
Strategies Shaping the Transcription of Carbohydrate-Active Enzyme Genes in Aspergillus nidulans

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
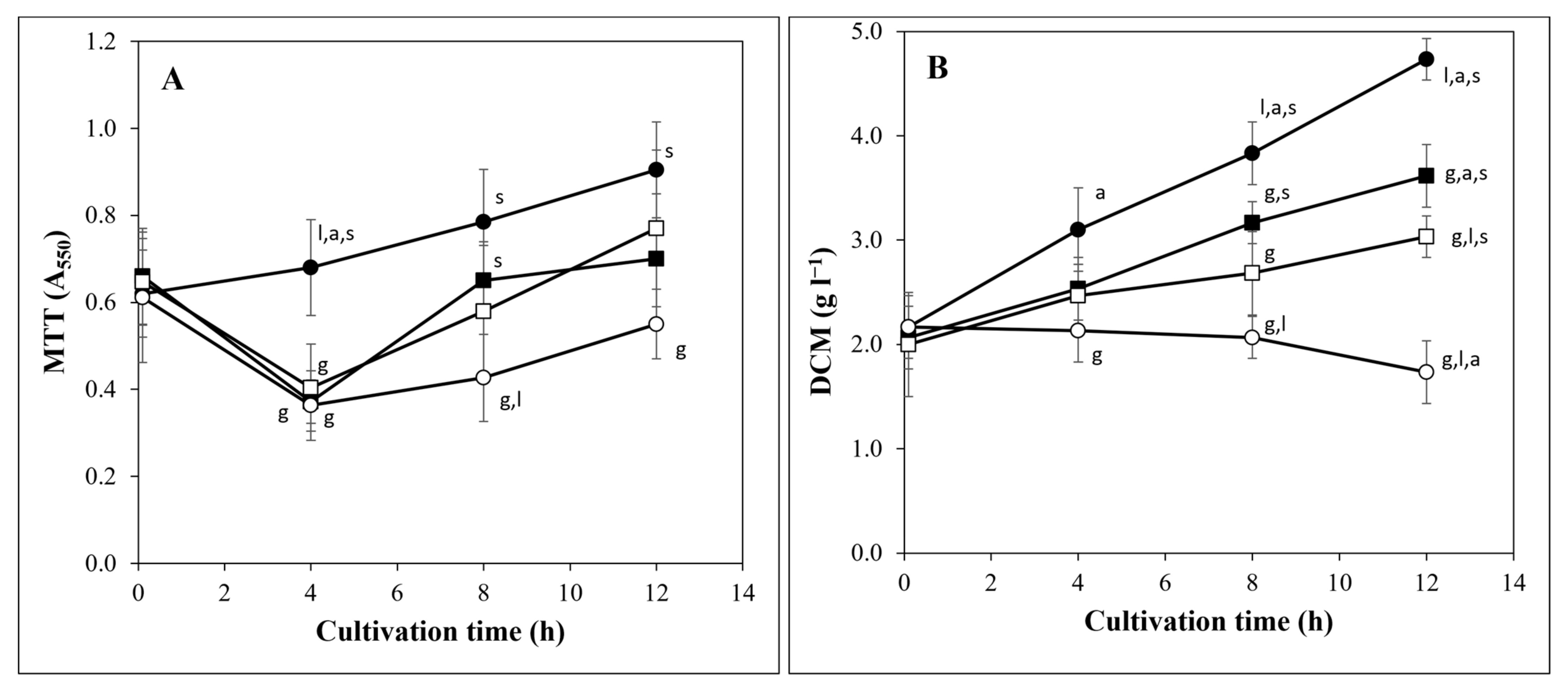
2.2. Detecting Growth, Metabolic Activity, Carbohydrate Utilization, and Formation of Sterigmatocystin
2.3. Enzyme Assays
2.4. Reverse-Transcription Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR) Assays
2.5. High throughput RNA Sequencing
2.6. Evaluation of the Transcriptome Data
2.7. Identification of Extracellular Proteins
3. Results
3.1. Three Carbon Stress Types Caused Similar Physiological Changes
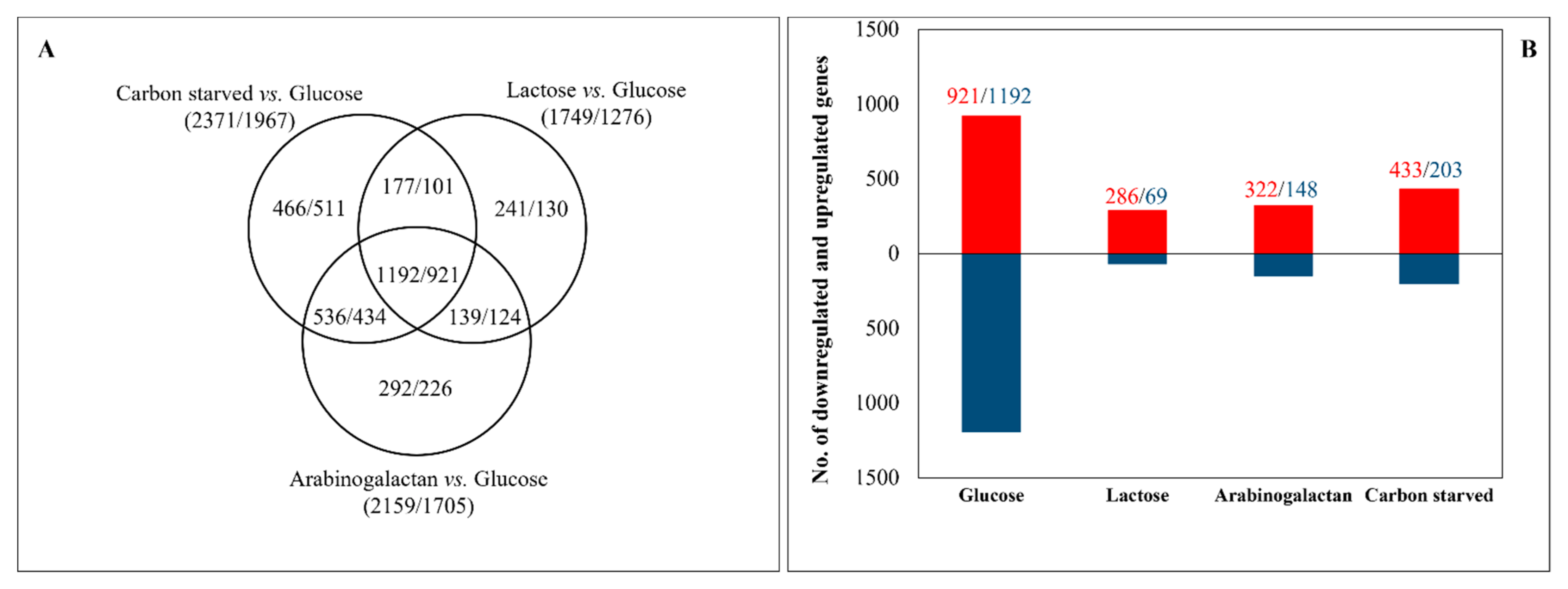
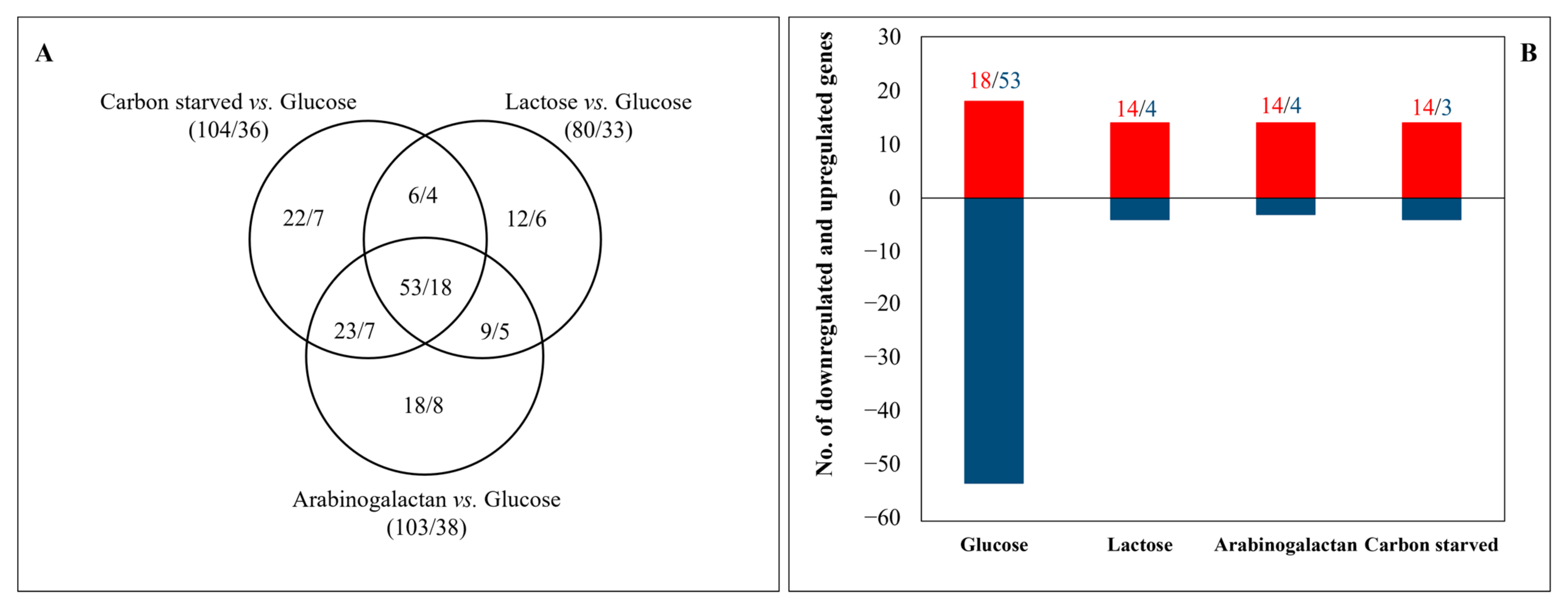
3.2. Transcriptome Analyses Revealed Important Differences among the Carbon-Stressed Cultures
4. Discussion
4.1. Carbon-Starved Cultures Produce Many Different “Scouting” Enzymes
4.2. Adaptive Prediction Can Be Important in the Regulation of CAZyme Genes on Arabinogalactan
4.3. High Lactose Concentration May Activate Strategies to Control the Cheaters
4.4. High Glucose Concentration Can Lead to the “Tragedy of the Commons” Scenario
4.5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Winderickx, J.; Holsbeeks, I.; Lagatie, O.; Giots, F.; Thevelein, J.; De Winde, H. From feast to famine; adaptation to nutrient availability in yeast. In Yeast Stress Responses; Hohmann, S., Mager, W.H., Eds.; Topics in Current Genetics; Springer: Berlin/Heidelberg, Germany, 2003; pp. 305–386. ISBN 978-3-540-45611-7. [Google Scholar]
- Van Munster, J.M.; Burggraaf, A.-M.; Pocsi, I.; Szilágyi, M.; Emri, T.; Ram, A.F. Post-genomic Approaches to Dissect Carbon Starvation Responses in Aspergilli. In Aspergillus and Penicillium in the Post-Genomic Era; Caister Academic Press: Norfolk, UK, 2016; pp. 89–112. ISBN 978-1-910190-39-5. [Google Scholar]
- Ho, A.; Di Lonardo, D.P.; Bodelier, P.L.E. Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiol. Ecol. 2017, 93, fix006. [Google Scholar] [CrossRef] [PubMed]
- Glass, N.L.; Schmoll, M.; Cate, J.H.; Coradetti, S. Plant Cell Wall Deconstruction by Ascomycete Fungi. Annu. Rev. Microbiol. 2013, 67, 477–498. [Google Scholar] [CrossRef] [PubMed]
- Brakhage, A.A. Regulation of fungal secondary metabolism. Nat. Rev. Genet. 2012, 11, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef]
- Shapiro, R.; Robbins, N.; Cowen, L.E. Regulatory Circuitry Governing Fungal Development, Drug Resistance, and Disease. Microbiol. Mol. Biol. Rev. 2011, 75, 213–267. [Google Scholar] [CrossRef] [PubMed]
- Ene, I.V.; Adya, A.K.; Wehmeier, S.; Brand, A.C.; MacCallum, D.M.; Gow, N.A.R.; Brown, A.J.P. Host carbon sources modulate cell wall architecture, drug resistance and virulence in a fungal pathogen. Cell. Microbiol. 2012, 14, 1319–1335. [Google Scholar] [CrossRef]
- Brown, N.A.; Goldman, G.H. The contribution of Aspergillus fumigatus stress responses to virulence and antifungal resistance. J. Microbiol. 2016, 54, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.Y.; Ho, K.L.; Cheah, Y.K.; Sandai, D.; Brown, A.J.P.; Than, L.T.L. Physiologically Relevant Alternative Carbon Sources Modulate Biofilm Formation, Cell Wall Architecture, and the Stress and Antifungal Resistance of Candida glabrata. Int. J. Mol. Sci. 2019, 20, 3172. [Google Scholar] [CrossRef]
- Held, M.A.; Jiang, N.; Basu, D.; Showalter, A.M.; Faik, A. Plant Cell Wall Polysaccharides: Structure and Biosynthesis. In Polysaccharides: Bioactivity and Biotechnology; Ramawat, K.G., Mérillon, J.-M., Eds.; Springer International Publishing: Cham, Germany, 2015; pp. 3–54. ISBN 978-3-319-16298-0. [Google Scholar]
- Culleton, H.; McKie, V.; De Vries, R.P. Physiological and molecular aspects of degradation of plant polysaccharides by fungi: What have we learned from Aspergillus? Biotechnol. J. 2013, 8, 884–894. [Google Scholar] [CrossRef] [PubMed]
- A Znameroski, E.; Glass, N.L. Using a model filamentous fungus to unravel mechanisms of lignocellulose deconstruction. Biotechnol. Biofuels 2013, 6, 6. [Google Scholar] [CrossRef]
- Smith, P.; Schuster, M. Public goods and cheating in microbes. Curr. Biol. 2019, 29, R442–R447. [Google Scholar] [CrossRef]
- Chundawat, S.P.; Beckham, G.T.; Himmel, M.E.; Dale, B.E. Deconstruction of Lignocellulosic Biomass to Fuels and Chemicals. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Chettri, D.; Verma, A.K.; Verma, A.K. Innovations in CAZyme gene diversity and its modification for biorefinery applications. Biotechnol. Rep. 2020, 28, e00525. [Google Scholar] [CrossRef] [PubMed]
- Contesini, F.J.; Frandsen, R.J.N.; Damasio, A. Editorial: CAZymes in Biorefinery: From Genes to Application. Front. Bioeng. Biotechnol. 2021, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Saykhedkar, S.; Ray, A.; Ayoubi-Canaan, P.; Hartson, S.D.; Prade, R.; Mort, A.J. A time course analysis of the extracellular proteome of Aspergillus nidulans growing on sorghum stover. Biotechnol. Biofuels 2012, 5, 52. [Google Scholar] [CrossRef]
- Martins, I.; Garcia, H.; Varela, A.; Núñez, O.; Planchon, S.; Galceran, M.T.; Renaut, J.; Rebelo, L.P.; Pereira, C.S. Investigating Aspergillus nidulans secretome during colonisation of cork cell walls. J. Proteom. 2014, 98, 175–188. [Google Scholar] [CrossRef]
- De Assis, L.J.; Ries, L.N.A.; Savoldi, M.; Dos Reis, T.F.; Brown, N.A.; Goldman, G.H. Aspergillus nidulans protein kinase A plays an important role in cellulase production. Biotechnol. Biofuels 2015, 8, 213. [Google Scholar] [CrossRef]
- Kumar, A. Aspergillus nidulans: A Potential Resource of the Production of the Native and Heterologous Enzymes for Industrial Applications. Int. J. Microbiol. 2020, 2020, 8894215. [Google Scholar] [CrossRef]
- Martens-Uzunova, E.; Schaap, P.J. Assessment of the pectin degrading enzyme network of Aspergillus niger by functional genomics. Fungal Genet. Biol. 2009, 46, S170–S179. [Google Scholar] [CrossRef]
- Delmas, S.; Pullan, S.T.; Gaddipati, S.; Kokolski, M.; Malla, S.; Blythe, M.J.; Ibbett, R.; Campbell, M.; Liddell, S.; Aboobaker, A.; et al. Uncovering the Genome-Wide Transcriptional Responses of the Filamentous Fungus Aspergillus niger to Lignocellulose Using RNA Sequencing. PLoS Genet. 2012, 8, e1002875. [Google Scholar] [CrossRef]
- van Munster, J.M.; Daly, P.; Delmas, S.; Pullan, S.T.; Blythe, M.J.; Malla, S.; Kokolski, M.; Noltorp, E.C.; Wennberg, K.; Fetherston, R.; et al. The role of carbon starvation in the induction of enzymes that degrade plant-derived carbohydrates in Aspergillus niger. Fungal Genet. Biol. 2014, 72, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.; Romano, G.H.; Groisman, B.; Yona, A.; Dekel, E.; Kupiec, M.; Dahan, O.; Pilpel, Y. Adaptive prediction of environmental changes by microorganisms. Nature 2009, 460, 220–224. [Google Scholar] [CrossRef]
- Brown, A.J.; Gow, N.A.; Warris, A.; Brown, G.D. Memory in Fungal Pathogens Promotes Immune Evasion, Colonisation, and Infection. Trends Microbiol. 2019, 27, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Gila, B.C.; Moon, H.; Antal, K.; Hajdu, M.; Kovács, R.; Jónás, A.P.; Pusztahelyi, T.; Yu, J.-H.; Pócsi, I.; Emri, T. The DUG Pathway Governs Degradation of Intracellular Glutathione in Aspergillus nidulans. Appl. Environ. Microbiol. 2021, 87, e01321-20. [Google Scholar] [CrossRef]
- Barratt, R.W.; Johnson, G.B.; Ogata, W.N. Wild type and mutant stocks of Aspergillus nidulans. Genetics 1965, 52, 233–246. [Google Scholar] [CrossRef]
- Emri, T.; Molnár, Z.; Pócsi, I. The appearances of autolytic and apoptotic markers are concomitant but differently regulated in carbon-starving Aspergillus nidulans cultures. FEMS Microbiol. Lett. 2005, 251, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, T.; Hartland, R.P.; Beauvais, A.; Diaquin, M.; Latge, J.-P. Purification and Characterization of an Endo-1,3-beta-Glucanase from Aspergillus fumigatus. JBIC J. Biol. Inorg. Chem. 1997, 243, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Klich, M.; Mendoza, C.; Mullaney, E.; Keller, N.; Bennett, J. A New Sterigmatocystin-producing Emericella Variant from Agricultural Desert Soils. Syst. Appl. Microbiol. 2001, 24, 131–138. [Google Scholar] [CrossRef]
- Roggenkamp, R.; Sahm, H.; Wagner, F. Microbial assimilation of methanol induction and function of catalase in Candida boidinii. FEBS Lett. 1974, 41, 283–286. [Google Scholar] [CrossRef]
- Oberley, L.W.; Spitz, D.R. Assay of superoxide dismutase activity in tumor tissue. Methods Enzymol. 1984, 105, 457–464. [Google Scholar] [CrossRef]
- Emri, T.; Molnár, Z.; Pusztahelyi, T.; Pócsi, I. Physiological and morphological changes in autolyzing Aspergillus nidulans cultures. Folia Microbiol. 2004, 49, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Mata, A.M.; Lopez-Barea, J. Reversible inactivation of Saccharomyces cerevisiae glutathione reductase under reducing conditions. Arch. Biochem. Biophys. 1984, 228, 1–12. [Google Scholar] [CrossRef]
- Bruinenberg, P.M.; Van Dijken, J.P.; Scheffers, W.A. An Enzymic Analysis of NADPH Production and Consumption in Candida utilis. Microbiology 1983, 129, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.; Kiss, T.; Szentirmai, A.; Biró, S. β-Galactosidase of Penicillium chrysogenum: Production, Purification, and Characterization of the Enzyme. Protein Expr. Purif. 2001, 21, 24–29. [Google Scholar] [CrossRef]
- Pusztahelyi, T.; Pócsi, I.; Szentirmai, A. Aging of Penicillium chrysogenum cultures under carbon starvation: II: Protease and N-acetyl-b-D-hexosaminidase production. Biotechnol. Appl. Biochem. 1997, 25, 87–93. [Google Scholar] [CrossRef]
- Spitzmüller, Z.; Kwon, N.-J.; Szilágyi, M.; Keserű, J.; Tóth, V.; Yu, J.-H.; Pócsi, I.; Emri, T. γ-Glutamyl transpeptidase (GgtA) of Aspergillus nidulans is not necessary for bulk degradation of glutathione. Arch. Microbiol. 2014, 197, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Tomarelli, R.M.; Charney, J.; Harding, M.L. The use of azoalbumin as a substrate in the colorimetric determination or peptic and tryptic activity. J. Lab. Clin. Med. 1949, 34, 428–433. [Google Scholar]
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 1993, 15, 532–534. [Google Scholar]
- Szilágyi, M.; Miskei, M.; Karányi, Z.; Lenkey, B.; Pócsi, I.; Emri, T. Transcriptome changes initiated by carbon starvation in Aspergillus nidulans. Microbiology 2013, 159, 176–190. [Google Scholar] [CrossRef]
- Emri, T.; Gila, B.; Antal, K.; Fekete, F.; Moon, H.; Yu, J.-H.; Pócsi, I. AtfA-Independent Adaptation to the Toxic Heavy Metal Cadmium in Aspergillus nidulans. Microorganisms 2021, 9, 1433. [Google Scholar] [CrossRef]
- Christensen, U.; Gruben, B.S.; Madrid, S.; Mulder, H.; Nikolaev, I.; de Vries, R.P. Unique Regulatory Mechanism for d-Galactose Utilization in Aspergillus nidulans. Appl. Environ. Microbiol. 2011, 77, 7084–7087. [Google Scholar] [CrossRef] [PubMed]
- Fekete, E.; Karaffa, L.; Seiboth, B.; Fekete, Éva; Kubicek, C.P.; Flipphi, M. Identification of a permease gene involved in lactose utilisation in Aspergillus nidulans. Fungal Genet. Biol. 2012, 49, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Fekete, E.; Orosz, A.; Kulcsár, L.; Kavalecz, N.; Flipphi, M.; Karaffa, L. Characterization of a second physiologically relevant lactose permease gene (lacpB) in Aspergillus nidulans. Microbiology 2016, 162, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fuoli, L.; Mellado, E. Ergosterol biosynthesis in Aspergillus fumigatus: Its relevance as an antifungal target and role in antifungal drug resistance. Front. Microbiol. 2013, 3, 439. [Google Scholar] [CrossRef]
- Flipphi, M.; Sun, J.; Robellet, X.; Karaffa, L.; Fekete, E.; Zeng, A.-P.; Kubicek, C.P. Biodiversity and evolution of primary carbon metabolism in Aspergillus nidulans and other Aspergillus spp. Fungal Genet. Biol. 2009, 46, S19–S44. [Google Scholar] [CrossRef]
- De Groot, P.W.; Brandt, B.W.; Horiuchi, H.; Ram, A.F.; de Koster, C.; Klis, F.M. Comprehensive genomic analysis of cell wall genes in Aspergillus nidulans. Fungal Genet. Biol. 2009, 46, S72–S81. [Google Scholar] [CrossRef]
- Inglis, D.; Binkley, J.; Skrzypek, M.S.; Arnaud, M.B.; Cerqueira, G.C.; Shah, P.; Wymore, F.; Wortman, J.R.; Sherlock, G. Comprehensive annotation of secondary metabolite biosynthetic genes and gene clusters of Aspergillus nidulans, A. fumigatus, A. niger and A. oryzae. BMC Microbiol. 2013, 13, 91. [Google Scholar] [CrossRef]
- Keserű, J.S.; Szabó, I.; Gál, Z.; Massidda, O.; Mingoia, M.; Kaszanyitzky, É.; Jánosi, S.; Hulvely, J.; Csorba, A.; Buzás, K.; et al. Identification of β-lactamases in human and bovine isolates of Staphylococcus aureus strains having borderline resistance to penicillinase-resistant penicillins (PRPs) with proteomic methods. Vet. Microbiol. 2011, 147, 96–102. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gruben, B.S.; Zhou, M.; de Vries, R. GalX regulates thed-galactose oxido-reductive pathway in Aspergillus niger. FEBS Lett. 2012, 586, 3980–3985. [Google Scholar] [CrossRef]
- Kowalczyk, J.E.; Gruben, B.S.; Battaglia, E.; Wiebenga, A.; Majoor, E.; De Vries, R.P. Genetic Interaction of Aspergillus nidulans galR, xlnR and araR in Regulating D-Galactose and L-Arabinose Release and Catabolism Gene Expression. PLoS ONE 2015, 10, e0143200. [Google Scholar] [CrossRef]
- Emri, T.; Molnár, Z.; Szilágyi, M.; Pócsi, I. Regulation of Autolysis in Aspergillus nidulans. Appl. Biochem. Biotechnol. 2008, 151, 211–220. [Google Scholar] [CrossRef]
- Yamazaki, H.; Tanaka, A.; Kaneko, J.-I.; Ohta, A.; Horiuchi, H. Aspergillus nidulans ChiA is a glycosylphosphatidylinositol (GPI)-anchored chitinase specifically localized at polarized growth sites. Fungal Genet. Biol. 2008, 45, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Nevalainen, H.; Peterson, R. Making recombinant proteins in filamentous fungi are we expecting too much? Front. Microbiol. 2014, 5, 75. [Google Scholar] [CrossRef]
- Coutinho, P.M.; Andersen, M.R.; Kolenova, K.; Vankuyk, P.A.; Benoit, I.; Gruben, B.S.; Trejo-Aguilar, B.; Visser, H.; Van Solingen, P.; Pakula, T. Post-genomic insights into the plant polysaccharide degradation potential of Aspergillus nidulans and comparison to Aspergillus niger and Aspergillus oryzae. Fungal Genet. Biol. 2009, 46, S161–S169. [Google Scholar] [CrossRef]
- Coradetti, S.T.; Craig, J.P.; Xiong, Y.; Shock, T.; Tian, C.; Glass, N.L. Conserved and essential transcription factors for cellulase gene expression in ascomycete fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 7397–7402. [Google Scholar] [CrossRef]
- Pardo, E.; Orejas, M. The Aspergillus nidulans Zn(II)2Cys6transcription factor AN5673/RhaR mediates L-rhamnose utilization and the production of α-L-rhamnosidases. Microb. Cell Factories 2014, 13, 161. [Google Scholar] [CrossRef] [PubMed]
- E Katz, M.; Braunberger, K.; Yi, G.; Cooper, S.; Nonhebel, H.; Gondro, C. A p53-like transcription factor similar to Ndt80 controls the response to nutrient stress in the filamentous fungus, Aspergillus nidulans. F1000Research 2013, 2, 72. [Google Scholar] [CrossRef] [PubMed]
- Saloheimo, M.; Valkonen, M.; Penttilä, M. Activation mechanisms of the HACI-mediated unfolded protein response in filamentous fungi. Mol. Microbiol. 2003, 47, 1149–1161. [Google Scholar] [CrossRef]
- Ruiz-Villafán, B.; Cruz-Bautista, R.; Manzo-Ruiz, M.; Passari, A.K.; Villarreal-Gómezs, K.; Rodríguez-Sanoja, R.; Sánchez, S. Carbon catabolite regulation of secondary metabolite formation, an old but not well-established regulatory system. Microb. Biotechnol. 2021, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kurucz, V.; Krüger, T.; Antal, K.; Dietl, A.-M.; Haas, H.; Pócsi, I.; Kniemeyer, O.; Emri, T. Additional oxidative stress reroutes the global response of Aspergillus fumigatus to iron depletion. BMC Genom. 2018, 19, 357. [Google Scholar] [CrossRef]
- Vivek-Ananth, R.P.; Mohanraj, K.; Vandanashree, M.; Jhingran, A.; Craig, J.P.; Samal, A. Comparative systems analysis of the secretome of the opportunistic pathogen Aspergillus fumigatus and other Aspergillus species. Sci. Rep. 2018, 8, 6617. [Google Scholar] [CrossRef]
- Yoshimi, Y.; Yaguchi, K.; Kaneko, S.; Tsumuraya, Y.; Kotake, T. Properties of two fungal endo-β-1,3-galactanases and their synergistic action with an exo-β-1,3-galactanase in degrading arabinogalactan-proteins. Carbohydr. Res. 2017, 453–454, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Gielkens, M.M.C.; Dekkers, E.; Visser, J.; de Graaff, L.H. Two Cellobiohydrolase-Encoding Genes from Aspergillus niger Require d -Xylose and the Xylanolytic Transcriptional Activator XlnR for Their Expression. Appl. Environ. Microbiol. 1999, 65, 4340–4345. [Google Scholar] [CrossRef] [PubMed]
- Margolles-Clark, E.; Ihnen, M.; Penttilä, M. Expression patterns of ten hemicellulase genes of the filamentous fungus Trichoderma reesei on various carbon sources. J. Biotechnol. 1997, 57, 167–179. [Google Scholar] [CrossRef]
- Morikawa, Y.; Ohashi, T.; Mantani, O.; Okada, H. Cellulase induction by lactose in Trichoderma reesei PC-3-Appl. Microbiol. Biotechnol. 1995, 44, 106–111. [Google Scholar] [CrossRef]
- Fang, X.; Yano, S.; Inoue, H.; Sawayama, S. Lactose enhances cellulase production by the filamentous fungus Acremonium cellulolyticus. J. Biosci. Bioeng. 2008, 106, 115–120. [Google Scholar] [CrossRef]
- Wang, M.; Lu, X. Exploring the Synergy between Cellobiose Dehydrogenase from Phanerochaete chrysosporium and Cellulase from Trichoderma reesei. Front. Microbiol. 2016, 7, 620. [Google Scholar] [CrossRef]
- Garcia-Rubio, R.; De Oliveira, H.C.; Rivera, J.; Trevijano-Contador, N. The Fungal Cell Wall: Candida, Cryptococcus, and Aspergillus Species. Front. Microbiol. 2020, 10, 2993. [Google Scholar] [CrossRef]
- Dragosits, M.; Pflügl, S.; Kurz, S.; Razzazi-Fazeli, E.; Wilson, I.; Rendic, D. Recombinant Aspergillus β-galactosidases as a robust glycomic and biotechnological tool. Appl. Microbiol. Biotechnol. 2014, 98, 3553–3567. [Google Scholar] [CrossRef]
- de Vries, R.; Visser, J.; de Graaff, L.H. CreA modulates the XlnR-induced expression on xylose of Aspergillus niger genes involved in xylan degradation. Res. Microbiol. 1999, 150, 281–285. [Google Scholar] [CrossRef]
- Ali, S.; Sayed, A. Regulation of the cellulase biosynthesis in Aspergillus terreus. World J. Microbiol. Biotechnol. 1992, 8, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, S.; Vallier, L.G.; Flick, J.S.; Carlson, M.; Johnston, M. Expression of the SUC2 Gene of Saccharomyces cerevisiae Is Induced by Low Levels of Glucose. Yeast 1997, 13, 127–137. [Google Scholar] [CrossRef]
- Gore, J.; Youk, H.; Van Oudenaarden, A. Snowdrift game dynamics and facultative cheating in yeast. Nature 2009, 459, 253–256. [Google Scholar] [CrossRef]
- Kato, N.; Suyama, S.; Shirokane, M.; Kato, M.; Kobayashi, T.; Tsukagoshi, N. Novel α-Glucosidase from Aspergillus nidulans with Strong Transglycosylation Activity. Appl. Environ. Microbiol. 2002, 68, 1250–1256. [Google Scholar] [CrossRef]
- Bauer, S.; Vasu, P.; Mort, A.J.; Somerville, C.R. Cloning, expression, and characterization of an oligoxyloglucan reducing end-specific xyloglucanobiohydrolase from Aspergillus nidulans. Carbohydr. Res. 2005, 340, 2590–2597. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Brumbauer, A.; Fehér, E.; Réczey, K.; Kiss, L. Production and characterization of β-glucosidases from different Aspergillus strains. World J. Microbiol. Biotechnol. 2001, 17, 455–461. [Google Scholar] [CrossRef]
- Hallsworth, J.E. Stress-free microbes lack vitality. Fungal Biol. 2018, 122, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Antal, K.; Gila, B.; Pócsi, I.; Emri, T. General stress response or adaptation to rapid growth in Aspergillus nidulans? Fungal Biol. 2020, 124, 376–386. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glucose | Lactose | Arabinogalactan | Carbon-Starved | |
|---|---|---|---|---|
| Intracellular enzyme activities | ||||
| SOD (U mg−1 protein) | 81 ± 10 a,s | 97 ± 15 a,s | 156 ± 21 g,l,s | 210 ± 30 g,l,a |
| Catalase (kat kg−1 protein) | 2.2 ± 0.3 | 1.6 ± 0.3 | 1.7 ± 0.2 | 1.7 ± 0.2 |
| Nitrate reductase (mkat kg−1 protein) | 1.40 ± 0.09 l,a,s | 1.09 ± 0.05 g,a,s | 0.62 ± 0.12 g,l | 0.56 ± 0.15 g,l |
| Glutathione reductase (mkat kg−1 protein) | 9.3 ± 0.3 l,a,s | 6.4 ± 0.8 g,a | 4.0 ± 0.5 g,l,s | 5.3 ± 0.6 g,a |
| β-Galactosidase (µkat kg−1 protein) | <0.1 l,a,s | 10.2 ± 1.1 g,a,s | 3.7 ± 0.3 g,l,s | 0.6 ± 0.02 g,l,a |
| Extracellular enzyme activities | ||||
| SOD (U mL−1) | 4.3 ± 1.1 l,a,s | 9.7 ± 1.5 g | 8.1 ± 1.0 g | 9.3 ± 0.9 g |
| Catalase (µkat mL−1) | <8 l,a,s | 11 ± 1 g,a,s | 16 ± 1 g,l,s | 52 ± 10 g,l,a |
| β-Galactosidase (nkat mL−1) | <0.006 l,a | 0.012 ± 0.001 g,a,s | 0.018 ± 0.002 g,l,s | <0.006 l,a |
| β-Glucosidase (nkat mL−1) | <0.006 l,a,s | 0.09 ± 0.02 g,a | 0.18 ± 0.01 g,l,s | 0.11 ± 0.02 g,a |
| Cellulase (nkat mL−1) | 0.7 ± 0.1 l,a,s | 2.5 ± 0.3 g,a,s | 1.2 ± 0.3 g,l,s | <0.3 g,l,a |
| Chitinase (U mL−1) | <0.1 l,a,s | 0.6 ± 0.2 g,a,s | 1.1 ± 0.2 g,l | 1.5 ± 0.3 g,l |
| Proteinase (U mL−1) | <0.1 l,a,s | 3.1 ± 0.3 g,a | 0.8 ± 0.3 g,l,s | 2.7 ± 0.3 g,a |
| γGT (nkat mL−1) | <0.01 l,a,s | 0.12 ± 0.02 g,a,s | 0.84 ± 0.05 g,l,s | 0.35 ± 0.05 g,l,a |
| Comparison a | Significantly Enriched Terms | |
|---|---|---|
| In Upregulated Gene Set | In Downregulated Gene Set | |
| Lactose vs. Glucose (1749/1276) | glycerolipid metabolism, fatty acid metabolism, polysaccharide catabolic process, mannan catabolic process, xylan catabolic process, cellulose catabolic process, arabinose metabolic process, D-xylose metabolic process, xylulose metabolic process, galactose metabolic process, fructose and mannose metabolism; alpha-galactosidase activity, secondary metabolism, sterigmatocystin biosynthetic process, metabolism of melanins, phenylalanine metabolism, tryptophan metabolism, tyrosine metabolism, | glycolytic process, gluconeogenesis, respiration, mitochondrion, steroid biosynthesis, biosynthesis of vitamins, cofactors, and prosthetic groups, biosynthesis of secondary metabolites |
| Arabinogalactan vs. Glucose (2159/1705) | glycerolipid metabolism, oxidation of fatty acids, cell wall organization, extracellular polysaccharide degradation, pectin catabolic process, xylan catabolic process, glucan catabolic process, cellulose catabolic process, mannan catabolic process, pentose-phosphate pathway, arabinose metabolic process, D-xylose metabolic process, galactose metabolism, beta-glucosidase activity, secondary metabolism, sterigmatocystin biosynthetic process, phenylalanine metabolism, tryptophan metabolism, tyrosine metabolism | glycolysis and gluconeogenesis, respiration, citrate cycle (TCA cycle), mitochondrion, biosynthesis of amino acids, biosynthesis of vitamins, cofactors, and prosthetic groups, steroid biosynthesis, pentose phosphate pathway, fructose and mannose metabolism, galactose metabolism, biosynthesis of secondary metabolites, response to stress, cellular response to osmotic stress |
| Carbon-starved vs. Glucose (2371/1967) | glycerolipid metabolism, oxidation of fatty acids, peroxisome, cell wall organization, extracellular polysaccharide degradation, chitin catabolism, xylan catabolic process, pectin catabolic process, secondary metabolism, sterigmatocystin biosynthetic process, metabolism of melanins, phenylalanine metabolism, tryptophan metabolism, tyrosine metabolism | translation, ribosome biogenesis, nucleotide-sugar metabolism, glycolysis and gluconeogenesis, respiration, Fe/S binding, mitochondrion, cellular amino acid biosynthetic process, cysteine biosynthetic process, sulfate assimilation, steroid biosynthesis, biosynthesis of vitamins, cofactors, and prosthetic groups, heavy metal ion transport (Cu2+, Fe3+, etc.), biosynthesis of secondary metabolites, oxidative stress response, heat shock response, cellular response to osmotic stress |
| Behavior of the Genes a | |||
|---|---|---|---|
| Lactose vs. Glucose | Arabinogalactan vs. Glucose | Carbon-Starved vs. Glucose | |
| Lactose utilization genes | up | up | - |
| β-galactosidases—lactose permeases | up | up | up |
| Leloir pathway | - | - | down |
| D-galactose oxidoreductive pathway | up | - | - |
| Glycolysis genes | down | down | down |
| Oxidative pentose-phosphate shunt genes | - | down | down |
| TCA cycle genes | - | down | down |
| Autophagy genes | - | - | up |
| Cell wall genes | down | down | both |
| Synthases, transglycosylases, and regulatory proteins | - | down | down |
| Hydrolases | down | up | up |
| Extracellular peptidase genes | - | up | up |
| CAZyme genes | both | up | up |
| α-Glucosidase | - | - | - |
| β-Glucosidase | - | up | - |
| β-1,4-Endoglucanase | - | up | - |
| Cellobiohydrolase—cellobiose dehydrogenase | - | up | - |
| α-Galactosidase | up | up | - |
| β-Galactosidase | up | up | - |
| Arabinofuranosidase and endo-arabinosidase | - | up | - |
| Xylanase | - | - | - |
| Xylosidase | up | up | up |
| Mannan degradation | - | up | - |
| Galacturonan degradation | - | up | up |
| Rhamnogalacturonan degradation | up | up | up |
| Antioxidant enzyme genes | - | - | down |
| Squalene—ergosterol pathway genes | down | down | down |
| Ribosome biogenesis genes | - | - | down |
| Mitotic cell cycle genes | - | - | - |
| Transcription factor genes | up | up | up |
| Group a | Glucose b | Lactose b | Arabinogalactan b | Carbon-Starved b |
|---|---|---|---|---|
| CAZyme genes (317) | 77 (29) | 81 (6) | 138 (62) | 107 (16) |
| α-Glucosidases (9) | 2 (1) | 0 (0) | 4 (2) | 1 (1) |
| Cellulolytic enzymes (32) | 10 (5) | 11 (0) | 19 (14) | 8 (1) |
| β-Glucosidases (13) | 3 (1) | 4 (0) | 8 (5) | 5 (0) |
| β-1,4-Endoglucanases (14) | 5 (4) | 5 (0) | 7 (5) | 2 (1) |
| Cellobiohydrolases—cellobiose dehydrogenases (5) | 2 (0) | 2 (0) | 4 (4) | 1 (0) |
| Galactoside-degrading enzymes (16) | 1 (0) | 10 (4) | 11 (4) | 5 (0) |
| α-Galactosidases (7) | 1 (0) | 5 (3) | 5 (1) | 2 (0) |
| β-Galactosidases (7) | 0 (0) | 5 (1) | 6 (2) | 3 (0) |
| Galactanases (2) | 0 (0) | 0 (0) | 0 (1) | 0 (0) |
| Arabinofuranosidases and endo-arabinosidases (14) | 4 (1) | 5 (0) | 10 (7) | 6 (0) |
| Xylanolytic enzymes (24) | 2 (0) | 17 (1) | 19 (13) | 14 (2) |
| Xylanases (5) | 1 (0) | 2 (0) | 3 (3) | 2 (1) |
| Xylosidases (12) | 1 (0) | 10 (1) | 11 (8) | 7 (0) |
| Further xylan-degrading enzymes (7) | 0 (0) | 5 (0) | 5 (2) | 5 (1) |
| Mannan degradation enzymes (19) | 4 (1) | 5 (0) | 9 (4) | 6 (0) |
| Xyloglucan degradation enzymes (3) | 1 (1) | 1 (0) | 2 (1) | 2 (0) |
| Pectinolytic enzymes (40) | 4 (1) | 12 (1) | 24 (7) | 23 (3) |
| Galacturonan degradation enzymes (24) | 4 (1) | 4 (0) | 12 (4) | 11 (2) |
| Rhamnogalacturonan degradation enzymes (16) | 0 (0) | 8 (1) | 12 (3) | 12 (1) |
| Cluster a | Lactose vs. Glucose | Arabinogalactan vs. Glucose | Carbon-Starved vs. Glucose |
|---|---|---|---|
| Number of Upregulated/Downregulated Cluster Genes | |||
| Sterigmatocystin cluster (26) | 26/0 | 26/0 | 26/0 |
| Monodictyphenone cluster (12) | 12/0 | 12/0 | 11/0 |
| AN8105 cluster (10) | 8/1 | 9/1 | 8/1 |
| Pkb cluster (9) | 6/0 | 7/0 | 8/0 |
| Pkg cluster (6) | 6/0 | 3/0 | 6/0 |
| Emericellamide cluster (5) | 0/0 | 5/0 | 1/1 |
| Terriquinone cluster (5) | 5/0 | 5/0 | 5/0 |
| AN1680 cluster (4) | 1/0 | 4/0 | 4/0 |
| Penicillin cluster (3) | 0/1 | 3/0 | 3/0 |
| Ivo cluster (2) | 2/0 | 2/0 | 2/0 |
| AN9129 cluster (2) | 2/0 | 2/0 | 2/0 |
| AN9314 cluster (2) | 2/0 | 2/0 | 2/0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gila, B.C.; Antal, K.; Birkó, Z.; Keserű, J.S.; Pócsi, I.; Emri, T. Strategies Shaping the Transcription of Carbohydrate-Active Enzyme Genes in Aspergillus nidulans. J. Fungi 2022, 8, 79. https://doi.org/10.3390/jof8010079
Gila BC, Antal K, Birkó Z, Keserű JS, Pócsi I, Emri T. Strategies Shaping the Transcription of Carbohydrate-Active Enzyme Genes in Aspergillus nidulans. Journal of Fungi. 2022; 8(1):79. https://doi.org/10.3390/jof8010079
Chicago/Turabian StyleGila, Barnabás Cs., Károly Antal, Zsuzsanna Birkó, Judit Sz. Keserű, István Pócsi, and Tamás Emri. 2022. "Strategies Shaping the Transcription of Carbohydrate-Active Enzyme Genes in Aspergillus nidulans" Journal of Fungi 8, no. 1: 79. https://doi.org/10.3390/jof8010079
APA StyleGila, B. C., Antal, K., Birkó, Z., Keserű, J. S., Pócsi, I., & Emri, T. (2022). Strategies Shaping the Transcription of Carbohydrate-Active Enzyme Genes in Aspergillus nidulans. Journal of Fungi, 8(1), 79. https://doi.org/10.3390/jof8010079