequence Accession Number
3. Results
3.1. Genome Sequencing and Assembly
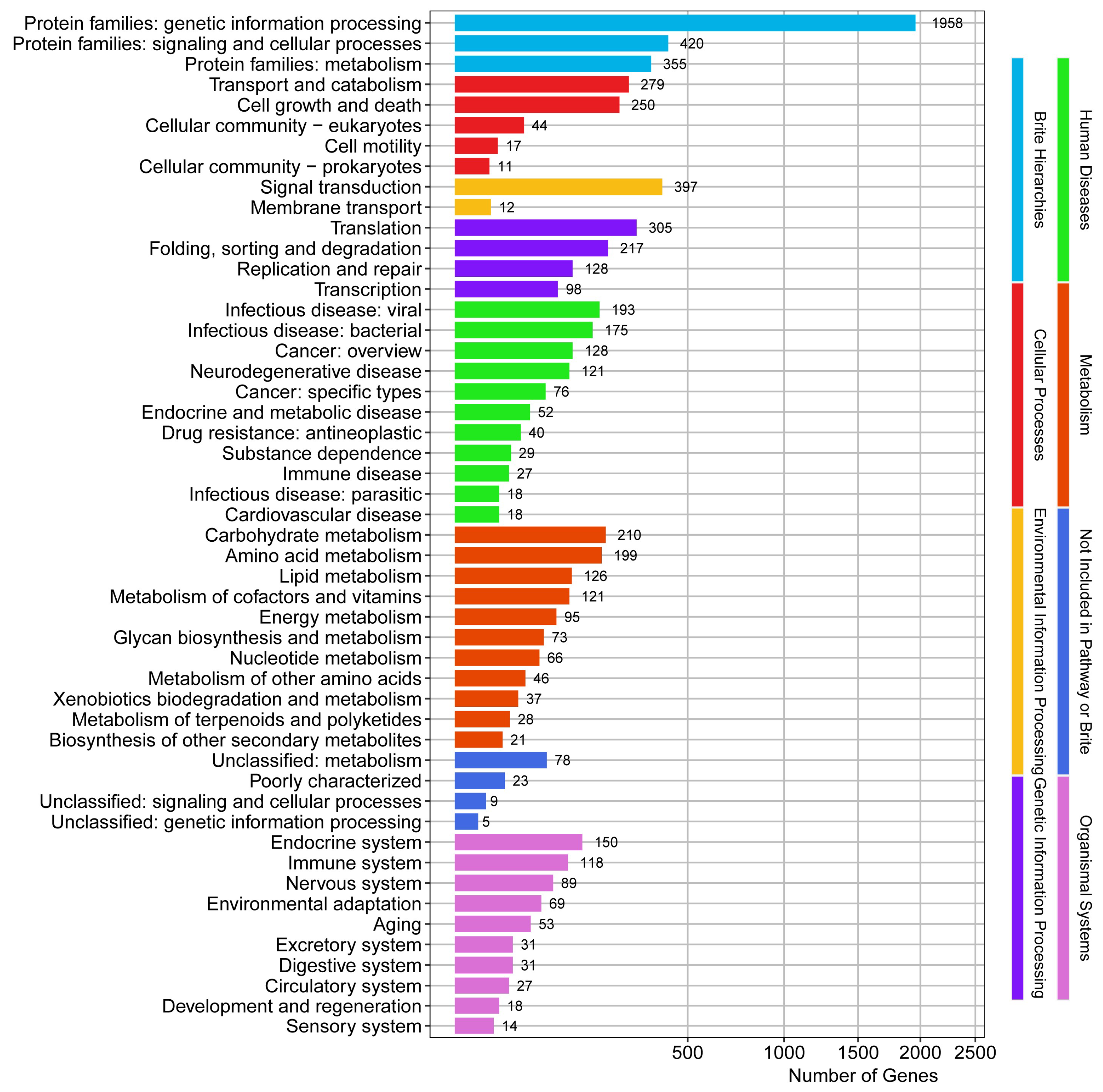
3.2. Genomic Functional Element Profiling
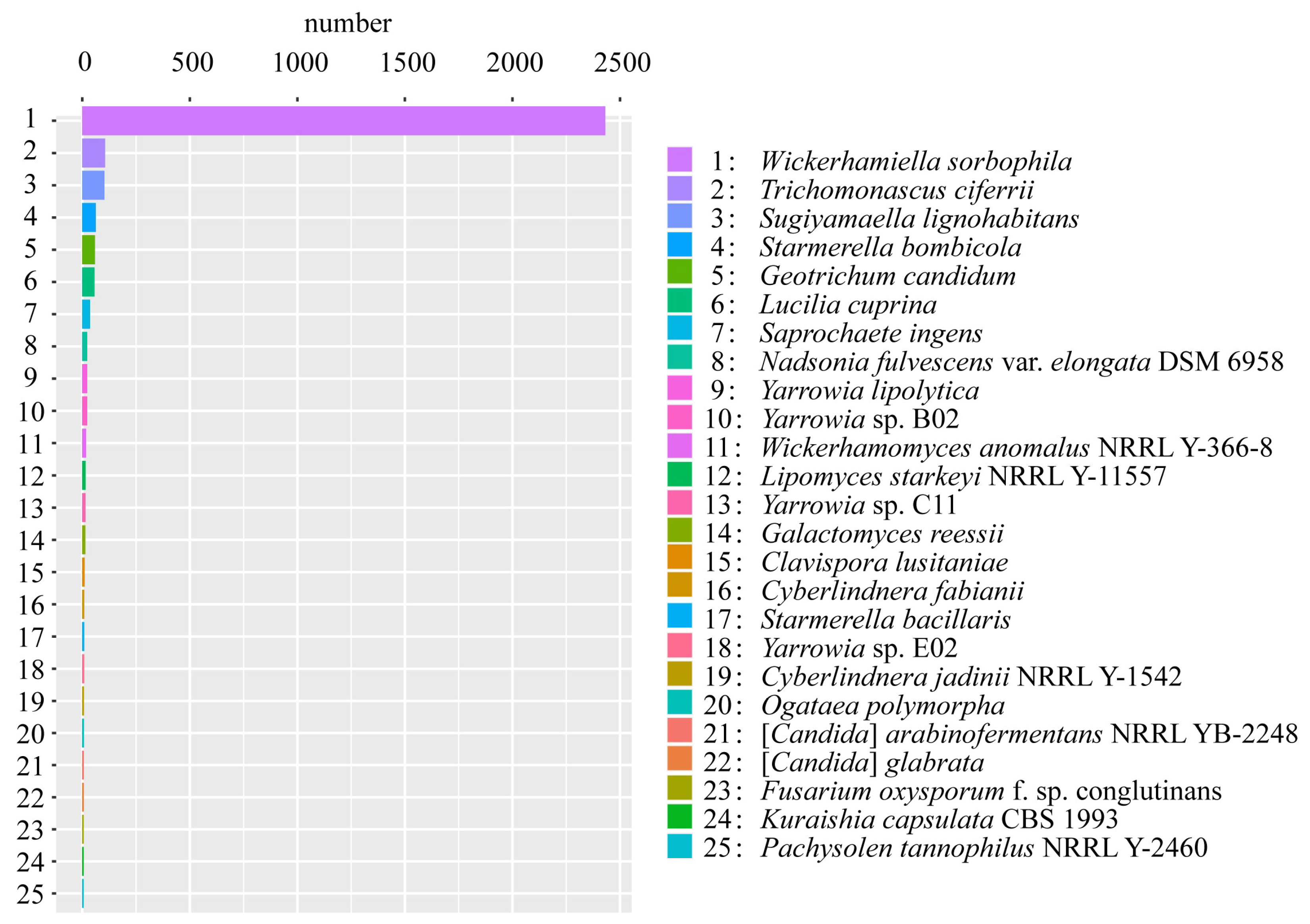
3.3. Gene Annotation
3.4. CAZy Analysis
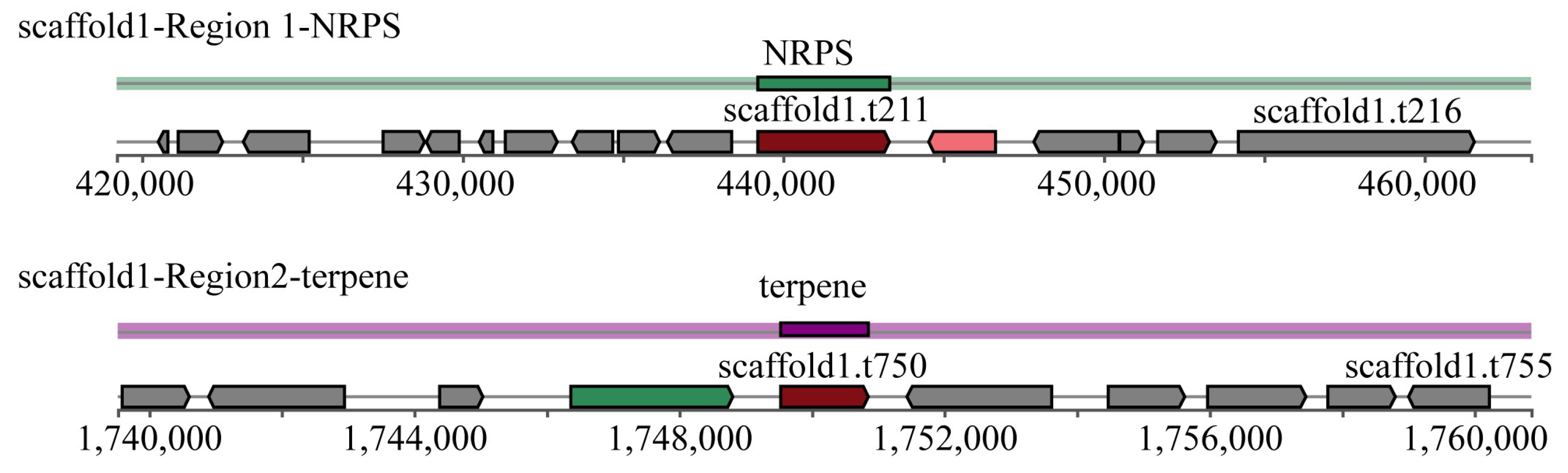
3.5. Secondary Metabolite Biosynthesis Gene Clusters
3.6. Killer Toxin and Lysostaphin Encoding Genes
3.7. Adaptation to Stress Analysis
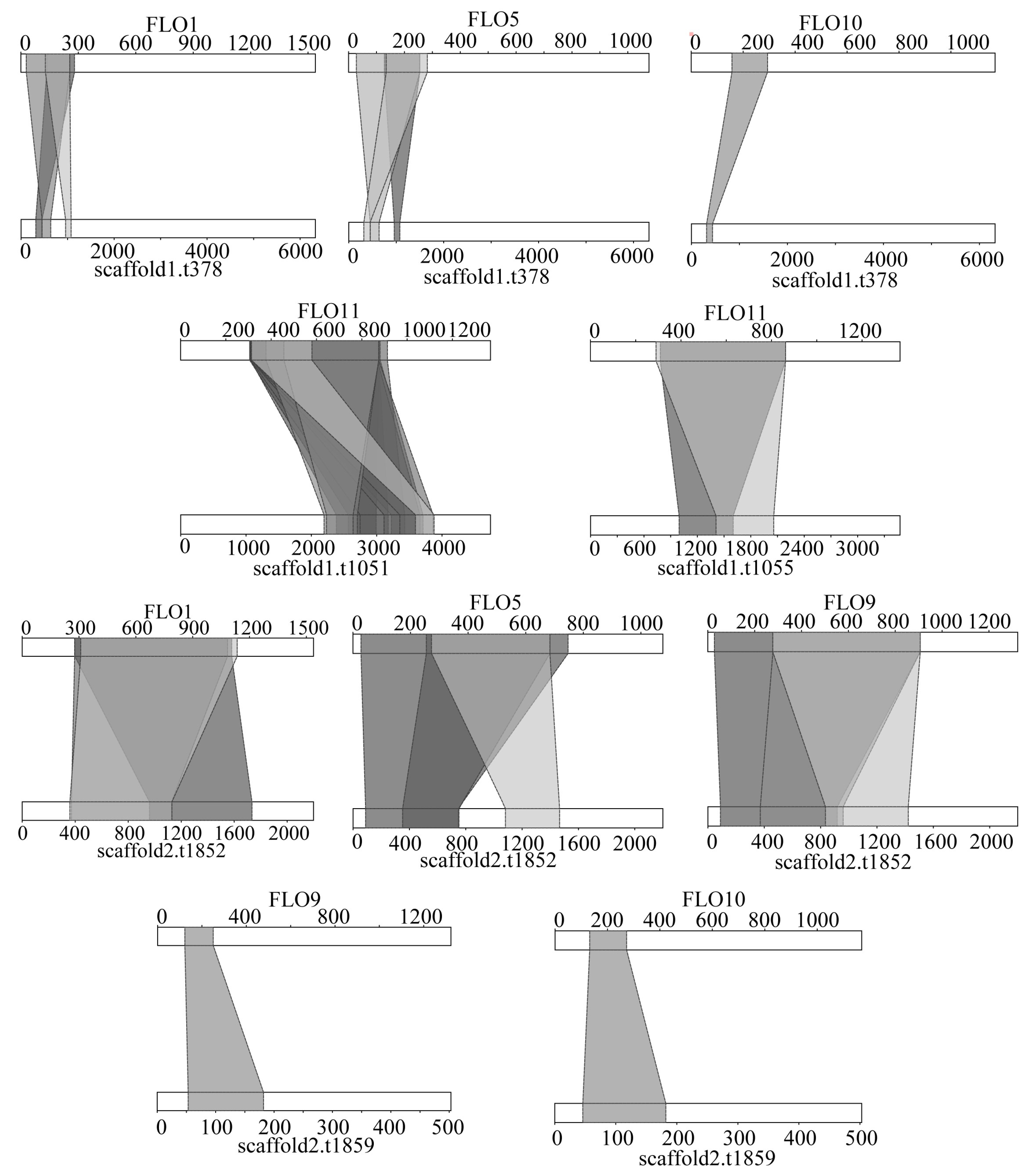
3.8. Flocculation and Adhesion Analysis
3.9. Drug Resistance Gene Analysis
3.10. Pathogenicity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raymond Eder, M.L.; Conti, F.; Bely, M.; Masneuf-Pomarède, I.; Albertin, W.; Rosa, A.L. Vitis species, vintage, and alcoholic fermentation do not drive population structure in Starmerella bacillaris (synonym Candida zemplinina) species. Yeast 2019, 36, 411–420. [Google Scholar] [CrossRef]
- Lemos, W.J.F., Jr.; Bovo, B.; Nadai, C.; Crosato, G.; Carlot, M.; Favaron, F.; Giacomini, A.; Corich, V. Biocontrol ability and action mechanism of Starmerella bacillaris (Synonym Candida zemplinina) isolated from wine musts against gray mold disease agent Botrytis cinerea on grape and their effects on alcoholic fermentation. Front. Microbiol. 2016, 7, 1249. [Google Scholar] [CrossRef]
- Mills, D.A.; Johannsen, E.A.; Cocolin, L. Yeast diversity and persistence in botrytis-affected wine fermentations. Appl. Environ. Microbiol. 2002, 68, 4884–4893. [Google Scholar] [CrossRef] [PubMed]
- Sipiczki, M. Candida zemplinina sp. nov., an osmotolerant and psychrotolerant yeast that ferments sweet botrytized wines. Int. J. Syst. Evol. Microbiol. 2003, 53, 2079–2083. [Google Scholar] [CrossRef] [PubMed]
- Duarte, F.L.; Pimentel, N.H.; Teixeira, A.; Fonseca, Á. Saccharomyces bacillaris is not a synonym of Candida stellata: Reinstatement as Starmerella bacillaris comb. nov. Antonie Van Leeuwenhoek 2012, 102, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Englezos, V.; Rantsiou, K.; Torchio, F.; Rolle, L.; Gerbi, V.; Cocolin, L. Exploitation of the non-Saccharomyces yeast Starmerella bacillaris (synonym Candida zemplinina) in wine fermentation: Physiological and molecular characterizations. Int. J. Food Microbiol. 2015, 199, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mas, A.; Esteve-Zarzoso, B. The interaction between Saccharomyces cerevisiae and non-Saccharomyces yeast during alcoholic fermentation is species and strain specific. Front. Microbiol. 2016, 7, 502. [Google Scholar] [CrossRef]
- Englezos, V.; Rantsiou, K.; Cravero, F.; Torchio, F.; Pollon, M.; Fracassetti, D.; Ortiz-Julien, A.; Gerbi, V.; Rolle, L.; Cocolin, L. Volatile profile of white wines fermented with sequential inoculation of Starmerella bacillaris and Saccharomyces cerevisiae. Food Chem. 2018, 257, 350–360. [Google Scholar] [CrossRef]
- Englezos, V.; Rantsiou, K.; Cravero, F.; Torchio, F.; Giacosa, S.; Ortiz-Julien, A.; Gerbi, V.; Rolle, L.; Cocolin, L. Volatile profiles and chromatic characteristics of red wines produced with Starmerella bacillaris and Saccharomyces cerevisiae. Food Res. Int. 2018, 109, 298–309. [Google Scholar] [CrossRef]
- Sadoudi, M.; Tourdot-Maréchal, R.; Rousseaux, S.; Steyer, D.; Gallardo-Chacón, J.-J.; Ballester, J.; Vichi, S.; Guérin-Schneider, R.; Caixach, J.; Alexandre, H. Yeast-yeast interactions revealed by aromatic profile analysis of Sauvignon Blanc wine fermented by single or co-culture of non-Saccharomyces and Saccharomyces yeasts. Food Microbiol. 2012, 32, 243–253. [Google Scholar] [CrossRef]
- Hahn, M. The rising threat of fungicide resistance in plant pathogenic fungi: Botrytis as a case study. J. Chem. Biol. 2014, 7, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Yu, H.H.; Song, Y.J.; Park, Y.J.; Lee, N.-K.; Paik, H.-D. Anti-biofilm effect of the cell-free supernatant of probiotic Saccharomyces cerevisiae against Listeria monocytogenes. Food Control 2021, 121, 107667. [Google Scholar] [CrossRef]
- Lacotte, P.-A.; Simons, A.; Bouttier, S.; Malet-Villemagne, J.; Nicolas, V.; Janoir, C. Inhibition of in vitro Clostridioides difficile biofilm formation by the probiotic yeast Saccharomyces boulardii CNCM I-745 through modification of the extracellular matrix composition. Microorganisms 2022, 10, 1082. [Google Scholar] [CrossRef] [PubMed]
- Malka, O.; Kalson, D.; Yaniv, K.; Shafir, R.; Rajendran, M.; Ben-David, O.; Kushmaro, A.; Meijler, M.M.; Jelinek, R. Cross-kingdom inhibition of bacterial virulence and communication by probiotic yeast metabolites. Microbiome 2021, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Lemos Junior, W.J.F.; Binati, R.L.; Felis, G.E.; Slaghenaufi, D.; Ugliano, M.; Torriani, S. Volatile organic compounds from Starmerella bacillaris to control gray mold on apples and modulate cider aroma profile. Food Microbiol. 2020, 89, 103446. [Google Scholar] [CrossRef] [PubMed]
- Nadai, C.; Fernandes Lemos, W.J.J.; Favaron, F.; Giacomini, A.; Corich, V. Biocontrol activity of Starmerella bacillaris yeast against blue mold disease on apple fruit and its effect on cider fermentation. PLoS ONE 2018, 13, e0204350. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Bai, X.; Zhang, Y.; Gao, Q.; Bu, X.; Xu, Y.; Guo, N. Evaluation of the potential probiotic yeast characteristics with anti-MRSA abilities. Probiotics Antimicrob. Proteins 2022, 14, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Haidar, R.; Fermaud, M.; Calvo-Garrido, C.; Roudet, J.; Deschamps, A. Modes of action for biological control of Botrytis cinerea by antagonistic bacteria. Phytopathol. Mediterr. 2016, 55, 301–322. [Google Scholar]
- Ribes, S.; Fuentes, A.; Talens, P.; Barat, J.M. Prevention of fungal spoilage in food products using natural compounds: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 2002–2016. [Google Scholar] [CrossRef]
- Villalba, M.L.; Mazzucco, M.B.; Lopes, C.A.; Ganga, M.A.; Sangorrín, M.P. Purification and characterization of Saccharomyces eubayanus killer toxin: Biocontrol effectiveness against wine spoilage yeasts. Int. J. Food Microbiol. 2020, 331, 108714. [Google Scholar] [CrossRef]
- Masneuf-Pomarede, I.; Juquin, E.; Miot-Sertier, C.; Renault, P.; Laizet, Y.; Salin, F.; Alexandre, H.; Capozzi, V.; Cocolin, L.; Colonna-Ceccaldi, B.; et al. The yeast Starmerella bacillaris (synonym Candida zemplinina) shows high genetic diversity in winemaking environments. FEMS Yeast Res. 2015, 15, fov045. [Google Scholar] [CrossRef] [PubMed]
- Lemos, W.J.F., Jr.; Treu, L.; Duarte, V.D.S.; Campanaro, S.; Nadai, C.; Giacomini, A.; Corich, V. Draft genome sequence of the yeast Starmerella bacillaris (syn., Candida zemplinina) FRI751 isolated from fermenting must of dried raboso grapes. Genome Announc. 2017, 5, e00224-17. [Google Scholar]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Tempel, S. Using and understanding RepeatMasker. Methods Mol. Biol. 2012, 859, 29–51. [Google Scholar] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S. Annotating non-coding RNAs with Rfam. In Current Protocols in Bioinformatics; Editorial Board, Baxevanis, A.D., Eds.; Wiley: Hoboken, NJ, USA, 2005; Chapter 12: Unit 12.5. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using evidence modeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema Marnix, H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef] [PubMed]
- Khatri, I.; Tomar, R.; Ganesan, K.; Prasad, G.S.; Subramanian, S. Complete genome sequence and comparative genomics of the probiotic yeast Saccharomyces boulardii. Sci. Rep. 2017, 7, 371. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Leguina, A.C.D.V.; Nieto, C.; Pajot, H.F.; Bertini, E.V.; Mac Cormack, W.; Castellanos de Figueroa, L.I.; Nieto-Peñalver, C.G. Inactivation of bacterial quorum sensing signals N-acyl homoserine lactones is widespread in yeasts. Fungal Biol. 2018, 122, 52–62. [Google Scholar] [CrossRef]
- Kulakovskaya, E.; Zvonarev, A.; Farofonova, V. Characteristics of killer toxin of the yeast Cryptococcus pinus. J. Biosci. Med. 2019, 7, 73–82. [Google Scholar]
- Jayakumar, J.; Kumar, V.A.; Biswas, L.; Biswas, R. Therapeutic applications of lysostaphin against Staphylococcus aureus. J. Appl. Microbiol. 2021, 131, 1072–1082. [Google Scholar] [CrossRef]
- Englezos, V.; Giacosa, S.; Rantsiou, K.; Rolle, L.; Cocolin, L. Starmerella bacillaris in winemaking: Opportunities and risks. Curr. Opin. Food Sci. 2017, 17, 30–35. [Google Scholar] [CrossRef]
- Sabaghian, S.; Braschi, G.; Vannini, L.; Patrignani, F.; Samsulrizal, N.H.; Lanciotti, R. Isolation and identification of wild yeast from malaysian grapevine and evaluation of their potential antimicrobial activity against grapevine fungal pathogens. Microorganisms 2021, 9, 2582. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Klenk, H.-P.; Göker, M. Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int. J. Syst. Evol. Microbiol. 2014, 64, 352–356. [Google Scholar] [CrossRef]
- Rosa Alberto, L.; Miot-Sertier, C.; Laizet Yh Salin, F.; Sipiczki, M.; Bely, M.; Masneuf-Pomarede, I.; Albertin, W.; Dennehy John, J. Draft genome sequence of the Starmerella bacillaris (syn., Candida zemplinina) type strain CBS 9494. Microbiol. Resour. Ann. 2018, 7, e00872-18. [Google Scholar]
- Lemos Junior, W.J.F.; Treu, L.; da Silva Duarte, V.; Carlot, M.; Nadai, C.; Campanaro, S.; Giacomini, A.; Corich, V. Whole-genome sequence of starmerella bacillaris pas13, a nonconventional enological yeast with antifungal activity. Genome Announc. 2017, 5, e00788-17. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhou, X.; Kominek, J.; Kurtzman, C.P.; Hittinger, C.T.; Rokas, A. Reconstructing the backbone of the Saccharomycotina yeast phylogeny using genome-scale data. G3 Genes Genomes Genet. 2016, 6, 3927–3939. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, C.; Ferreira, C.; Gonçalves, L.G.; Turner, D.L.; Leandro, M.J.; Salema-Oom, M.; Santos, H.; Gonçalves, P. A new pathway for mannitol metabolism in yeasts suggests a link to the evolution of alcoholic fermentation. Front. Microbiol. 2019, 10, 2510. [Google Scholar] [CrossRef]
- Gonçalves, P.; Gonçalves, C.; Brito, P.H.; Sampaio, J. The Wickerhamiella/Starmerella clade-A treasure trove for the study of the evolution of yeast metabolism. Yeast 2020, 37, 3463. [Google Scholar] [CrossRef]
- Liu, D.-M.; Huang, Y.-Y.; Liang, M.-H. Analysis of the probiotic characteristics and adaptability of Lactiplantibacillus plantarum DMDL 9010 to gastrointestinal environment by complete genome sequencing and corresponding phenotypes. LWT 2022, 158, 113129. [Google Scholar] [CrossRef]
- Haskins, R.H.; Thorn, J.A. Biochemistry of the ustilaginales: Vii. antibiotic activity of ustilagic acid. Can. J. Bot. 1951, 29, 585–592. [Google Scholar] [CrossRef]
- Hasumi, K.; Tachikawa, K.; Sakai, K.; Murakawa, S.; Yoshikawa, N.; Kumazawa, S.; Endo, A. Competitive inhibition of squalene synthetase by squalestatin 1. J. Antibiot. 1993, 46, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Yarborough, R.; Schwartz, J.; Patel, M.; Horan, A.; Gullo, V.; Das, P.; Puar, M. Sch 47554 AND Sch 47555, Two novel antifungal antibiotics produced rom a Streptomyces sp. J. Antibiot. 1993, 46, 861–865. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Allen, L.L.; Heng, N.C.K.; Tompkins, G.R. Streptococcus salivarius isolates of varying acid tolerance exhibit F1F0-ATPase conservation. Caries Res. 2021, 55, 288–291. [Google Scholar] [CrossRef]
- Whitehead, K.; Versalovic, J.; Roos, S.; Britton Robert, A. Genomic and genetic characterization of the bile stress response of probiotic Lactobacillus reuteri ATCC 55730. Appl. Environ. Microbiol. 2008, 74, 1812–1819. [Google Scholar] [CrossRef]
- Bustos, A.Y.; Font de Valdez, G.; Fadda, S.; Taranto, M.P. New insights into bacterial bile resistance mechanisms: The role of bile salt hydrolase and its impact on human health. Food Res. Int. 2018, 112, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Goossens, K.; Willaert, R. Flocculation protein structure and cell-cell adhesion mechanism in Saccharomyces cerevisiae. Biotechnol. Lett. 2010, 32, 1571–1585. [Google Scholar] [CrossRef]
- Paulsen, I.T.; Brown, M.H.; Skurray, R.A. Proton-dependent multidrug efflux systems. Microbiol. Rev. 1996, 60, 575–608. [Google Scholar] [CrossRef]
- Calabrese, D.; Bille, J.; Sanglard, D. A novel multidrug efflux transporter gene of the major facilitator superfamily from Candida albicans (FLU1) conferring resistance to fluconazole. Microbiology 2000, 146, 2743–2754. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Query_Name | Hit_Name | Hit_Species | Identity (%) |
|---|---|---|---|
| scaffold1.t310 | ALK02041.1 | Starmerella bacillaris | 100 |
| scaffold1.t1149 | AGQ04602.1 | Starmerella bacillaris | 100 |
| scaffold1.t1341 | AGQ04605.1 | Starmerella bacillaris | 100 |
| scaffold2.t730 | AGQ04603.1 | Starmerella bacillaris | 100 |
| scaffold1.t293 | AGQ04604.1 | Starmerella bacillaris | 99.93 |
| scaffold1.t322 | AGQ04600.1 | Starmerella bacillaris | 99.88 |
| scaffold1.t583 | AGQ04601.1 | Starmerella bacillaris | 99.83 |
| scaffold2.t1627 | AJE25530.1 | Starmerella bombicola | 98.67 |
| scaffold1.t311 | ALK02042.1 | Starmerella bacillaris | 98.60 |
| scaffold4.t1 | YP_052724.2 | Starmerella bacillaris | 97 |
| Type | Number of Genes | Percentage (%) |
|---|---|---|
| Glycosyl Transferases | 44 | 1.06 |
| Glycoside Hydrolases | 28 | 0.67 |
| Carbohydrate Esterases | 12 | 0.29 |
| Auxiliary Activities | 9 | 0.22 |
| Carbohydrate-Binding Modules | 2 | 0.05 |
| Polysaccharide Lyases | 0 | 0 |
| Query_Name | MIBIG Cluster | BLAST Score | Main Product |
|---|---|---|---|
| scaffold1.t210 | BGC0001281 | 253.0 | ustilagic acid |
| scaffold1.t211 | BGC0000397 | 420.0 | nostocyclopeptide A2 |
| scaffold1.t212 | BGC0000268 | 527.0 | Sch-47554, Sch-47555 |
| scaffold1.t749 | BGC0001281 | 116.0 | ustilagic acid |
| scaffold1.t750 | BGC0001839 | 346.0 | squalestatin S1 |
| Query_Name | Hit_Name | Hit_Description | Database |
|---|---|---|---|
| scaffold1.t1525 | O94474 | Superkiller protein 3 | Swiss-Prot |
| scaffold2.t307 | P09805 | Killer toxin subunits alpha/beta | Swiss-Prot |
| scaffold2.t1859 | P10548 | Lysostaphin | Swiss-Prot |
| Query_Name | PHI a | DFVF b | ||
|---|---|---|---|---|
| Hit_Name | Identity (%) | Hit_Name | Identity (%) | |
| scaffold1.t37 | Q6FVH8 | 31.45 | OPSB_ASPFU | 31.78 |
| scaffold1.t92 | A4VWD1 | 27.96 | ||
| scaffold1.t160 | Q8Y8N0 | 28.79 | F2QYD1_PICP7 | 26.64 |
| scaffold1.t163 | K8DPN9 | 33.94 | ||
| scaffold1.t212 | Q8MPM3 | 50.76 | ||
| scaffold1.t352 | P08179 | 32.47 | ||
| scaffold1.t675 | G8B6Y8 | 33.33 | OPSB_ASPFU | 28.89 |
| scaffold1.t729 | A0A2R6UD96 | 38.98 | ||
| scaffold1.t922 | A5IXN3 | 35.91 | ||
| scaffold1.t977 | Q8Y8N0 | 38.10 | DHH1_CRYNV | 34.17 |
| scaffold1.t1086 | Q8Y8N0 | 37.43 | DHH1_CRYNV | 40.71 |
| scaffold1.t1160 | C4YFX2 | 26.13 | TUP1_CANAL | 26.13 |
| scaffold1.t1414 | Q8Y8N0 | 26.85 | MAK5_COCIM | 27.98 |
| scaffold1.t1501 | C4YFX2 | 36.00 | TUP1_CANAL | 36.00 |
| scaffold1.t1597 | Q8Y7M8 | 29.50 | C1GMG4_PARBD | 28.41 |
| scaffold1.t1605 | C4YFX2 | 25.20 | TUP1_CANAL | 25.20 |
| scaffold2.t288 | C4YFX2 | 24.77 | TUP1_CANAL | 24.77 |
| scaffold2.t751 | Q8Z4H3 | 36.23 | ||
| scaffold2.t774 | Q8Y8N0 | 36.15 | DHH1_CRYNV | 32.02 |
| scaffold2.t863 | A0A1D8PS71 | 36.42 | ||
| scaffold2.t1078 | Q5AC08 | 34.96 | Q5ACC9_CANAL | 34.96 |
| scaffold2.t1137 | P0DJM0 | 29.55 | B9WMV3_CANDC | 34.83 |
| scaffold2.t1188 | Q8Y8N0 | 40.57 | DHH1_CRYNV | 34.90 |
| scaffold2.t1236 | P19786 | 39.52 | ||
| scaffold2.t1305 | Q59M00 | 40.84 | ||
| scaffold2.t1501 | Q8Y8N0 | 34.24 | DHH1_CRYNV | 29.53 |
| scaffold2.t1544 | Q59M00 | 28.57 | ||
| scaffold2.t1811 | Q8Z4H3 | 30.47 | Q3Y5V5_MAGGR | 28.96 |
| scaffold3.t273 | C4YFX2 | 22.91 | TUP1_CANAL | 22.91 |
| scaffold3.t300 | Q8Y6G5 | 20.72 | ||
| scaffold3.t352 | A0A2R6UD96 | 38.92 | ||
| scaffold3.t406 | Q5A7S7 | 31.80 | Q5A7S7_CANAL | 36.16 |
| scaffold3.t454 | Q6FVH9 | 35.71 | OPSB_ASPFU | 35.88 |
| scaffold3.t461 | Q6FVH9 | 24.89 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.; Bai, X.; Zhou, X.; Wang, J.; Guo, N.; Deng, Y. Whole-Genome Analysis of Starmerella bacillaris CC-PT4 against MRSA, a Non-Saccharomyces Yeast Isolated from Grape. J. Fungi 2022, 8, 1255. https://doi.org/10.3390/jof8121255
Shen Y, Bai X, Zhou X, Wang J, Guo N, Deng Y. Whole-Genome Analysis of Starmerella bacillaris CC-PT4 against MRSA, a Non-Saccharomyces Yeast Isolated from Grape. Journal of Fungi. 2022; 8(12):1255. https://doi.org/10.3390/jof8121255
Chicago/Turabian StyleShen, Yong, Xue Bai, Xiran Zhou, Jiaxi Wang, Na Guo, and Yanhong Deng. 2022. "Whole-Genome Analysis of Starmerella bacillaris CC-PT4 against MRSA, a Non-Saccharomyces Yeast Isolated from Grape" Journal of Fungi 8, no. 12: 1255. https://doi.org/10.3390/jof8121255
APA StyleShen, Y., Bai, X., Zhou, X., Wang, J., Guo, N., & Deng, Y. (2022). Whole-Genome Analysis of Starmerella bacillaris CC-PT4 against MRSA, a Non-Saccharomyces Yeast Isolated from Grape. Journal of Fungi, 8(12), 1255. https://doi.org/10.3390/jof8121255