

The First Telomere-to-Telomere Chromosome-Level Genome Assembly of Stagonospora tainanensis Causing Sugarcane Leaf Blight


 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Sequencing
2.2. Genome Size Estimation
2.3. De Novo Genome Assembly
2.4. Genome Completeness Assessment
2.5. Repeat Masking
2.6. Annotation of Protein-Coding Genes
2.7. Identification of Non-Coding RNAs
2.8. Functional Annotation of Protein-Coding Genes
2.8.1. General Functional Annotation
2.8.2. Fungal Pathogenicity-Related Gene Annotation
2.8.3. Secondary Metabolite Biosynthetic Gene Clusters Analysis
2.9. Comparative Genomic Analysis
3. Results and Discussion
3.1. The Morphology of the Pathological Lesions and Pathogenic S. tainanensis Used for Genome Sequencing
3.2. Genome Sequencing and Assembly
3.3. Genome Quality Assessment
3.4. Repeat Analysis
3.5. Gene Structural Annotation
3.6. General Gene Functional Annotation
3.7. Annotation of Pathogenicity-Related Genes
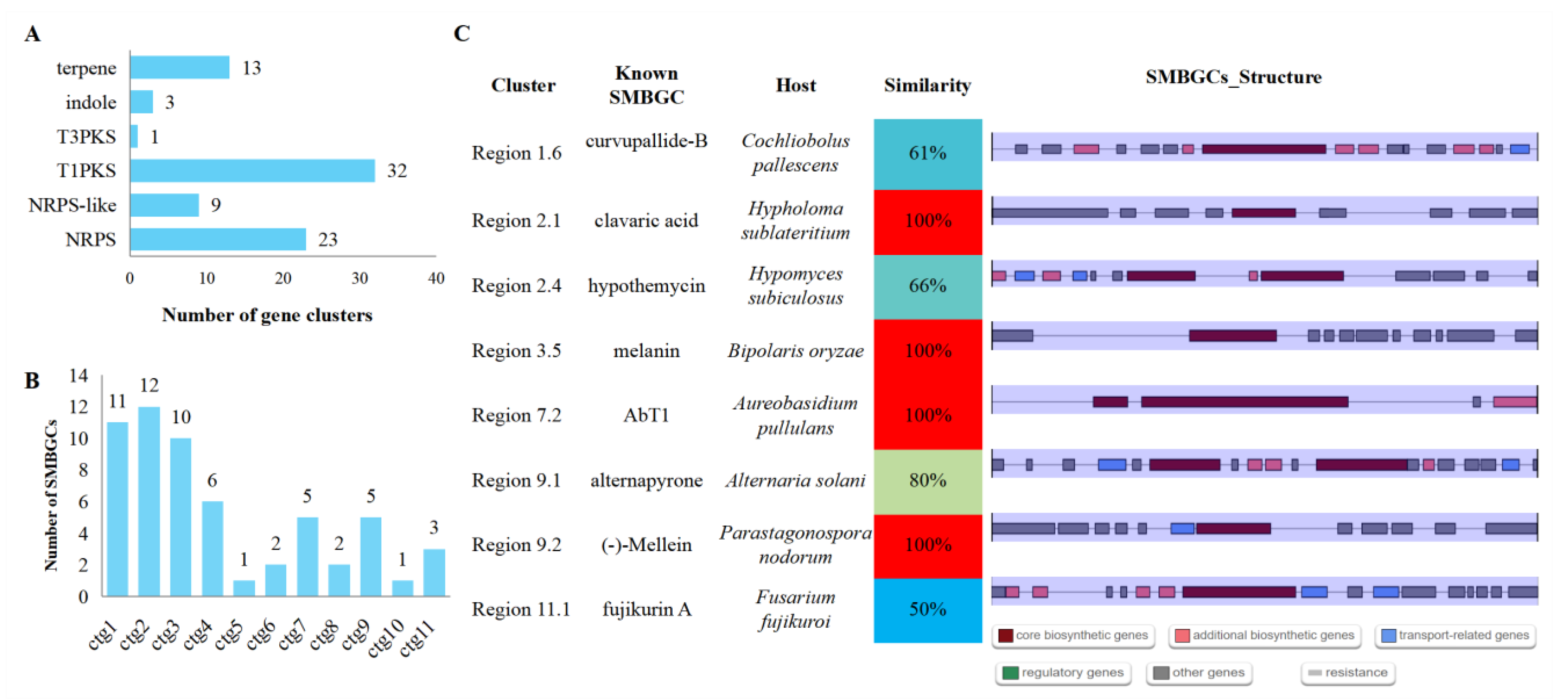
3.8. Secondary Metabolite Biosynthetic Gene Clusters (SMBGCs)
3.9. Comparative Genomic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Y.-R.; Yang, L.-T. Sugarcane agriculture and sugar industry in China. Sugar Tech 2015, 17, 1–8. [Google Scholar] [CrossRef]
- Xu, F.; Wang, Z.T.; Lu, G.L.; Zeng, R.S.; Que, Y.X. Sugarcane ratooning ability: Research status, shortcomings, and prospects. Biology 2021, 10, 1052. [Google Scholar] [CrossRef] [PubMed]
- Raid, R.N. Physiological specialization in sugarcane rust (Puccinia melanocephala) in Florida. Plant Dis. 1989, 73, 183. [Google Scholar] [CrossRef]
- Rajput, M.A.; Rajput, N.A.; Syed, R.N.; Lodhi, A.M.; Que, Y. Sugarcane smut: Current knowledge and the way forward for management. J. Fungi 2021, 7, 1095. [Google Scholar] [CrossRef]
- Hoy, J.W.; Hollier, C.A. Effect of brown rust on yield of sugarcane in Louisiana. Plant Dis. 2009, 93, 1171–1174. [Google Scholar] [CrossRef]
- Shan, H.; Li, W.; Zhang, R.; Wang, X.; Li, J.; Cang, X.; Yin, J.; Luo, Z.; Huang, Y. Analysis on epidemic reason of sugarcane pokahh boeng and its losses on yield and sucrose content. Sugar Crop China 2018, 40, 40–42, 45. [Google Scholar]
- Patil, A.S.; Hapase, D.G. Studies on pokkah boeng disease of sugarcane in Maharashtra. Indian Phytopathol. 1987, 40, 290. [Google Scholar]
- Vishwakarma, S.K.; Kumar, P.; Nigam, A.; Singh, A.; Kumar, A. Pokkah boeng: An emerging disease of sugarcane. J. Plant Pathol. Microb. 2013, 4, 1000170. [Google Scholar]
- Viswanathan, R. Changing scenario of sugarcane diseases in India since introduction of hybrid cane varieties: Path travelled for a century. J. Sugarcane Res. 2018, 8, 1–35. [Google Scholar]
- Comstock, J.C.; Glynn, N.C.; Davidson, R.W. Sugarcane rusts in Florida. Proc. Inter. Soc. Sugar Cane Tech. 2010, 27, 1–9. [Google Scholar]
- Selvakumar, R.; Viswanathan, R. History of sugarcane rusts in India. In Res Accomplishments in Sugarcane Pathology; Viswanathan, R., Sundar, R.A., Malathi, P., Selvakumar, R., Jayakumar, V., Nithya, K., Eds.; ICAR-Sugarcane Breeding Institute: Coimbatore, India, 2018; pp. 76–79. [Google Scholar]
- Comstock, J.C.; Sood, S.G.; Glynn, N.C.; Shine, J.M., Jr.; McKemy, J.M.; Castlebury, L.A. First report of Puccinia kuehnii, causal agent of orange rust of sugarcane, in the United States and western hemisphere. Plant Dis. 2008, 92, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Viswanathana, R.; Ashwin, N.M.R. Brown spot of sugarcane: An emerging disease in South Western region in India. J. Sugarcane Res. 2020, 10, 87–93. [Google Scholar] [CrossRef]
- Patel, R.R.; Patel, D.D.; Bhatt, J.; Thakor, P.; Triplett, L.R.; Thakkar, V.R. Induction of pre-chorismate, jasmonate and salicylate pathways by Burkholderia sp. RR18 in peanut seedlings. J. Appl. Microbiol. 2021, 131, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.R.; Thakkar, V.R.; Subramanian, B.R. A Pseudomonas guariconensis strain capable of promoting growth and controlling collar rot disease in Arachis hypogaea L. Plant Soil 2015, 390, 369–381. [Google Scholar] [CrossRef]
- Wang, X.Y.; Li, J.; Yang, K.; Shan, H.L.; Zhang, R.Y.; Wang, C.M.; Cang, X.Y.; Yin, J.; Luo, Z.M.; Li, W.F.; et al. Evaluation of resistance to brown streak disease in new and main cultivated sugarcane varieties. Acta Phytopathol. Sin. 2021, 51, 287–293. [Google Scholar]
- Yen, W.Y.; Chi, C.C. Studies on leaf blight of sugarcane (I). J. Sugarcane Res. Taiwan 1952, 6, 191–215. [Google Scholar]
- Hsieh, W.H. The Causal organism of sugarcane leaf blight. Mycologia 1997, 71, 892–898. [Google Scholar] [CrossRef]
- Shah, D.A.; Bergstrom, G.C. A rainfall-based model for predicting the regional incidence of wheat seed infection by Stagonospora nodorum in New York. Phytopathology 2002, 92, 511–518. [Google Scholar] [CrossRef]
- Abeysekara, N.S.; Friesen, T.L.; Keller, B.; Faris, J.D. Identification and characterization of a novel host-toxin interaction in the wheat-Stagonospora nodorum pathosystem. Theor. Appl. Genet. 2009, 120, 117–126. [Google Scholar] [CrossRef]
- Solomon, P.S.; Lowe, R.G.H.; Tan, K.-C.; Waters, O.D.C.; Oliver, R.P. Stagonospora nodorum: Cause of stagonospora nodorum blotch of wheat. Mol. Plant Pathol. 2006, 7, 147–156. [Google Scholar] [CrossRef]
- Hsieh, W.H.; Comstock, J.C. Leaf Blight. In A Guide to Sugarcane Diseases; Rott, P., Bailey, R.A., Comstock, J.C., Croft, A., Saumtally, A.S., Eds.; Production Cirad Publications: Paris, France, 2000; pp. 111–113. ISSN 1251-7224. [Google Scholar]
- O’Neill, N.R.; Farr, D.F. Miscanthus blight, a new foliar disease of ornamental grasses and sugarcane incited by Leptosphaeria sp. and its anamorphic state Stagonospora sp. Plant Dis. 1996, 80, 980–987. [Google Scholar] [CrossRef]
- Ren, H. Isolation, Genome Assembly and Molecular Detection Method of the Pathogen Causing Sugarcane Leaf Blight. Master’s Thesis, Fujian Agriculture and Forestry University, Fuzhou, China, April 2022. [Google Scholar]
- Wang, Z.; Ren, H.; Pang, C.; Lu, G.; Xu, F.; Cheng, W.; Que, Y.; Xu, L. An autopolyploid-suitable polyBSA-seq strategy for screening candidate genetic markers linked to leaf blight resistance in sugarcane. Theor. Appl. Genet. 2022, 135, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lu, G.; Wu, Q.; Li, A.; Que, Y.; Xu, L. Isolating QTL controlling sugarcane leaf blight resistance using a two-way pseudo-testcross strategy. Crop J. 2022, 10, 1131–1140. [Google Scholar] [CrossRef]
- Tedersoo, L.; Albertsen, M.; Anslan, S.; Callahan, B. Perspectives and benefits of high-throughput long-read sequencing in microbial ecology. Appl. Environ. Microbiol. 2021, 87, e0062621. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Haegi, A.; Valente, M.T.; Riccioni, L.; Orzali, L.; Vitale, S.; Luongo, L.; Infantino, A. New-generation sequencing technology in diagnosis of fungal plant pathogens: A dream comes true? J. Fungi 2022, 8, 737. [Google Scholar] [CrossRef]
- Guiglielmoni, N.; Houtain, A.; Derzelle, A.; Van Doninck, K.; Flot, J.F. Overcoming uncollapsed haplotypes in long-read assemblies of non-model organisms. BMC Bioinform. 2021, 22, 303. [Google Scholar] [CrossRef]
- Bao, J.; Chen, M.; Zhong, Z.; Tang, W.; Lin, L.; Zhang, X.; Jiang, H.; Zhang, D.; Miao, C.; Tang, H.; et al. PacBio sequencing reveals transposable elements as a key contributor to genomic plasticity and virulence variation in Magnaporthe oryzae. Mol. Plant 2017, 10, 1465–1468. [Google Scholar] [CrossRef]
- Zhong, Z.; Chen, M.; Lin, L.; Han, Y.; Bao, J.; Tang, W.; Lin, L.; Lin, Y.; Somai, R.; Lu, L.; et al. Population genomic analysis of the rice blast fungus reveals specific events associated with expansion of three main clades. ISME J. 2018, 12, 1867–1878. [Google Scholar] [CrossRef]
- Kelly, A.C.; Ward, T.J. Population genomics of Fusarium graminearum reveals signatures of divergent evolution within a major cereal pathogen. PLoS ONE 2018, 13, e0194616. [Google Scholar]
- Alouane, T.; Rimbert, H.; Bormann, J.; Gonzalez-Montiel, G.A.; Loesgen, S.; Schafer, W.; Freitag, M.; Langin, T.; Bonhomme, L. Comparative genomics of eight Fusarium graminearum strains with contrasting aggressiveness reveals an expanded open pangenome and extended effector content signatures. Int. J. Mol. Sci. 2021, 22, 6257. [Google Scholar] [CrossRef]
- Feng, Z.; Hsiang, T.; Liang, X.; Zhang, R.; Sun, G. Draft genome sequence of cumin blight pathogen Alternaria burnsii. Plant Dis. 2021, 105, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Haridas, S.; Albert, R.; Binder, M.; Bloem, J.; LaButti, K.; Salamov, A.; Andreopoulos, B.; Baker, S.E.; Barry, K.; Bills, G.; et al. 101 Dothideomycetes genomes: A test case for predicting lifestyles and emergence of pathogens. Stud. Mycol. 2020, 96, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Zeiner, C.A.; Purvine, S.O.; Zink, E.M.; Pasa-Tolic, L.; Chaput, D.L.; Haridas, S.; Wu, S.; LaButti, K.; Grigoriev, I.V.; Henrissat, B.; et al. Comparative analysis of secretome profiles of manganese (II)-oxidizing ascomycete fungi. PLoS ONE 2016, 11, e0157844. [Google Scholar] [CrossRef]
- Hane, J.K.; Lowe, R.G.; Solomon, P.S.; Tan, K.C.; Schoch, C.L.; Spatafora, J.W.; Crous, P.W.; Kodira, C.; Birren, B.W.; Galagan, J.E. Dothideomycete plant interactions illuminated by genome sequencing and EST analysis of the wheat pathogen Stagonospora nodorum. Plant Cell 2007, 19, 3347–3368. [Google Scholar] [CrossRef]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [PubMed]
- Kokot, M.; Dlugosz, M.; Deorowicz, S. KMC 3: Counting and manipulating k-mer statistics. Bioinformatics 2017, 33, 2759–2761. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simao, F.A.; Zdobnov, E.M. BUSCO Update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Jung, Y.; Han, D. BWA-MEME: BWA-MEM emulated with a machine learning approach. Bioinformatics 2022, 38, 2404–2413. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Bruna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [PubMed]
- Hoff, K.J.; Stanke, M. Predicting genes in single genomes with AUGUSTUS. Curr. Protoc. Bioinform. 2019, 65, e57. [Google Scholar] [CrossRef]
- Bruna, T.; Lomsadze, A.; Borodovsky, M. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2020, 2, lqaa026. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernandez-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Moller, S.; Croning, M.D.; Apweiler, R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 2001, 17, 646–653. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of apoplastic and cytoplasmic effectors in fungi and Oomycetes. Mol. Plant Microbe Interact. 2022, 35, 146–156. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kang, H.; Yao, J.; Li, Z.; Xia, X.; Zhou, S. An improved genome sequence resource of Bipolaris maydis, causal agent of Southern corn leaf blight. Phytopathology 2022, 112, 1386–1390. [Google Scholar] [CrossRef]
- O’Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N.; et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012, 44, 1060–1065. [Google Scholar] [CrossRef]
- Syauqi, J.; Chen, R.K.; Cheng, A.H.; Wu, Y.F.; Chung, C.L.; Lin, C.C.; Chou, H.P.; Wu, H.Y.; Jian, J.Y.; Liao, C.T.; et al. Surveillance of rice blast resistance effectiveness and emerging virulent isolates in Taiwan. Plant Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.; Mehta, S.; Prakash, G.; Sheoran, N.; Kumar, A. Structured framework and genome analysis of Magnaporthe grisea inciting pearl millet blast disease reveals versatile metabolic pathways, protein families, and virulence factors. J. Fungi 2022, 8, 614. [Google Scholar] [CrossRef] [PubMed]
- Ellwood, S.R.; Liu, Z.; Syme, R.A.; Lai, Z.; Hane, J.K.; Keiper, F.; Moffat, C.S.; Oliver, R.P.; Friesen, T.L. A first genome assembly of the barley fungal pathogen Pyrenophora Teres F. Teres. Genome Biol. 2010, 11, R109. [Google Scholar] [CrossRef]
- Grandaubert, J.; Lowe, R.G.T.; Soyer, J.L.; Schoch, C.L.; Van de Wouw, A.P.; Fudal, I.; Robbertse, B.; Lapalu, N.; Links, M.G.; Ollivier, B.; et al. Transposable element-assisted evolution and adaptation to host plant within the Leptosphaeria maculans-Leptosphaeria biglobosa species complex of fungal pathogens. BMC Genom. 2014, 15, 891. [Google Scholar] [CrossRef]
- Devanna, B.N.; Jain, P.; Solanke, A.U.; Das, A.; Thakur, S.; Singh, P.K.; Kumari, M.; Dubey, H.; Jaswal, R.; Pawar, D.; et al. Understanding the dynamics of blast resistance in rice-Magnaporthe oryzae interactions. J. Fungi 2022, 8, 584. [Google Scholar] [CrossRef]
- Hane, J.K.; Williams, A.; Oliver, R.P. Genomic and comparative analysis of the class Dothideomycetes. In The Mycota; Poggeler, S., Wostemeyer, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 14, pp. 205–226. [Google Scholar]
- Bertazzoni, S.; Jones, D.A.B.; Phan, H.T.; Tan, K.C.; Hane, J.K. Chromosome-level genome assembly and manually-curated proteome of model necrotroph Parastagonospora nodorum Sn15 reveals a genome-wide trove of candidate effector homologs, and redundancy of virulence-related functions within an accessory chromosome. BMC Genom. 2021, 22, 382. [Google Scholar] [CrossRef]
- Faino, L.; Seidl, M.F.; Shi-Kunne, X.; Pauper, M.; van den Berg, G.C.; Wittenberg, A.H.; Thomma, B.P. Transposons passively and actively contribute to evolution of the two-speed genome of a fungal pathogen. Genome Res. 2016, 26, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Ma, Z.; Zhou, X. Comparative genomic data provide new insight on the evolution of pathogenicity in Sporothrix species. Front. Microbiol. 2020, 11, 565439. [Google Scholar] [CrossRef] [PubMed]
- Jayasuriya, H.; Silverman, K.C.; Zink, D.L.; Jenkins, R.G.; Sanchez, M.; Pelaez, F.; Vilella, D.; Lingham, R.B.; Singh, S.B. Clavaric acid: A triterpenoid inhibitor of farnesyl-protein transferase from Clavariadelphus truncatus. J. Nat. Prod. 1998, 61, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Lingham, R.B.; Silverman, K.C.; Jayasuriya, H.; Kim, B.M.; Amo, S.E.; Wilson, F.R.; Rew, D.J.; Schaber, M.D.; Bergstrom, J.D.; Koblan, K.S.; et al. Clavaric acid and steroidal analogues as Ras-and FPP-directed inhibitors of human farnesyl-protein transferase. J. Med. Chem. 1998, 41, 4492–4501. [Google Scholar] [CrossRef]
- Godio, R.P.; Fouces, R.; Martin, J.F. A squalene epoxidase is involved in biosynthesis of both the antitumor compound clavaric acid and sterols in the basidiomycete H. sublateritium. Chem. Biol. 2007, 14, 1334–1346. [Google Scholar] [CrossRef]
- Du, X.; Li, H.; Qi, J.; Chen, C.; Lu, Y.; Wang, Y. Genome mining of secondary metabolites from a marine-derived Aspergillus terreus B12. Arch. Microbiol. 2021, 203, 5621–5633. [Google Scholar] [CrossRef]
- Zhu, S.; Yan, Y.; Qu, Y.; Wang, J.; Feng, X.; Liu, X.; Lin, F.; Lu, J. Role refinement of melanin synthesis genes by gene knockout reveals their functional diversity in Pyricularia oryzae strains. Microbiol. Res. 2021, 242, 126620. [Google Scholar] [CrossRef]
- Freitas, D.F.; Rocha, I.M.; Vieira-da-Motta, O.; de Paula Santos, C. The role of melanin in the biology and ecology of nematophagous Fungi. J. Chem. Ecol. 2021, 47, 597–613. [Google Scholar] [CrossRef]
- Members, C.-N.; Partners. Database resources of the National Genomics Data Center, China National Center for Bioinformation in 2021. Nucleic Acids Res. 2021, 49, D18–D28. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | StFZ01 |
|---|---|
| Assembly size (bp) | 38,252,541 |
| Contig number | 12 |
| Contig N50 (bp) | 2,858,663 |
| L50 | 4 |
| Contig N90 (bp) | 2,113,312 |
| L90 | 10 |
| Average contig length (bp) | 3,187,712 |
| Maximum contig length (bp) | 7,120,155 |
| GC content | 51.49% |
| Repeat sequences | 13.20% |
| Protein-coding genes | 12,206 |
| tRNAs | 162 |
| rRNAs | 142 |
| Other ncRNA | 43 |
| Features | Count | Length (bp) | Percentage (%) | |
|---|---|---|---|---|
| Interspersed repeats | SINEs 1 | 62 | 12,515 | 0.03 |
| LINEs 2 | 288 | 498,662 | 1.30 | |
| LTR 3 elements | 2592 | 1,927,917 | 5.04 | |
| DNA transposons | 1101 | 1,242,571 | 3.25 | |
| Unclassified | 3246 | 882,135 | 2.31 | |
| Tandem repeats | Small RNA | 137 | 111,320 | 0.29 |
| Simple repeats | 7496 | 312,397 | 0.82 | |
| Low complexity | 1175 | 60,609 | 0.16 | |
| Total repeats | - | 5,048,126 | 13.20 |
| Annotation | Gene Number | Percentage (%) |
|---|---|---|
| Pfam | 8452 | 69.24% |
| GO 1 | 5121 | 41.95% |
| KEGG 2 | 4443 | 36.40% |
| KOG 3 | 9078 | 74.37% |
| CAZys 4 | 599 | 4.91% |
| PHIs 5 | 2379 | 19.49% |
| Cytochrome P450 enzymes | 191 | 1.56% |
| Membrane transport proteins | 248 | 2.03% |
| Putative secreted proteins | 606 | 4.96% |
| Effectors | 332 | 2.72% |
| SMBGCs 6 | 58 | 0.48% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, F.; Li, X.; Ren, H.; Zeng, R.; Wang, Z.; Hu, H.; Bao, J.; Que, Y. The First Telomere-to-Telomere Chromosome-Level Genome Assembly of Stagonospora tainanensis Causing Sugarcane Leaf Blight. J. Fungi 2022, 8, 1088. https://doi.org/10.3390/jof8101088
Xu F, Li X, Ren H, Zeng R, Wang Z, Hu H, Bao J, Que Y. The First Telomere-to-Telomere Chromosome-Level Genome Assembly of Stagonospora tainanensis Causing Sugarcane Leaf Blight. Journal of Fungi. 2022; 8(10):1088. https://doi.org/10.3390/jof8101088
Chicago/Turabian StyleXu, Fu, Xiuxiu Li, Hui Ren, Rensen Zeng, Zhoutao Wang, Hongli Hu, Jiandong Bao, and Youxiong Que. 2022. "The First Telomere-to-Telomere Chromosome-Level Genome Assembly of Stagonospora tainanensis Causing Sugarcane Leaf Blight" Journal of Fungi 8, no. 10: 1088. https://doi.org/10.3390/jof8101088
APA StyleXu, F., Li, X., Ren, H., Zeng, R., Wang, Z., Hu, H., Bao, J., & Que, Y. (2022). The First Telomere-to-Telomere Chromosome-Level Genome Assembly of Stagonospora tainanensis Causing Sugarcane Leaf Blight. Journal of Fungi, 8(10), 1088. https://doi.org/10.3390/jof8101088