
Genome-Wide Analysis of Four Pathotypes of Wheat Rust Pathogen (Puccinia graminis) Reveals Structural Variations and Diversifying Selection


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Genomic DNA Isolation
2.3. Genome Sequence and Assembly
2.4. Gene Prediction and Annotations
2.5. Identification of Repeat Elements within the Genomes of P. graminis Pathotypes
2.6. SNP and InDel Analysis
2.7. Secretome Analysis
2.8. Identification of Homologous Genes within and across the P. graminis Pathotypes
2.9. Phylogenetic Analysis
2.10. Diversifying Selection Analysis and Estimation of Substitution Rates
3. Results
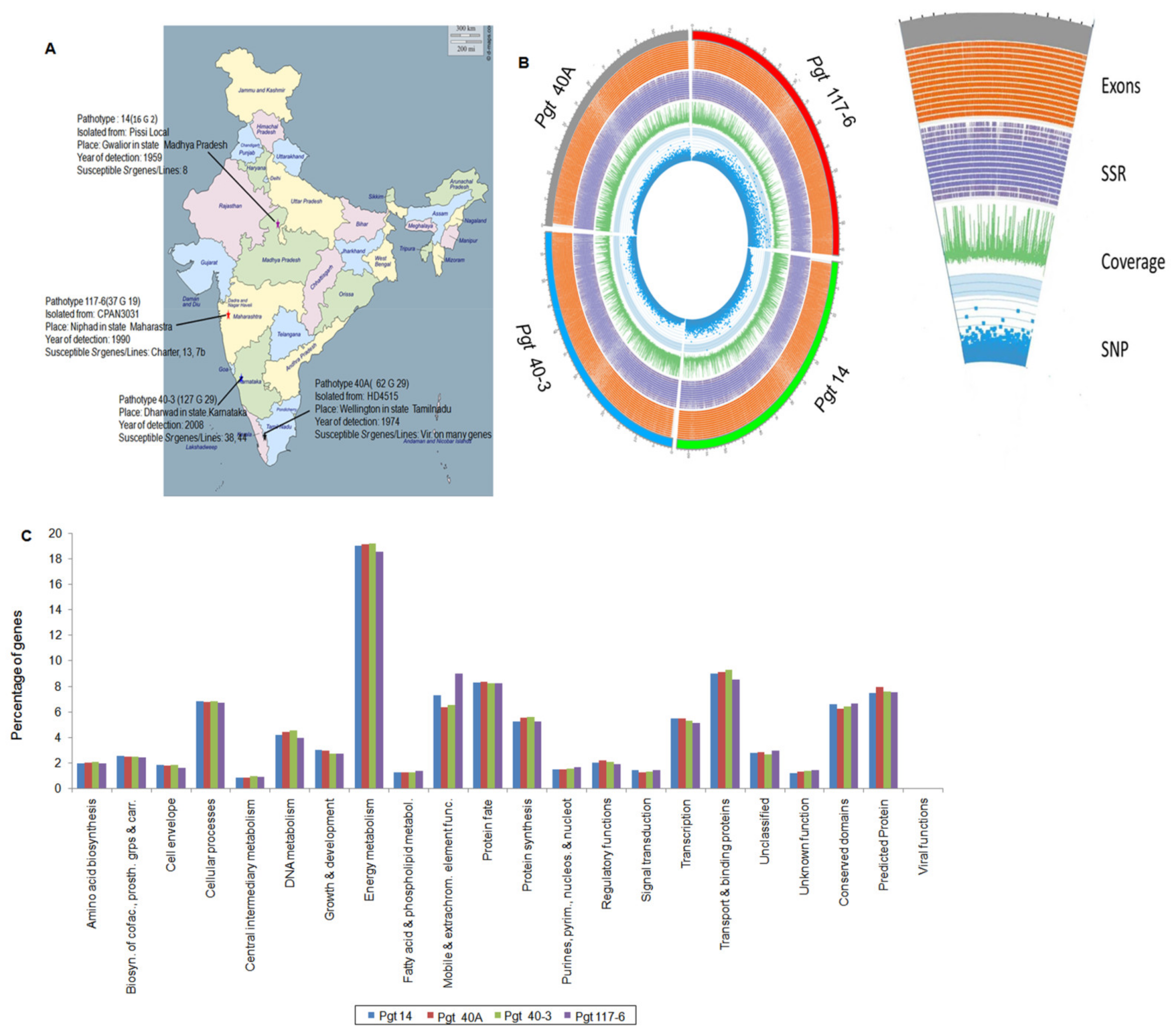
3.1. Genome Sequencing and Assembly of the P. graminis Pathotypes
3.2. Gene Prediction and Annotations
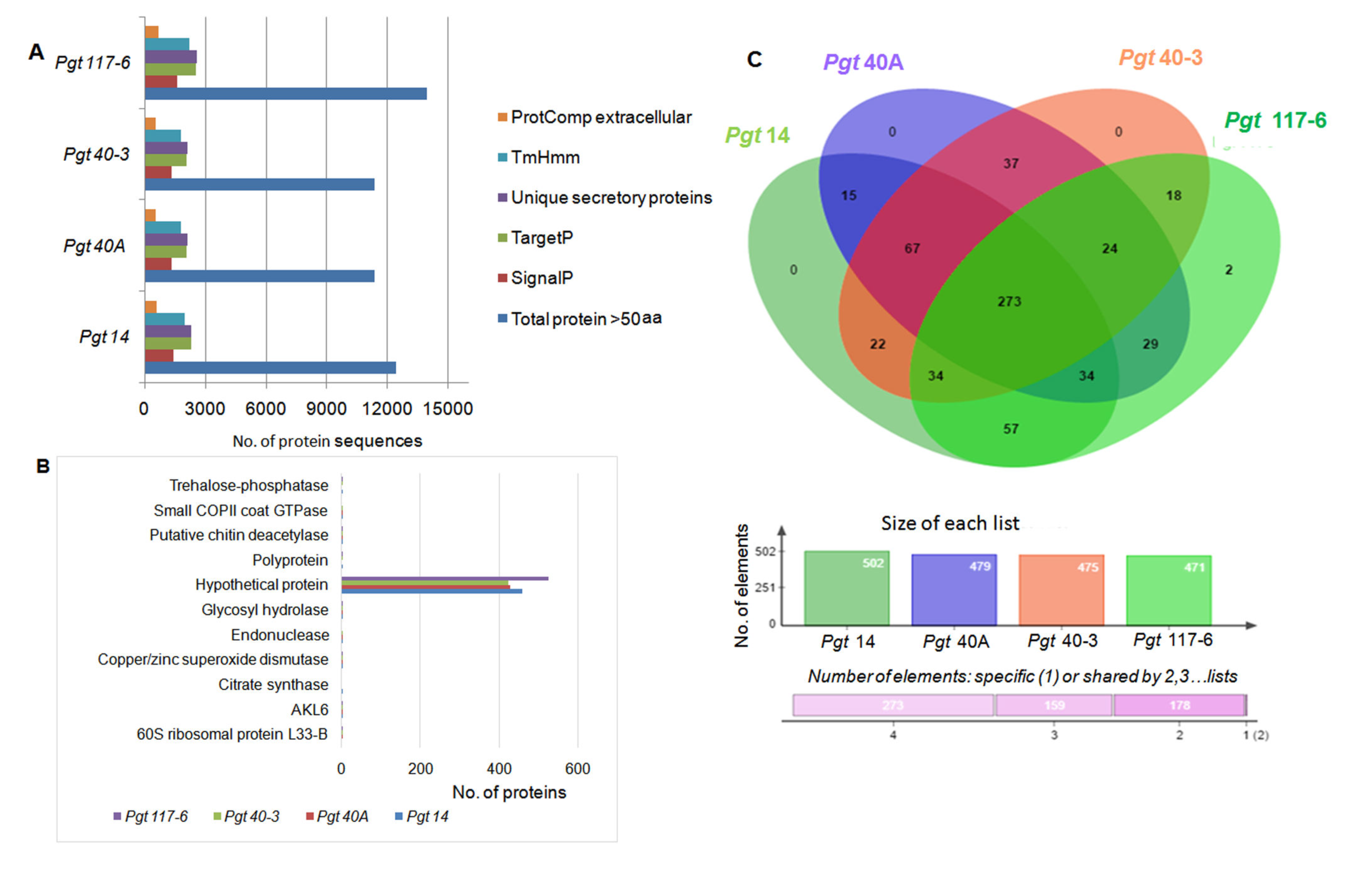
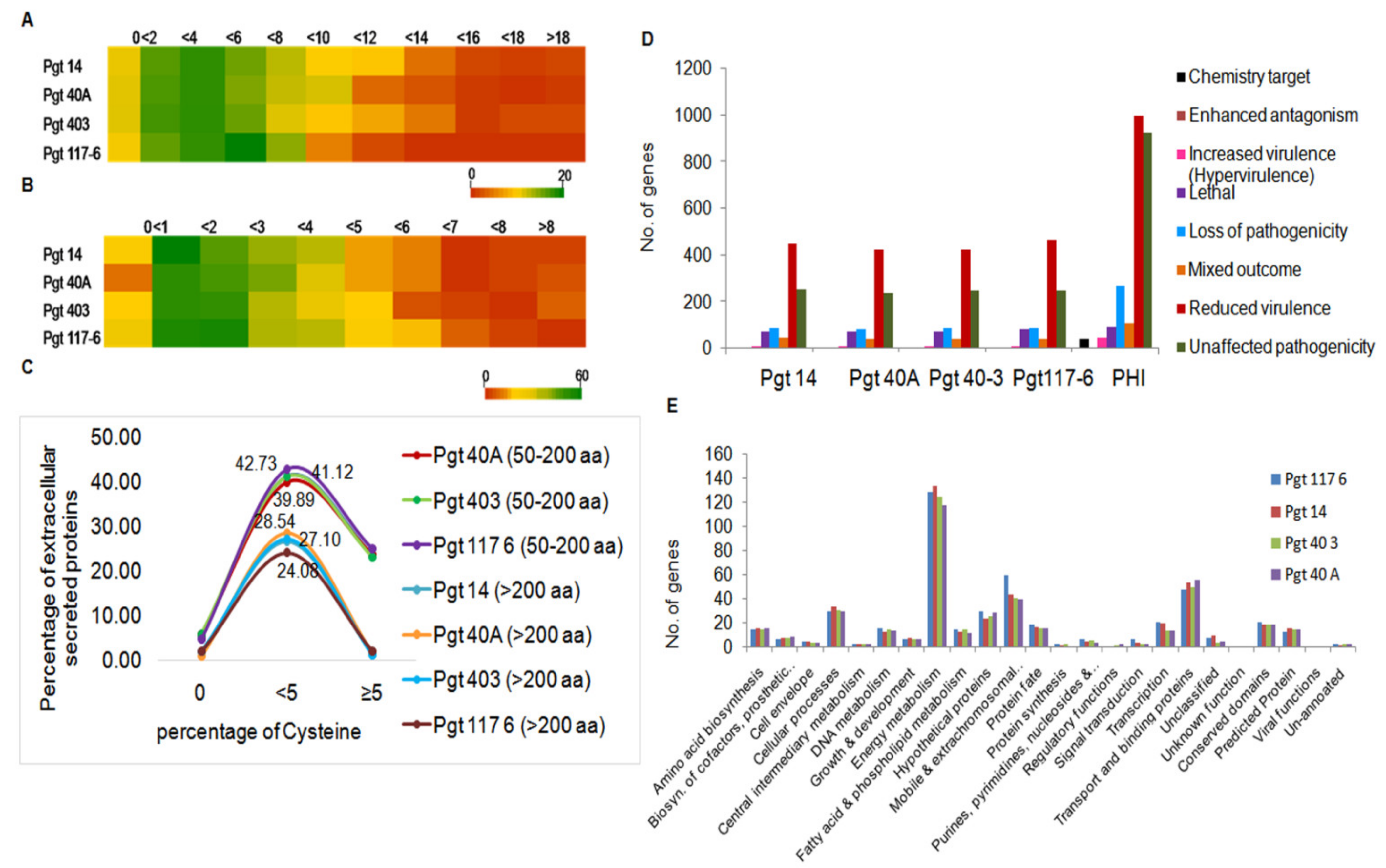
3.3. Identification and Analysis of Secretory Proteins
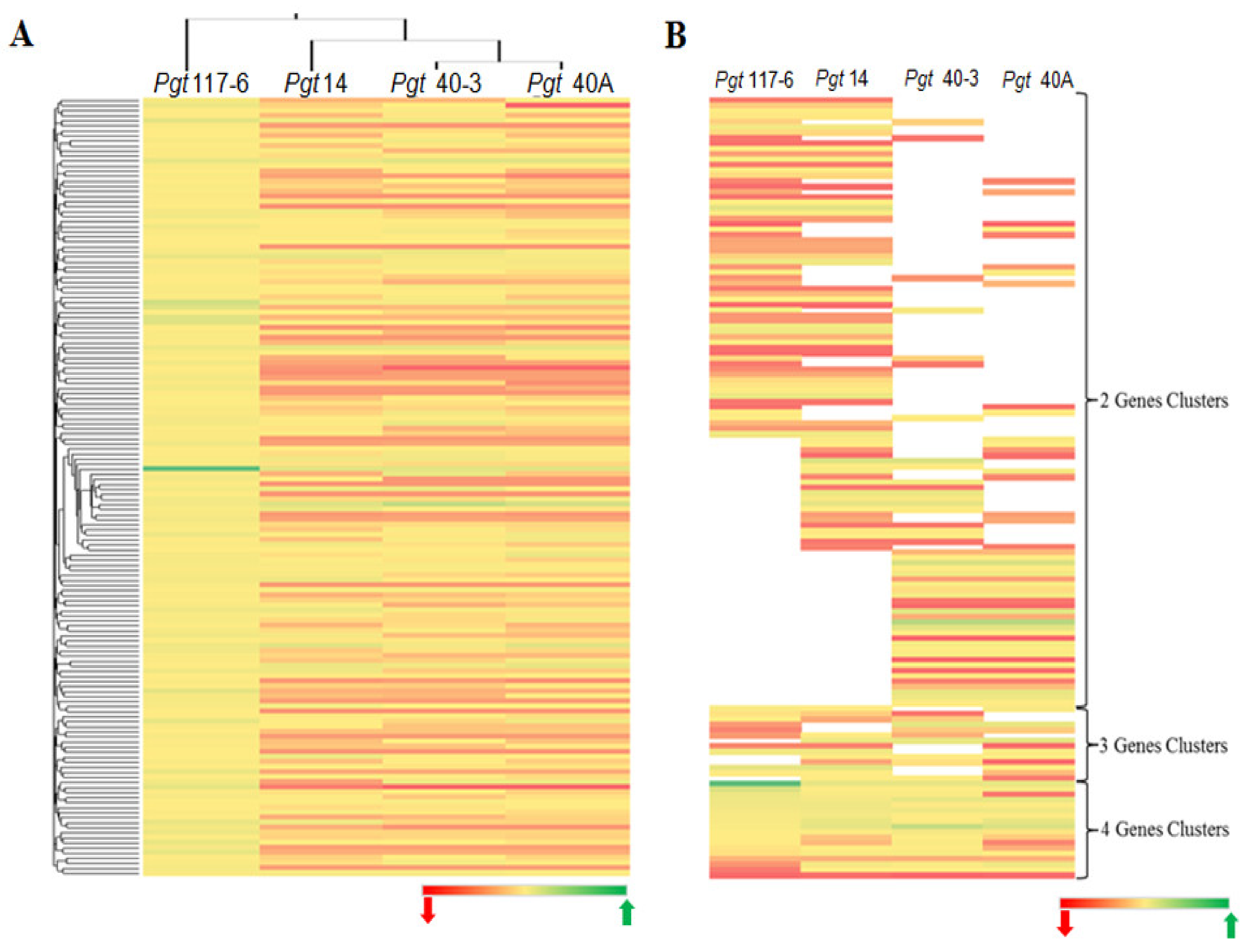
3.4. Genome-Wide Analysis for Cysteine-Rich Genes
3.5. Identification of Pathogenicity-Related Genes
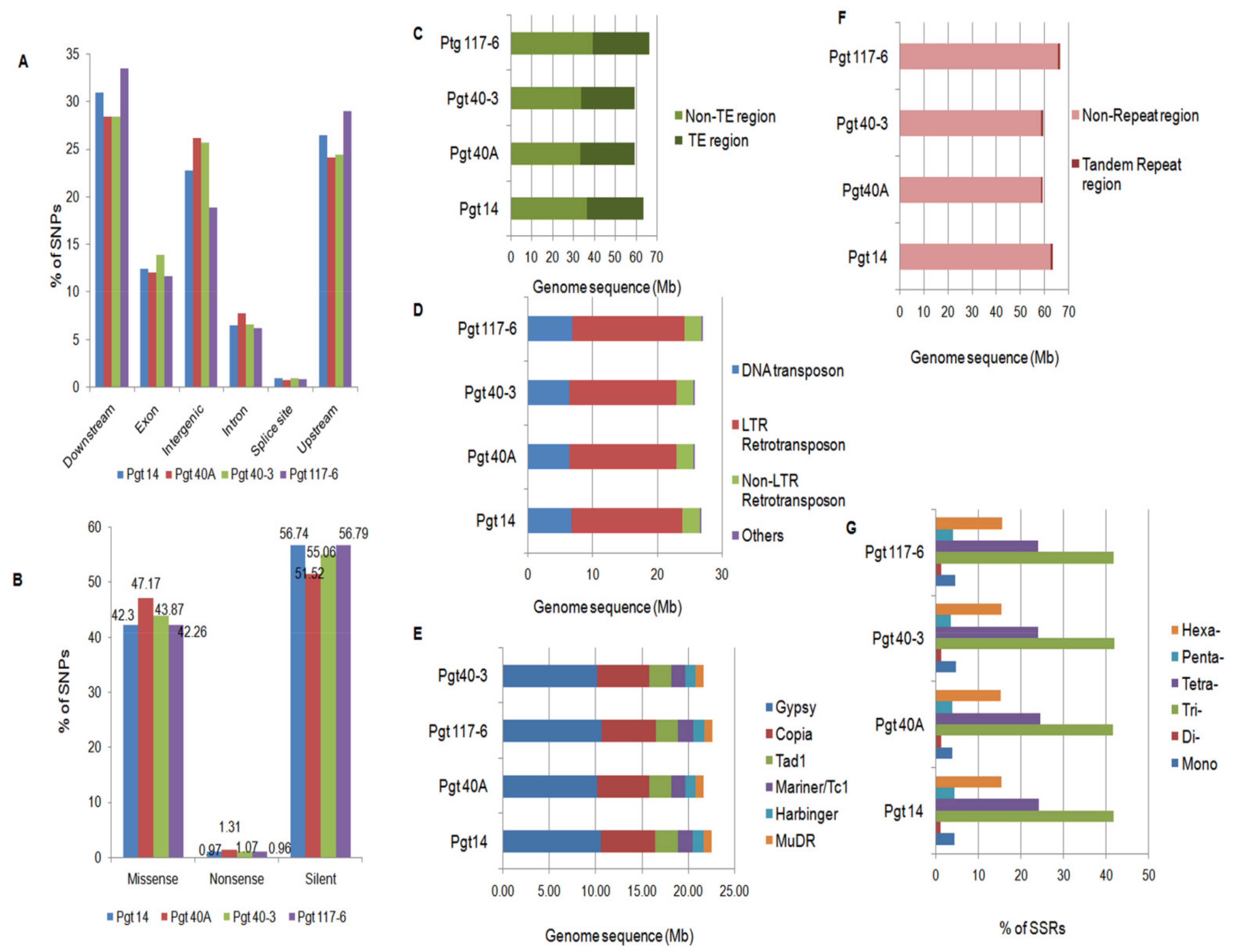
3.6. Genome-Wide Identification of SNPs
3.7. Identification of Repetitive Elements
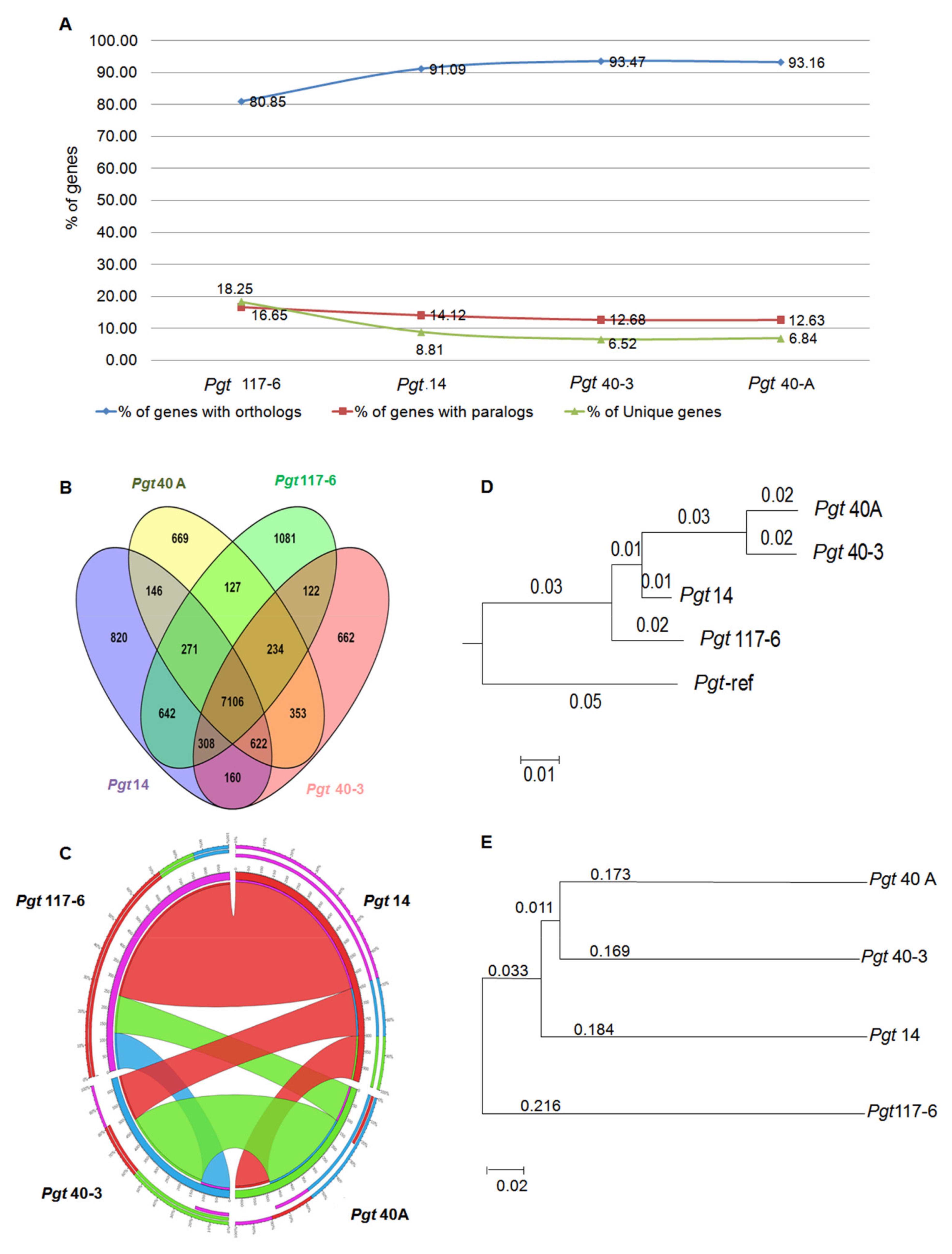
3.8. Conservation of Orthology and Paralogy Genes in the Pathotypes
3.9. Evolutionary Relationship among the P. graminis Pathotypes
3.10. Genome-Wide Analysis of Diversifying Selection
3.11. Analysis of Substitution Rates of Sequence Divergence
3.12. Identification of Genes under Site-Specific Diversifying Selection and Their Functional Categorization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kolmer, J.A. Tracking wheat rust on a continental scale. Curr. Opin. Plant Biol. 2005, 8, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. Pathogens which threaten food security: Puccinia striiformis, the wheat stripe rust pathogen. Food Secur. 2020, 12, 239–251. [Google Scholar] [CrossRef]
- Kleinhofs, A.; Brueggeman, R.; Nirmala, J.; Zhang, L.; Mirlohi, A.; Druka, A.; Rostoks, N.; Steffenson, B.J. Barley Stem Rust Resistance Genes: Structure and Function. Plant Genome 2009, 2, 109–120. [Google Scholar] [CrossRef]
- Zheng, W.; Huang, L.; Huang, J.; Wang, X.; Chen, X.; Zhao, J.; Guo, J.; Zhuang, H.; Qiu, C.; Liu, J.; et al. High genome heterozygosity and endemic genetic recombination in the wheat stripe rust fungus. Nat. Commun. 2013, 4, 2673. [Google Scholar] [CrossRef]
- Cantu, D.; Govindarajulu, M.; Kozik, A.; Wang, M.; Chen, X.; Kojima, K.; Jurka, J.; Michelmore, R.W.; Dubcovsky, J. Next Generation Sequencing Provides Rapid Access to the Genome of Puccinia striiformis f. sp. tritici, the Causal Agent of Wheat Stripe Rust. PLoS ONE 2011, 6, e24230. [Google Scholar] [CrossRef]
- Cantu, D.; Segovia, V.; MacLean, D.; Bayles, R.; Chen, X.; Kamoun, S.; Dubcovsky, J.; Saunders, D.G.; Uauy, C. Genome analyses of the wheat yellow (stripe) rust pathogen Puccinia striiformis f. sp. tritici reveal poly-morphic and haustorial expressed secreted proteins as candidate effectors. BMC Genom. 2013, 14, 1–18. [Google Scholar] [CrossRef]
- Cuomo, C.A.; Bakkeren, G.; Khalil, H.B.; Panwar, V.; Joly, D.; Linning, R.; Sakthikumar, S.; Song, X.; Adiconis, X.; Fan, L.; et al. Comparative Analysis Highlights Variable Genome Content of Wheat Rusts and Divergence of the Mating Loci. Genes Genomes Genet. 2017, 7, 361–376. [Google Scholar] [CrossRef]
- Duplessis, S.; Cuomo, C.A.; Lin, Y.-C.; Aerts, A.; Tisserant, E.; Veneault-Fourrey, C.; Joly, D.; Hacquard, S.; Amselem, J.; Cantarel, B.L.; et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. USA 2011, 108, 9166–9171. [Google Scholar] [CrossRef] [PubMed]
- Kiran, K.; Rawal, H.C.; Dubey, H.; Jaswal, R.; Bhardwaj, S.C.; Prasad, P.; Pal, D.; Devanna, B.N.; Sharma, T.R. Dissection of genomic features and variations of three pathotypes of Puccinia striiformis through whole genome sequencing. Sci. Rep. 2017, 7, srep42419. [Google Scholar] [CrossRef]
- Kiran, K.; Rawal, H.C.; Dubey, H.; Jaswal, R.; Devanna, B.; Gupta, D.K.; Bhardwaj, S.C.; Prasad, P.; Pal, D.; Chhuneja, P.; et al. Draft Genome of the Wheat Rust Pathogen (Puccinia triticina) Unravels Genome-Wide Structural Variations during Evolution. Genome Biol. Evol. 2016, 8, 2702–2721. [Google Scholar] [CrossRef]
- Rutter, W.B.; Salcedo, A.; Akhunova, A.; He, F.; Wang, S.; Liang, H.; Bowden, R.L.; Akhunov, E. Divergent and convergent modes of interaction between wheat and Puccinia graminis f. sp. tritici isolates revealed by the comparative gene co-expression network and genome analyses. BMC Genom. 2017, 18, 291. [Google Scholar] [CrossRef]
- Upadhyaya, N.M.; Garnica, D.P.; Ekaraoglu, H.; Sperschneider, J.; Enemri, A.; Exu, B.; Mago, R.; Cuomo, C.; Rathjen, J.; Park, R.; et al. Comparative genomics of Australian isolates of the wheat stem rust pathogen Puccinia graminis f. sp. tritici reveals extensive polymorphism in candidate effector genes. Front. Plant Sci. 2015, 5, 759. [Google Scholar] [CrossRef] [PubMed]
- Edae, E.A.; Rouse, M.N. Association mapping of resistance to emerging stem rust pathogen races in spring wheat using genotyping-by-sequencing. Plant Genome 2020, 13, e20050. [Google Scholar] [CrossRef]
- Jain, S.K.; Prashar, M.; Bhardwaj, S.C.; Singh, S.B.; Sharma, Y.P. Emergence of Virulence to Sr25 of Puccinia graminis f. sp. tritici on Wheat in India. Plant Dis. 2009, 93, 840. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, S.C. Puccinia-Triticum interaction: An update. Indian Phytopath. 2013, 66, 14–19. [Google Scholar]
- Pretorius, Z.A.; Singh, R.P.; Wagoire, W.W.; Payne, T.S. Detection of Virulence to Wheat Stem Rust Resistance Gene Sr31 in Puccinia graminis f. sp. tritici in Uganda. Plant Dis. 2000, 84, 203. [Google Scholar] [CrossRef]
- Singh, R.P.; Hodson, D.P.; Huerta-Espino, J.; Jin, Y.; Bhavani, S.; Njau, P.; Herrera-Foessel, S.; Singh, P.; Singh, S.; Govindan, V. The Emergence of Ug99 Races of the Stem Rust Fungus is a Threat to World Wheat Production. Annu. Rev. Phytopathol. 2011, 49, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Stukenbrock, E.H.; Jørgensen, F.G.; Zala, M.; Hansen, T.T.; McDonald, B.A.; Schierup, M.H. Whole-Genome and Chromosome Evolution Associated with Host Adaptation and Speciation of the Wheat Pathogen Mycosphaerella graminicola. PLoS Genet. 2010, 6, e1001189. [Google Scholar] [CrossRef]
- Raffaele, S.; Farrer, R.A.; Cano, L.M.; Studholme, D.J.; MacLean, D.; Thines, M.; Jiang, R.H.Y.; Zody, M.C.; Kunjeti, S.G.; Donofrio, N.M.; et al. Genome Evolution Following Host Jumps in the Irish Potato Famine Pathogen Lineage. Science 2010, 330, 1540–1543. [Google Scholar] [CrossRef]
- Wicker, T.; Oberhaensli, S.; Parlange, F.; Buchmann, J.P.; Shatalina, M.; Roffler, S.; Ben-David, R.; Doležel, J.; Šimková, H.; Schulze-Lefert, P. The wheat powdery mildew genome shows the unique evolution of an obligate biotroph. Nat. Genet. 2013, 45, 1092–1096. [Google Scholar] [CrossRef]
- Sperschneider, J.; Ying, H.; Dodds, P.N.; Gardiner, D.M.; Upadhyaya, N.M.; Singh, K.B.; Manners, J.M.; Taylor, J.M. Diversifying selection in the wheat stem rust fungus acts predominantly on pathogen-associated gene families and reveals candidate effectors. Front. Plant Sci. 2014, 5, 372. [Google Scholar] [CrossRef]
- Bhardwaj, S.C. Resistance Genes and Adult Plant Rust Resistance of Released Wheat Varieties of India; Regional Station, Directorate of Wheat Research: Shimla, India, 2011. [Google Scholar]
- Prasad, P.; Bhardwaj, S.C.; Savadi, S.; Kashyap, P.L.; Gangwar, O.P.; Khan, H.; Singh, S.B.; Kumar, S. Population distribution and differentiation of Puccinia graministritici detected in the Indian subcontinent during 2009–2015. Crop Protect. 2018, 108, 128–136. [Google Scholar] [CrossRef]
- Sharma, A.K.; Saharan, M.; Bhardwaj, S.C.; Prashar, M.; Chatrath, R.; Tiwari, V.; Singh, M.; Sharma, I. Evaluation of wheat (Triticum aestivum) germplasm and varieties against stem rust (Puccinia graminis f. sp. tritici) pathotype Ug99 and its variants. Indian Phytopathol. 2015, 88, 134–138. [Google Scholar]
- Jain, S.K.; Bhardwaj, S.C.; Prashar, M.; Singh, S.B. Physiologic specialization and new virulences of Puccinia graminis f. sp. tritici causing black rust of wheat (Triticum aestivum) in India during 2005–2009. Indian J. Agric. Sci. 2013, 83, 1058–1063. [Google Scholar]
- Bhardwaj, S.C.; Prashar, M.; Kumar, S.; Datta, D. Virulence and diversity of Puccinia triticina on wheat in India during 2002–2004. Indian J. Agric. Sci. 2006, 76, 302–306. [Google Scholar]
- Sai Prasad, S.V.; Singh, S.K.; Ambati, V.D.; Prakasha, T.L.; Singh, J.B.; Dubey, V.G.; Kantwa, S.R.; Mishra, A.N. Introgression of stem rust resistance gene Sr36 into durum wheat back ground using marker assisted backcross breeding. J. Wheat Res. 2014, 6, 21–24. [Google Scholar]
- Mishra, A.N.; Kaushal, K.; Dubey, V.G.; Prasad, S.V.S. New sources of stem rust resistance in durum wheat. Indian Phytopathol. 2014, 67, 199–200. [Google Scholar]
- Mishra, A.N.; Shirsekar, G.S.; Yadav, S.R.; Dubey, V.G.; Kaushal, K.; Prasad, S.V.S.; Pandey, H.N. Protocols for evaluating resistance to leaf and stem rusts in durum and bread wheats. Indian Phytopathol. 2009, 62, 461–468. [Google Scholar]
- Bahadur, P.; Nagarajan, S.; Nayar, S.K. A proposed system for virulence designation in India. 2. Puccinia graminis f sptritici. Proc. Plant Sci. 1985, 95, 29. [Google Scholar]
- Bhardwaj, S.C.; Gangwar, O.P.; Singh, S.B.; Saharan, M.S.; Sharma, S. Rust situation and pathotypes of Puccinia species in Leh Ladakh in relation to recurrence of wheat rusts in India. Indian Phytopathol. 2012, 65, 230–232. [Google Scholar]
- Roose-Amsaleg, C.; De Vallavieille-Pope, C.; Brygoo, Y.; Levis, C. Characterisation of a length polymorphism in the two intergenic spacers of ribosomal RNA in Puccinia striiformis f. sp. tritici, the causal agent of wheat yellow rust. Mycol. Res. 2002, 106, 918–924. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Ning, Z.; Keane, T.; Korf, I. Assessing the gene space in draft genomes. Nucleic Acids Res. 2008, 37, 289–297. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Hulsen, T.; Huynen, M.A.; De Vlieg, J.; Groenen, P.M.A. Bench marking ortholog identification methods using functional genomics data. Genome Biol. 2006, 7, R31. [Google Scholar] [CrossRef] [PubMed]
- Heidel, A.J.; Lawal, H.M.; Felder, M.; Schilde, C.; Helps, N.R.; Tunggal, B.; Rivero, F.; John, U.; Schleicher, M.; Eichinger, L.; et al. Phylogeny-wide analysis of social amoeba genomes highlights ancient origins for complex intercellular communication. Genome Res. 2011, 21, 1882–1891. [Google Scholar] [CrossRef]
- Rawal, H.C.; Singh, N.K.; Sharma, T.R. Conservation, divergence, and genome-wide distribution of PAL and POX A gene families in plants. Int. J. Genom. 2013, 2013, 678969. [Google Scholar]
- Winnenburg, R.; Baldwin, T.K.; Urban, M.; Rawlings, C.; Köhler, J.; Hammond-Kosack, K.E. PHI-base: A new database for pathogen host interactions. Nucleic Acids Res. 2006, 34, D459–D464. [Google Scholar] [CrossRef]
- Baugh, L.; Gallagher, L.A.; Patrapuvich, R.; Clifton, M.C.; Gardberg, A.S.; Edwards, T.E.; Armour, B.; Begley, D.W.; Dieterich, S.H.; Dranow, D.M.; et al. Combining Functional and Structural Genomics to Sample the Essential Burkholderia Structome. PLoS ONE 2013, 8, e53851. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C.; VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 17 December 2017).
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Yang, Z. PAMLX: A Graphical User Interface for PAML. Mol. Biol. Evol. 2013, 30, 2723–2724. [Google Scholar] [CrossRef] [PubMed]
- Djamei, A.; Kahmann, R. Ustilago maydis: Dissecting the Molecular Interface between Pathogen and Plant. PLoS Pathog. 2012, 8, e1002955. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Yang, F.; Rong, W.; Wu, X.; Zhang, J.; Chen, S.; He, C.; Zhou, J.-M. A Xanthomonas uridine 5’-monophosphate tra nsferase inhibits plant immune kinases. Nature 2012, 485, 114–118. [Google Scholar] [CrossRef]
- Houterman, P.M.; Speijer, D.; Dekker, H.L.; de Koster, C.G.; Cornelissen, B.J.C.; Rep, M. The mixed xylem sap proteome of Fusarium oxysporum-infected tomato plants X veMol. Plant Pathol. 2007, 8, 215–221. [Google Scholar]
- Ridout, C.; Skamnioti, P.; Porritt, O.; Sacristan, S.; Jones, J.; Brown, J.K. Multiple Avirulence Paralogues in Cereal Powdery Mildew Fungi May Contribute to Parasite Fitness and Defeat of Plant Resistance. Plant Cell 2006, 18, 2402–2414. [Google Scholar] [CrossRef]
- Lerat, E. Identifying repeats and transposable elements in sequenced genomes: How to find your way through the dense forest of programs. Heredity 2010, 104, 520–533. [Google Scholar] [CrossRef]
- Gladieux, P.; Ropars, J.; Badouin, H.; Branca, A.; Aguileta, G.; de Vienne, D.; De La Vega, R.C.R.; Branco, S.M.; Giraud, T. Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol. Ecol. 2014, 23, 753–773. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Estimating Synonymous and Nonsynonymous Substitution Rates Under Realistic Evolutionary Models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef]
- Chaves, M.; Martinelli, J.A.; Wesp-Guterres, C.; Graichen, F.A.S.; Brammer, S.P.; Scagliusi, S.M.; Da Silva, P.R.; Wiethölter, P.; Torres, G.A.M.; Yamazaki-Lau, E.; et al. The importance for food security of maintaining rust resistance in wheat. Food Secur. 2013, 5, 157–176. [Google Scholar] [CrossRef]
- Braun, H.J.; Atlin, G.; Payne, T. Multi-location testing as a tool to identify plant response to global climate change. Clim. Chang. Crop Prod. 2011, 1, 115–138. [Google Scholar] [CrossRef]
- Sheikh, F.A.; Dar, Z.A.; Sofi, P.A.; Lone, A.A.; Shiekh, N.A. Stem Rust of Wheat—A Basic Review. Int. J. Pure App. Biosci. 2017, 5, 316–328. [Google Scholar] [CrossRef]
- Patpour, M.; Justesen, A.F.; Tecle, A.W.; Yazdani, M.; Yasaie, M.; Hovmøller, M.S. First Report of Race TTRTF of Wheat Stem Rust (Puccinia graminis f. sp. tritici) in Eritrea. Plant Dis. 2020, 104, 973. [Google Scholar] [CrossRef]
- Bhardwaj, S.C.; Prashar, M.; Prasad, P. Ug99-Future Challenges. In Future Challenges in Crop Protection against Fungal Pathogens; Springer: Berlin/Heidelberg, Germany, 2014; pp. 231–247. [Google Scholar]
- Bhardwaj, S.C. Wheat rust pathotypes in Indian subcontinent then and now. In Wheat-Productivity Enhancement under Changing Climate, 1st ed.; Singh, S.S., Hanchinal, R.R., Singh, G., Sharma, R.K., Saharan, M.S., Sharma, I., Eds.; Narosa Publishing House Private Limited: New Delhi, India, 2012; pp. 227–238. Available online: https://www.researchgate.net/publication/317401824 (accessed on 6 September 2018).
- Sperschneider, J.; Dodds, P.; Gardiner, D.; Manners, J.M.; Singh, K.; Taylor, J. Advances and Challenges in Computational Prediction of Effectors from Plant Pathogenic Fungi. PLoS Pathog. 2015, 11, e1004806. [Google Scholar] [CrossRef]
- Persoons, A.; Morin, E.; Delaruelle, C.; Payen, T.; Halkett, F.; Frey, P.; De Mita, S.; Duplessis, S. Patterns of genomic variation in the poplar rust fungus Melampsora larici-populina identify pathogenesis-related factors. Front. Plant Sci. 2014, 5, 450. [Google Scholar] [CrossRef]
- Kemen, A.C.; Agler, M.T.; Kemen, E. Host–microbe and microbe–microbe interactions in the evolution of obligate plant parasitism. New Phytol. 2015, 206, 1207–1228. [Google Scholar] [CrossRef]
- Gauthier, N.W.; Maruthachalam, K.; Subbarao, K.V.; Brown, M.; Xiao, Y.; Robertson, C.L.; Schneider, R.W. Mycoparasitism of Phakopsora pachyrhizi, the soybean rust pathogen, by Simplicillium lanosoniveum. Biol. Control. 2014, 76, 87–94. [Google Scholar] [CrossRef]
- Giraldo, M.; Valent, B. Filamentous plant pathogen effectors in action. Nat. Rev. Genet. 2013, 11, 800–814. [Google Scholar] [CrossRef]
- Fernandez, D.; Tisserant, E.; Talhinhas, P.; Azinheira, H.; Vieira, A.; Petitot, A.; Loureiro, A.; Poulain, J.; Da Silva, C.; Silva, M.C.; et al. 454-pyrosequencing of Coffea arabica leaves infected by the rust fungus Hemileia vastatrix reveals in planta-expressed pathogen-secreted proteins and plant functions in a late compatible plant-rust interaction. Mol. Plant Pathol. 2011, 13, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Pretsch, K.; Kemen, A.; Kemen, E.; Geiger, M.; Mendgen, K.; Voegele, R. The rust transferred proteins-a new family of effector proteins exhibiting protease inhibitor function. Mol. Plant Pathol. 2012, 14, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Flor, H.H. Current Status of the Gene-For-Gene Concept. Annu. Rev. Phytopathol. 1971, 9, 275–296. [Google Scholar] [CrossRef]
- Wang, D.; Tian, L.; Zhang, D.; Song, J.; Song, S.; Yin, C.; Zhou, L.; Liu, Y.; Wang, B.; Kong, Z. Functional analyses of small secreted cysteine rich proteins identified candidate effectors in Verticillium dahliae. Mol. Plant Pathol. 2020, 21, 667–685. [Google Scholar] [CrossRef]
- Singh, S.K.; Prakasha, T.L.; Divya, A.; Kantwa, S.L.; Prasad, S.V.S.; Mishra, A.N. Evaluation of Indian durum wheat germplasm for the presence of Sr36 gene for resistance to pathotypes of stem rust race 117-group. Indian Phytopathol. 2013, 66, 308–310. [Google Scholar]
- Ellis, J.G.; Lagudah, E.; Spielmeyer, W.; Dodds, P. The past, present and future of breeding rust resistant wheat. Front. Plant Sci. 2014, 5, 641. [Google Scholar] [CrossRef]
- Möller, M.; Stukenbrock, E.H. Evolution and genome architecture in fungal plant pathogens. Nat. Rev. Genet. 2017, 15, 756–771. [Google Scholar] [CrossRef]
- Wellings, C.R. Puccinia striiformis in Australia: A review of the incursion, evolution, and adaptation of stripe rust in the period 1979–2006. Aust. J. Agric. Res. 2007, 58, 567–575. [Google Scholar] [CrossRef]
- Milus, E.A.; Kristensen, K.; Hovmoller, M. Evidence for Increased Aggressiveness in a Recent Widespread Strain of Puccinia striiformis f. sp. tritici Causing Stripe Rust of Wheat. Phytopathology 2009, 99, 89–94. [Google Scholar] [CrossRef]
- Hovmoller, M.; Walter, S.; Justesen, A.F. Escalating Threat of Wheat Rusts. Science 2010, 329, 369. [Google Scholar] [CrossRef] [PubMed]
- Ohm, R.A.; Feau, N.; Henrissat, B.; Schoch, C.L.; Horwitz, B.A.; Barry, K.W.; Condon, B.J.; Copeland, A.C.; Dhillon, B.; Glaser, F.; et al. Diverse Lifestyles and Strategies of Plant Pathogenesis Encoded in the Genomes of Eighteen Dothideomycetes Fungi. PLoS Pathog. 2012, 8, e1003037. [Google Scholar] [CrossRef]
- Stefansson, T.S.; McDonald, B.A.; Willi, Y. The Influence of Genetic Drift and Selection on Quantitative Traits in a Plant Pathogenic Fungus. PLoS ONE 2014, 9, e112523. [Google Scholar] [CrossRef]
- Ali, S.; Gladieux, P.; Leconte, M.; Gautier, A.; Justesen, A.F.; Hovmoller, M.; Enjalbert, J.; De Vallavieille-Pope, C. Origin, Migration Routes and Worldwide Population Genetic Structure of the Wheat Yellow Rust Pathogen Puccinia striiformis f. sp. tritici. PLoS Pathog. 2014, 10, e1003903. [Google Scholar] [CrossRef] [PubMed]
- Kiran, K.; Ansari, S.A.; Srivastava, R.; Lodhi, N.; Chaturvedi, C.P.; Sawant, S.V.; Tuli, R. The TATA-Box Sequence in the Basal Promoter Contributes to Determining Light-Dependent Gene Expression in Plants. Plant Physiol. 2006, 142, 364–376. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P.graminis Pathotypes | ||||
|---|---|---|---|---|
| Parameters | 14 | 40A | 40-3 | 117-6 |
| Input reads | 67,098,012 (6.57 Gb) | 61,276,376 (5.98 Gb) | 60,290,992 (5.90 Gb) | 65,648,262 (6.42 Gb) |
| Mapped Reads | 62.77% | 67.29% | 67.26% | 52.44% |
| Mapped Bases | 62.00% | 66.64% | 66.42% | 51.94% |
| Total contigs (assembled genome) | 68,622 (63.31 Mb) | 69,842 (59.15 Mb) | 70,264 (59.25 Mb) | 58,140 (66.25 Mb) |
| N50 (contigs) (bp) | 4153 | 3573 | 3584 | 5288 |
| Average contig length (bp) | 922 | 846 | 843 | 1139 |
| GC content of assembled genome (%) | 43.61 | 43.64 | 43.64 | 43.58 |
| Largest contig (bp) | 65,288 | 43,631 | 39,779 | 46,544 |
| Contigs ≥ 200 bases | 37,103 (59.08 Mb) | 36,874 (54.69 Mb) | 36,934 (54.76 Mb) | 33,359 (62.88 Mb) |
| Contigs ≥ 2Kb | 7352 (42.14 Mb) | 7364 (37.98 Mb) | 7363 (38.07 Mb) | 7499 (47.55 Mb) |
| Average contig length (>2 Kb) (bp) | 5732 | 5157 | 5170 | 6341 |
| N50 (>2 Kb contigs) (bp) | 7152 | 6013 | 6022 | 8342 |
| Depth coverage | 103× | 101× | 99× | 96× |
| Repeats (TE) (Mb) | 26.84 | 25.76 | 25.78 | 27.05 |
| Repeats (TE) % in assembled genome | 42.40 | 43.56 | 43.51 | 40.83 |
| Number of genes predicted | 13,854 | 12,636 | 12,670 | 15,401 |
| Mean gene length (bp) | 1132.44 | 1151.94 | 1152.51 | 1092.92 |
| Total number of exons | 65,082 | 59,820 | 59,942 | 71,844 |
| Mean number of exons per gene | 4.69 | 4.73 | 4.73 | 4.66 |
| Largest gene length (bp) | 17,007 | 17,040 | 17,040 | 16,833 |
| Genes (>150 bases) | 12,424 | 11,347 | 11,371 | 13,943 |
| Genes (>450 bases) | 9419 | 8665 | 8694 | 10,315 |
| Average gene length (bp) (>450 bases genes) | 1561.15 | 1578.23 | 1578.19 | 1548.67 |
| Mean number of exons per gene (>450 bases genes) | 5.72 | 5.75 | 5.74 | 5.65 |
| Pathotypes | 14 | 40A | 40-3 | 117-6 |
|---|---|---|---|---|
| SNPs | 780,353 | 686,441 | 710,804 | 1,001,341 |
| Insertions | 51,662 | 44,403 | 47,055 | 46,206 |
| Deletions | 24,105 | 22,336 | 23,630 | 26,882 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiran, K.; Rawal, H.C.; Dubey, H.; Jaswal, R.; Bhardwaj, S.C.; Deshmukh, R.; Sharma, T.R. Genome-Wide Analysis of Four Pathotypes of Wheat Rust Pathogen (Puccinia graminis) Reveals Structural Variations and Diversifying Selection. J. Fungi 2021, 7, 701. https://doi.org/10.3390/jof7090701
Kiran K, Rawal HC, Dubey H, Jaswal R, Bhardwaj SC, Deshmukh R, Sharma TR. Genome-Wide Analysis of Four Pathotypes of Wheat Rust Pathogen (Puccinia graminis) Reveals Structural Variations and Diversifying Selection. Journal of Fungi. 2021; 7(9):701. https://doi.org/10.3390/jof7090701
Chicago/Turabian StyleKiran, Kanti, Hukam C. Rawal, Himanshu Dubey, Rajdeep Jaswal, Subhash C. Bhardwaj, Rupesh Deshmukh, and Tilak Raj Sharma. 2021. "Genome-Wide Analysis of Four Pathotypes of Wheat Rust Pathogen (Puccinia graminis) Reveals Structural Variations and Diversifying Selection" Journal of Fungi 7, no. 9: 701. https://doi.org/10.3390/jof7090701
APA StyleKiran, K., Rawal, H. C., Dubey, H., Jaswal, R., Bhardwaj, S. C., Deshmukh, R., & Sharma, T. R. (2021). Genome-Wide Analysis of Four Pathotypes of Wheat Rust Pathogen (Puccinia graminis) Reveals Structural Variations and Diversifying Selection. Journal of Fungi, 7(9), 701. https://doi.org/10.3390/jof7090701