
A Multiomic Approach to Understand How Pleurotus eryngii Transforms Non-Woody Lignocellulosic Material

 ,
,  , , , ,
, , , , 
 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Organism and Culture Conditions
2.2. Library Preparation and RNA Sequencing
2.3. Protein Extraction and Detection
2.4. Automatic and Expert Gene Annotation
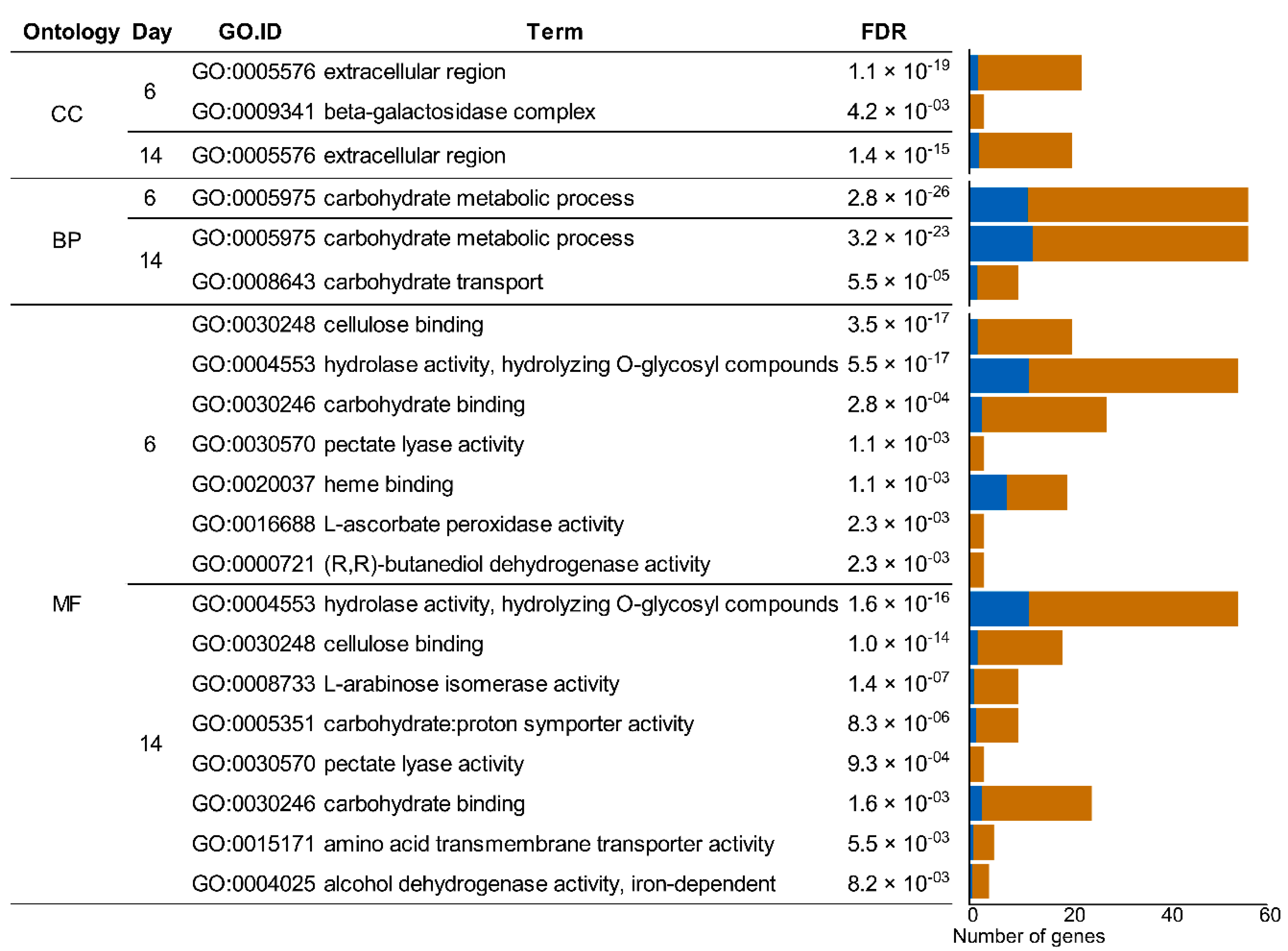
2.5. Gene Set Enrichment Analysis
2.6. Targeted Homology Searches for Aromatic Catabolism
2.7. Chemical Analyses
3. Results
3.1. Repertoire of Plant Cell-Wall Degrading Enzymes in the Genome of P. eryngii
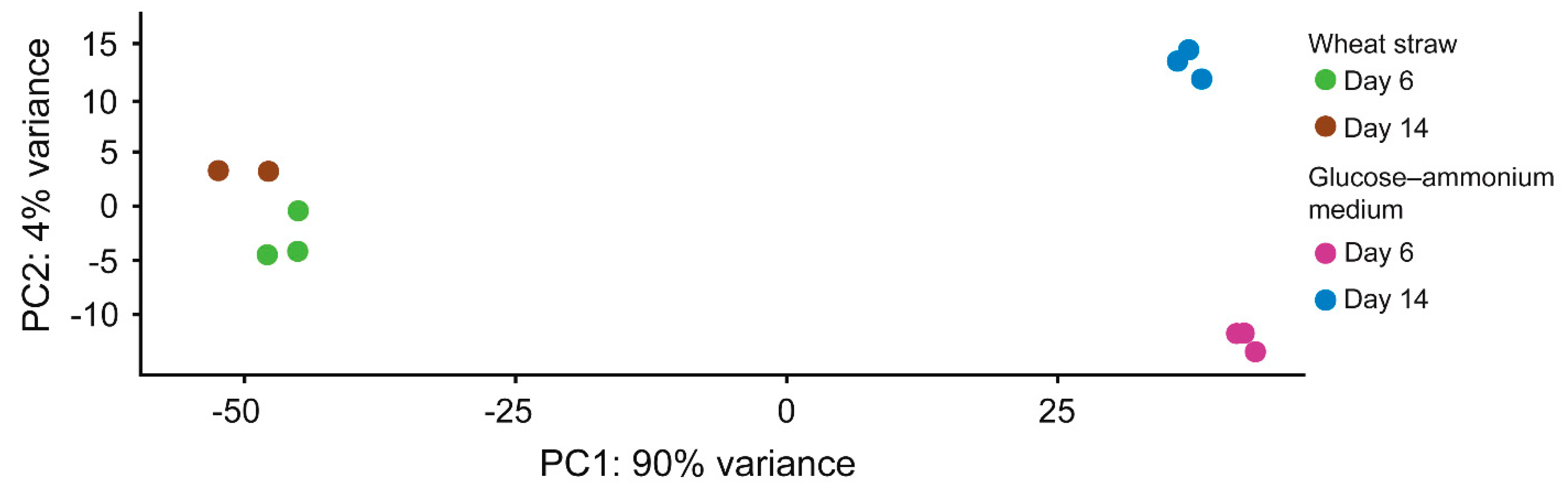
3.2. Proteins Mobilized by P. eryngii on Wheat–Straw and Glucose–Ammonium Cultures
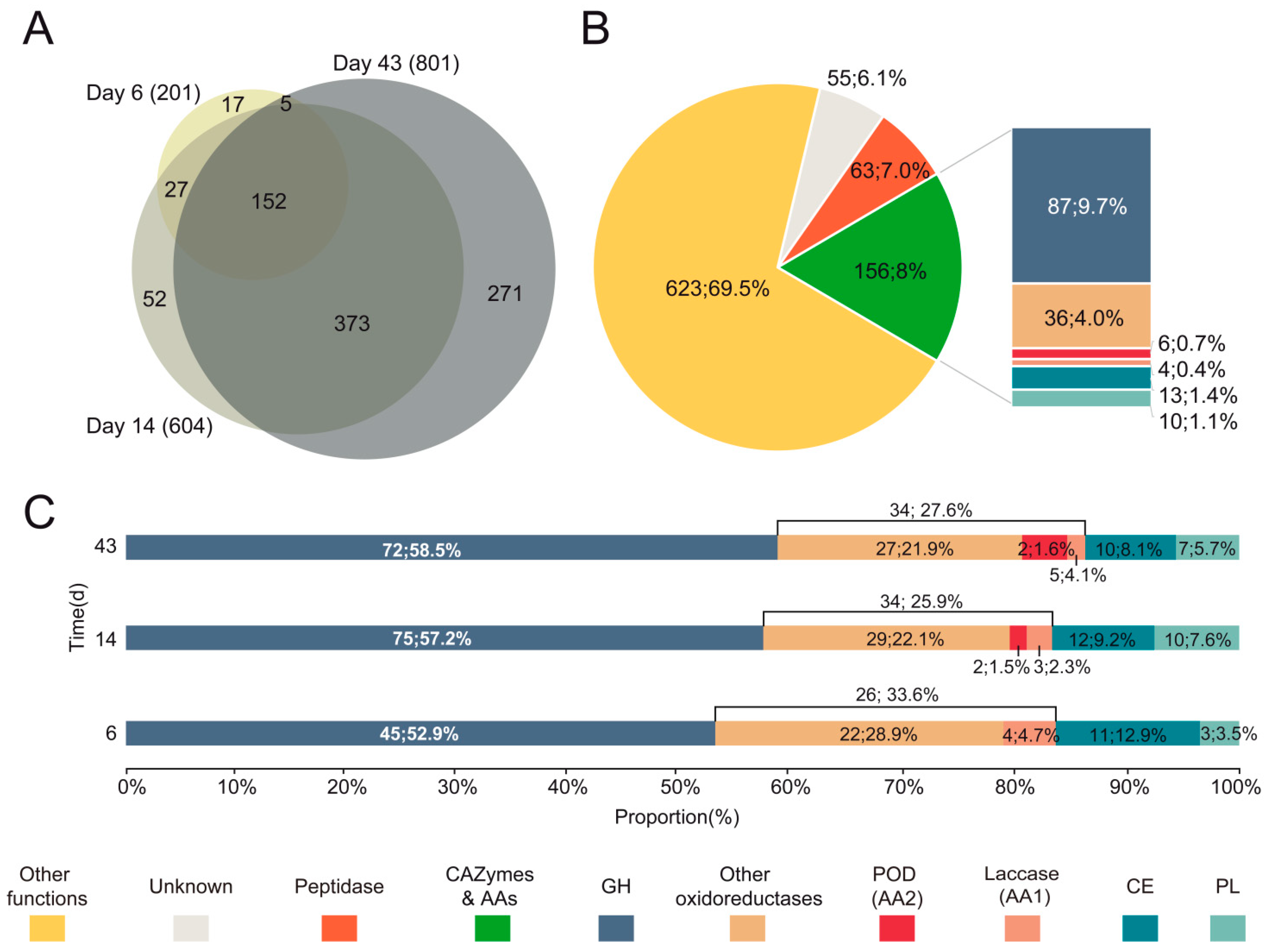
3.3. Diversity and Abundance of Main Enzyme Types in the Exoproteome of P. eryngii
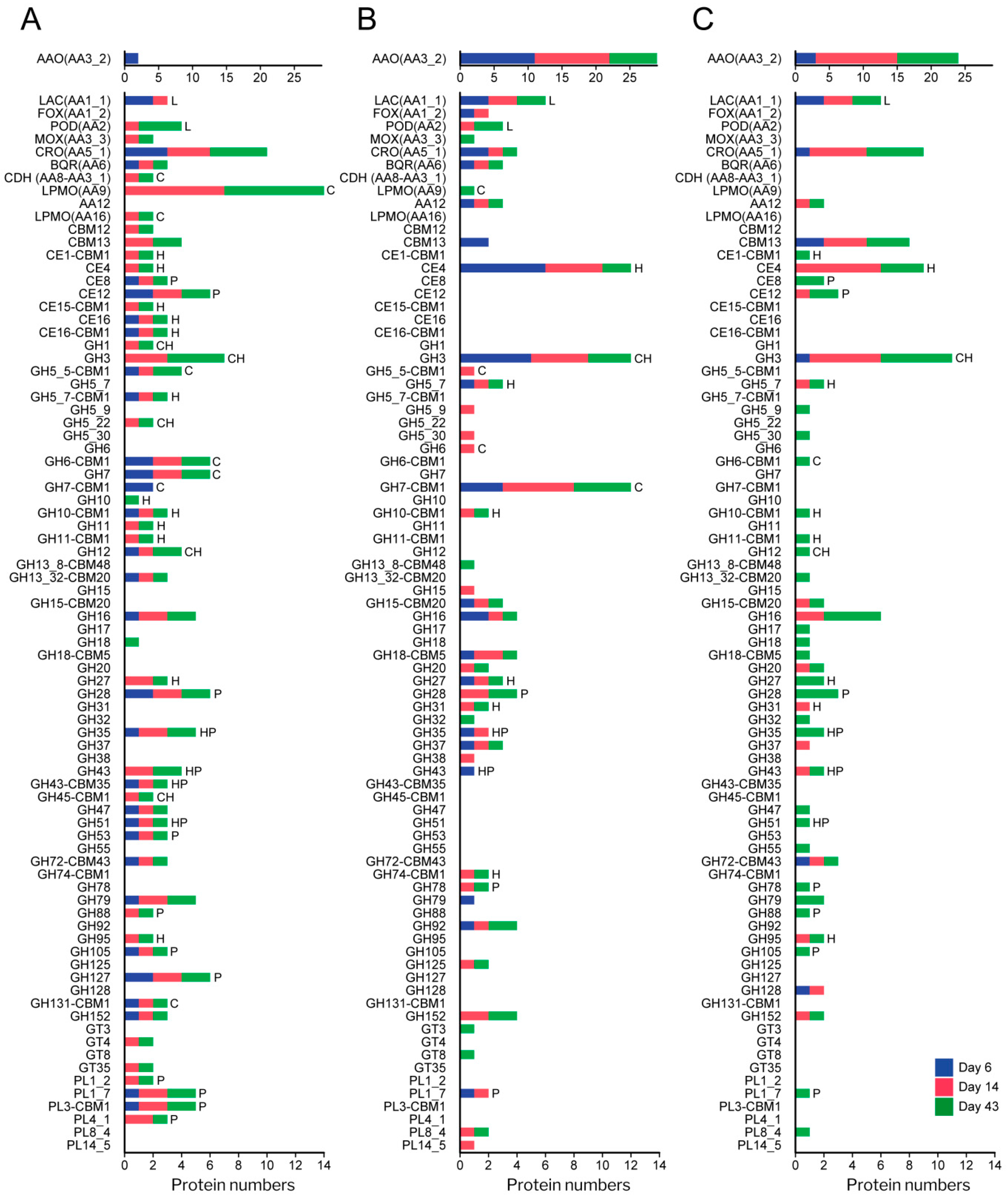
3.4. Analysis of the Enzymatic Arsenal Activated by P. eryngii Growing on Wheat–Straw
3.4.1. Polysaccharide-Decay Machinery
3.4.2. CBM-Containing Plant Cell-Wall Degrading Enzymes (PCWDEs)
3.4.3. Lignin-Decay Machinery
3.5. Catabolism of Simple Aromatic Compounds
3.6. Degradation of Wheat–Straw Components
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dahmen, N.; Lewandowski, I.; Zibek, S.; Weidtmann, A. Integrated lignocellulosic value chains in a growing bioeconomy: Status quo and perspectives. GCB Bioenergy 2019, 11, 107–117. [Google Scholar] [CrossRef]
- Himmel, M.E.; Ding, S.Y.; Johnson, D.K.; Adney, W.S.; Nimlos, M.R.; Brady, J.W.; Foust, T.D. Biomass recalcitrance: Engineering plants and enzymes for biofuels production. Science 2007, 315, 804–807. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Williams, C.K.; Davison, B.H.; Britovsek, G.; Cairney, J.; Eckert, C.A.; Frederick, W.J.; Hallett, J.P.; Leak, D.J.; Liotta, C.L.; et al. The path forward for biofuels and biomaterials. Science 2006, 311, 484–489. [Google Scholar] [CrossRef]
- Schutyser, W.; Renders, T.; Van den Bosch, S.; Koelewijn, S.F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Fukuoka, A. Synthesis and utilisation of sugar compounds derived from lignocellulosic biomass. Green Chem. 2013, 15, 1740–1763. [Google Scholar] [CrossRef]
- Rytioja, J.; Hilden, K.; Yuzon, J.; Hatakka, A.; de Vries, R.P.; Makela, M.R. Plant-polysaccharide-degrading enzymes from Basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.T.; Camarero, S.; Ruiz-Dueñas, F.J.; Martínez, M.J. Biological lignin degradation. In Lignin Valorization: Emerging Approaches; Beckham, G.T., Ed.; Royal Society of Chemistry: London, UK, 2018; pp. 199–225. [Google Scholar]
- Chen, S.; Su, L.; Chen, J.; Wu, J. Cutinase: Characteristics, preparation, and application. Biotechnol. Adv. 2013, 31, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Camarero, S.; Martínez, M.J.; Martínez, A.T. Understanding lignin biodegradation for the improved utilization of plant biomass in modern biorefineries. Biofuels Bioprod. Biorefining 2014, 8, 615–625. [Google Scholar] [CrossRef]
- Martínez, A.T.; Ruiz-Dueñas, F.J.; Martínez, M.J.; del Río, J.C.; Gutiérrez, A. Enzymatic delignification of plant cell wall: From nature to mill. COB 2009, 20, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fueyo, E.; Ruiz-Dueñas, F.J.; Ferreira, P.; Floudas, D.; Hibbett, D.S.; Canessa, P.; Larrondo, L.; James, T.Y.; Seelenfreund, D.; Lobos, S.; et al. Comparative genomics of Ceriporiopsis subvermispora and Phanerochaete chrysosporium provide insight into selective ligninolysis. Proc. Natl. Acad. Sci. USA 2012, 109, 5458–5463. [Google Scholar] [CrossRef]
- Hage, H.; Miyauchi, S.; Viragh, M.; Drula, E.; Min, B.; Chaduli, D.; Navarro, D.; Favel, A.; Norest, M.; Lesage-Meessen, L.; et al. Gene family expansions and transcriptome signatures uncover fungal adaptations to wood decay. Environ. Microbiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.; Larrondo, L.F.; Putnam, N.; Gelpke, M.D.; Huang, K.; Chapman, J.; Helfenbein, K.G.; Ramaiya, P.; Detter, J.C.; Larimer, F.; et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 2004, 22, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.; Challacombe, J.; Morgenstern, I.; Hibbett, D.S.; Schmoll, M.; Kubicek, C.P.; Ferreira, P.; Ruiz-Dueñas, F.J.; Martínez, A.T.; Kersten, P.; et al. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc. Natl. Acad. Sci. USA 2009, 106, 1954–1959. [Google Scholar] [CrossRef]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martínez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef]
- Suzuki, H.; MacDonald, J.; Syed, K.; Salamov, A.; Hori, C.; Aerts, A.; Henrissat, B.; Wiebenga, A.; vanKuyk, P.A.; Barry, K.; et al. Comparative genomics of the white-rot fungi, Phanerochaete carnosa and P. chrysosporium, to elucidate the genetic basis of the distinct wood types they colonize. BMC Genom. 2012, 13, 444. [Google Scholar] [CrossRef]
- Ruiz-Dueñas, F.J.; Barrasa, J.M.; Sánchez-García, M.; Camarero, S.; Miyauchi, S.; Serrano, A.; Linde, D.; Babiker, R.; Drula, E.; Ayuso-Fernández, I.; et al. Genomic analysis enlightens Agaricales lifestyle evolution and increasing peroxidase diversity. Mol. Biol. Evol. 2021, 38, 1428–1446. [Google Scholar] [CrossRef]
- Miyauchi, S.; Hage, H.; Drula, E.; Lesage-Meessen, L.; Berrin, J.G.; Navarro, D.; Favel, A.; Chaduli, D.; Grisel, S.; Haon, M.; et al. Conserved white rot enzymatic mechanism for wood decay in the Basidiomycota genus Pycnoporus. DNA Res. 2020. [Google Scholar] [CrossRef]
- Vanden Wymelenberg, A.; Gaskell, J.; Mozuch, M.; Bondurant, S.S.; Sabat, G.; Ralph, J.; Skyba, O.; Mansfield, S.D.; Blanchette, R.A.; Grigoriev, I.V.; et al. Significant alteration of gene expression in wood decay fungi Postia placenta and Phanerochaete chrysosporium by plant species. AEM 2011, 77, 4499–4507. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fueyo, E.; Ruiz-Dueñas, F.J.; Martínez, M.J.; Romero, A.; Hammel, K.E.; Medrano, F.J.; Martínez, A.T. Ligninolytic peroxidase genes in the oyster mushroom genome: Heterologous expression, molecular structure, catalytic and stability properties and lignin-degrading ability. Biotechnol. Biofuels 2014, 7, 2. [Google Scholar] [CrossRef]
- Fernández-Fueyo, E.; Ruiz-Dueñas, F.J.; Miki, Y.; Martínez, M.J.; Hammel, K.E.; Martínez, A.T. Lignin-degrading peroxidases from genome of selective ligninolytic fungus Ceriporiopsis subvermispora. JBC 2012, 287, 16903–16916. [Google Scholar] [CrossRef]
- Kües, U.; Ruhl, M. Multiple multi-copper oxidase gene families in basidiomycetes—What for? Curr. Genom. 2011, 12, 72–94. [Google Scholar] [CrossRef] [PubMed]
- Castanera, R.; Pérez, G.; Omarini, A.; Alfaro, M.; Pisabarro, A.G.; Faraco, V.; Amore, A.; Ramírez, L. Transcriptional and enzymatic profiling of Pleurotus ostreatus laccase genes in submerged and solid-state fermentation cultures. AEM 2012, 78, 4037–4045. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.; Carro, J.; Serrano, A.; Martínez, A.T. A survey of genes encoding H2O2-producing GMC oxidoreductases in 10 Polyporales genomes. Mycologia 2015, 107, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Dueñas, F.J.; Lundell, T.; Floudas, D.; Nagy, L.G.; Barrasa, J.M.; Hibbett, D.S.; Martínez, A.T. Lignin-degrading peroxidases in Polyporales: An evolutionary survey based on ten sequenced genomes. Mycologia 2013, 105, 1428–1444. [Google Scholar] [CrossRef]
- Couturier, M.; Ladeveze, S.; Sulzenbacher, G.; Ciano, L.; Fanuel, M.; Moreau, C.; Villares, A.; Cathala, B.; Chaspoul, F.; Frandsen, K.E.; et al. Lytic xylan oxidases from wood-decay fungi unlock biomass degradation. Nat. Chem. Biol. 2018, 14, 306–310. [Google Scholar] [CrossRef]
- Yamanaka, K. Cultivation of mushrooms in plastic bottles and small bags. In Edible and Medicinal Mushrooms: Technology and Applications; Zied, D.C., Pardo-Giménez, A., Eds.; Wiley-Blackwell: Oxford, UK, 2017; pp. 309–338. [Google Scholar]
- Camarero, S.; Barrasa, J.M.; Pelayo, M.; Martínez, A.T. Evaluation of Pleurotus species for wheat-straw biopulping. JPPS 1998, 24, 197–203. [Google Scholar]
- López-Abelairas, M.; Pallín, M.A.; Salvachúa, D.; Lú-Chau, T.; Martínez, M.J.; Lema, J.M. Optimisation of the biological pretreatment of wheat straw with white-rot fungi for ethanol production. Bioprocess Biosyst. Eng. 2013, 36, 1251–1260. [Google Scholar] [CrossRef]
- Sáez-Jiménez, V.; Baratto, M.C.; Pogni, R.; Rencoret, J.; Gutiérrez, A.; Santos, J.I.; Martínez, A.T.; Ruiz-Dueñas, F.J. Demonstration of lignin-to-peroxidase direct electron transfer: A transient-state kinetics, directed mutagenesis, EPR and NMR study. JBC 2015, 290, 23201–23213. [Google Scholar] [CrossRef]
- Guillén, F.; Martínez, A.T.; Martínez, M.J. Substrate specificity and properties of the aryl-alcohol oxidase from the ligninolytic fungus Pleurotus eryngii. Eur. J. Biochem. 1992, 209, 603–611. [Google Scholar] [CrossRef]
- Muñoz, C.; Guillén, F.; Martínez, A.T.; Martínez, M.J. Laccase isoenzymes of Pleurotus eryngii: Characterization, catalytic properties and participation in activation of molecular oxygen and Mn2+ oxidation. AEM 1997, 63, 2166–2174. [Google Scholar] [CrossRef] [PubMed]
- Caramelo, L.; Martínez, M.J.; Martínez, A.T. A search for ligninolytic peroxidases in the fungus Pleurotus eryngii involving α-keto-γ-thiomethylbutyric acid and lignin model dimers. AEM 1999, 65, 916–922. [Google Scholar] [CrossRef]
- Salvachúa, D.; Katahira, R.; Cleveland, N.S.; Khanna, P.; Resch, M.G.; Black, B.A.; Purvine, S.O.; Zink, E.M.; Prieto, A.; Martínez, M.J.; et al. Lignin depolymerization by fungal secretomes and a microbial sink. Green Chem. 2016, 18, 6046–6062. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. vegan: Community Ecology Package. R Package v.2.5-6. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 25 April 2021).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Cristobo, I.; Larriba, M.J.; de los Ríos, V.; García, F.; Muñoz, A.; Casal, J.I. Proteomic analysis of 1α,25-dihydroxyvitamin D3 action on human colon cancer cells reveals a link to splicing regulation. J. Proteom. 2011, 75, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Kall, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; Riley, R.; Salamov, A.; Zhao, X.; Korzeniewski, F.; et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014, 42, D699–D704. [Google Scholar] [CrossRef]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Cortazar, A.R.; Aransay, A.M.; Alfaro, M.; Oguiza, J.A.; Lavin, J.L. SECRETOOL: Integrated secretome analysis tool for fungi. Amino Acids 2014, 46, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Dodds, P.N.; Singh, K.B.; Taylor, J.M. ApoplastP: Prediction of effectors and plant proteins in the apoplast using machine learning. New Phytol. 2018, 217, 1764–1778. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology; R Package Version 2.42.0; R Packages: Vienna, Austria, 2020. [Google Scholar]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Del Cerro, C.; Erickson, E.; Dong, T.; Wong, A.R.; Eder, E.K.; Purvine, S.O.; Mitchell, H.D.; Weitz, K.K.; Markillie, L.M.; Burnet, M.C.; et al. Intracellular pathways for lignin catabolism in white-rot fungi. Proc. Natl. Acad. Sci. USA 2021, 118, e2017381118. [Google Scholar] [CrossRef]
- Camacho, C.; Madden, T.; Ma, N.; Tao, T.; Agarwala, R.; Morgulis, A. BLAST (r) Command Line Applications User Manual; National Center for Biotechnology Information: Bethesda, MD, USA, 2008.
- Tappi. 2006–2007 TAPPI Test Methods; TAPPI Press: Norcoss, GA, USA, 2006. [Google Scholar]
- Rencoret, J.; Marques, G.; Gutiérrez, A.; Nieto, L.; Santos, I.; Jiménez-Barbero, J.; Martínez, A.T.; del Río, J.C. HSQC-NMR analysis of lignin in woody (Eucalyptus globulus and Picea abies) and non-woody (Agave sisalana) ball-milled plant materials at the gel state. Holzforschung 2009, 63, 691–698. [Google Scholar]
- Del Río, J.C.; Rencoret, J.; Prinsen, P.; Martínez, A.T.; Ralph, J.; Gutiérrez, A. Structural characterization of wheat straw lignin as revealed by analytical pyrolysis, 2D-NMR, and reductive cleavage methods. JAFC 2012, 60, 5922–5935. [Google Scholar] [CrossRef]
- Wosten, H.A.; Schuren, F.H.; Wessels, J.G. Interfacial self-assembly of a hydrophobin into an amphipathic protein membrane mediates fungal attachment to hydrophobic surfaces. EMBO J. 1994, 13, 5848–5854. [Google Scholar] [CrossRef]
- Anasontzis, G.E.; Lebrun, M.H.; Haon, M.; Champion, C.; Kohler, A.; Lenfant, N.; Martin, F.; O’Connell, R.J.; Riley, R.; Grigoriev, I.V.; et al. Broad-specificity GH131 beta-glucanases are a hallmark of fungi and oomycetes that colonize plants. Environ. Microbiol. 2019, 21, 2724–2739. [Google Scholar] [CrossRef]
- Monrad, R.N.; Eklof, J.; Krogh, K.B.R.M.; Biely, P. Glucuronoyl esterases: Diversity, properties and biotechnological potential. A review. Crit. Rev. Biotechnol. 2018, 38, 1121–1136. [Google Scholar] [CrossRef]
- Kersten, P.; Cullen, D. Copper radical oxidases and related extracellular oxidoreductases of wood-decay Agaricomycetes. Fungal Genet. Biol. 2014, 72, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Korripally, P.; Hunt, C.G.; Houtman, C.J.; Jones, D.C.; Kitin, P.J.; Cullen, D.; Hammel, K.E. Regulation of gene expression during the onset of ligninolytic oxidation by Phanerochaete chrysosporium on spruce wood. AEM 2015, 81, 7802–7812. [Google Scholar] [CrossRef] [PubMed]
- Kracher, D.; Scheiblbrandner, S.; Felice, A.K.G.; Breslmays, E.; Preims, M.; Ludwicka, K.; Haltrich, D.; Eijsink, V.G.H.; Ludwig, R. Extracellular electron transfer systems fuel oxidative cellulose degradation. Science 2016, 352, 1098–1101. [Google Scholar] [CrossRef]
- Guillén, F.; Martínez, A.T.; Martínez, M.J.; Evans, C.S. Hydrogen peroxide-producing system of Pleurotus eryngii involving the extracellular enzyme aryl-alcohol oxidase. AMB 1994, 41, 465–470. [Google Scholar]
- Yasuda, S.; Fukushima, K.; Kakehi, A. Formation and chemical structures of acid-soluble lignin I: Sulfuric acid treatment time and acid-soluble lignin content of hardwood. J. Wood Sci. 2001, 47, 69–72. [Google Scholar] [CrossRef]
- Gold, M.H.; Youngs, H.L.; Gelpke, M.D. Manganese peroxidase. Met. Ions Biol. Syst. 2000, 37, 559–586. [Google Scholar]
- Watanabe, T.; Katayama, S.; Enoki, M.; Honda, Y.H.; Kuwahara, M. Formation of acyl radical in lipid peroxidation of linoleic acid by manganese-dependent peroxidase from Ceriporiopsis subvermispora and Bjerkandera adusta. Eur. J. Biochem. 2000, 267, 4222–4231. [Google Scholar] [CrossRef]
- Bao, W.L.; Fukushima, Y.; Jensen, K.A.; Moen, M.A.; Hammel, K.E. Oxidative degradation of non-phenolic lignin during lipid peroxidation by fungal manganese peroxidase. FEBS Lett. 1994, 354, 297–300. [Google Scholar] [CrossRef]
- Demmer, H.; Hinz, I.; Keller-Rudex, H.; Koeber, K.; Kottelbesch, H.; Schneider, D. Coordination Compounds of Manganese; Schleitzer-Ritz, E., Ed.; Springer: New York, NY, USA, 1980; pp. 1–85. [Google Scholar]
- Wariishi, H.; Valli, K.; Gold, M.H. Manganese(II) oxidation by manganese peroxidase from the basidiomycete Phanerochaete chrysosporium. Kinetic mechanism and role of chelators. JBC 1992, 267, 23688–23695. [Google Scholar] [CrossRef]
- Nelson, D.R.; Koymans, L.; Kamataki, T.; Stegeman, J.J.; Feyereisen, R.; Waxman, D.J.; Waterman, M.R.; Gotoh, O.; Coon, M.J.; Estabrook, R.W.; et al. P450 superfamily: Update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 1996, 6, 1–42. [Google Scholar] [CrossRef]
- Couturier, M.; Navarro, D.; Chevret, D.; Henrissat, B.; Piumi, F.; Ruiz-Dueñas, F.J.; Martínez, A.T.; Grigoriev, I.V.; Riley, R.; Lipzen, A.; et al. Enhanced degradation of softwood versus hardwood by the white-rot fungus Pycnoporus coccineus. Biotechnol. Biofuels 2015, 8, 1–16. [Google Scholar] [CrossRef]
- Gaskell, J.; Marty, A.; Mozuch, M.; Kersten, P.J.; Bondurant, S.S.; Sabat, G.; Azarpira, A.; Ralph, J.; Skyba, O.; Mansfield, S.D.; et al. Influence of Populus genotype on gene expression by the wood decay fungus Phanerochaete chrysosporium. AEM 2014, 80, 5828–5835. [Google Scholar] [CrossRef]
- Fernández-Fueyo, E.; Ruiz-Dueñas, F.J.; López-Lucendo, M.F.; Pérez-Boada, M.; Rencoret, J.; Gutiérrez, A.; Pisabarro, A.G.; Ramírez, L.; Martínez, A.T. A secretomic view of woody and nonwoody lignocellulose degradation by Pleurotus ostreatus. Biotechnol. Biofuels 2016, 9, 49. [Google Scholar] [CrossRef]
- Kuuskeri, J.; Hakkinen, M.; Laine, P.; Smolander, O.P.; Tamene, F.; Miettinen, S.; Nousiainen, P.; Kemell, M.; Auvinen, P.; Lundell, T. Time-scale dynamics of proteome and transcriptome of the white-rot fungus Phlebia radiata: Growth on spruce wood and decay effect on lignocellulose. Biotechnol. Biofuels 2016, 9, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Vanden Wymelenberg, A.; Gaskell, J.; Mozuch, M.; Sabat, G.; Ralph, J.; Skyba, O.; Mansfield, S.D.; Blanchette, R.A.; Martinez, D.; Grigoriev, I.; et al. Comparative transcriptome and secretome analysis of wood decay fungi Postia placenta and Phanerochaete chrysosporium. AEM 2010, 76, 3599–3610. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, S.; Navarro, D.; Grisel, S.; Chevret, D.; Berrin, J.G.; Rosso, M.N. The integrative omics of white-rot fungus Pycnoporus coccineus reveals co-regulated CAZymes for orchestrated lignocellulose breakdown. PLoS ONE 2017, 12, e0175528. [Google Scholar]
- Muñoz, C.; Guillén, F.; Martínez, A.T.; Martínez, M.J. Induction and characterization of laccase in the ligninolytic fungus Pleurotus eryngii. Curr. Microbiol. 1997, 34, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Cañas, A.I.; Camarero, S. Laccases and their natural mediators: Biotechnological tools for sustainable eco-friendly processes. Biotechnol. Adv. 2010, 28, 694–705. [Google Scholar] [CrossRef]
- Camarero, S.; Cañas, A.I.; Nousiainen, P.; Record, E.; Lomascolo, A.; Martínez, M.J.; Martínez, A.T. p-Hydroxycinnamic acids as natural mediators for laccase oxidation of recalcitrant compounds. Environ. Sci. Technol. 2008, 42, 6703–6709. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, A.; Caramelo, L.; Prieto, A.; Martínez, M.J.; Martínez, A.T. Anisaldehyde production and aryl-alcohol oxidase and dehydrogenase activities in ligninolytic fungi from the genus Pleurotus. AEM 1994, 60, 1783–1788. [Google Scholar] [CrossRef]
- Ruiz-Dueñas, F.J.; Guillén, F.; Camarero, S.; Pérez-Boada, M.; Martínez, M.J.; Martínez, A.T. Regulation of peroxidase transcript levels in liquid cultures of the ligninolytic fungus Pleurotus eryngii. AEM 1999, 65, 4458–4463. [Google Scholar] [CrossRef]
- Phillips, C.M.; Beeson, W.T.; Cate, J.H.; Marletta, M.A. Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 2011, 6, 1399–1406. [Google Scholar] [CrossRef]
- Kracher, D.; Ludwig, R. Cellobiose dehydrogenase: An essential enzyme for lignocellulose degradation in nature—A review. Die Bodenkult. J. Land Manag. Food Environ. 2016, 67, 145–163. [Google Scholar] [CrossRef]
- Ruiz-Dueñas, F.J.; Martínez, M.J.; Martínez, A.T. Molecular characterization of a novel peroxidase isolated from the ligninolytic fungus Pleurotus eryngii. Mol. Microbiol. 1999, 31, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Jiménez, V.; Rencoret, J.; Rodríguez-Carvajal, M.A.; Gutiérrez, A.; Ruiz-Dueñas, F.J.; Martínez, A.T. Role of surface tryptophan for peroxidase oxidation of nonphenolic lignin. Biotechnol. Biofuels 2016, 9, 198. [Google Scholar] [CrossRef]
- Camarero, S.; Sarkar, S.; Ruiz-Dueñas, F.J.; Martínez, M.J.; Martínez, A.T. Description of a versatile peroxidase involved in natural degradation of lignin that has both Mn-peroxidase and lignin-peroxidase substrate binding sites. JBC 1999, 274, 10324–10330. [Google Scholar] [CrossRef]
- Fernández-Fueyo, E.; Linde, D.; Almendral, D.; López-Lucendo, M.F.; Ruiz-Dueñas, F.J.; Martínez, A.T. Description of the first fungal dye-decolorizing peroxidase oxidizing manganese(II). AMB 2015, 99, 8927–8942. [Google Scholar] [CrossRef]
- Kinne, M.; Poraj-Kobielska, M.; Ullrich, R.; Nousiainen, P.; Sipila, J.; Scheibner, K.; Hammel, K.E.; Hofrichter, M. Oxidative cleavage of non-phenolic β-O-4 lignin model dimers by an extracellular aromatic peroxygenase. Holzforschung 2011, 65, 673–679. [Google Scholar] [CrossRef]
- Daniel, G.; Volc, J.; Filonova, L.; Plihal, O.; Kubátová, E.; Halada, P. Characteristics of Gloeophyllum trabeum alcohol oxidase, an extracellular source of H2O2 in brown rot decay of wood. AEM 2007, 73, 6241–6253. [Google Scholar] [CrossRef] [PubMed]
- Stajic, M.; Vukojevic, J.; Duletic-Lausevic, S. Biology of Pleurotus eryngii and role in biotechnological processes: A review. Crit. Rev. Biotechnol. 2009, 29, 55–66. [Google Scholar] [CrossRef]
- Valmaseda, M.; Almendros, G.; Martínez, A.T. Chemical transformation of wheat straw constituents after solid-state fermentation with selected lignocellulose-degrading fungi. Biomass Bioenergy 1991, 1, 261–266. [Google Scholar] [CrossRef]
- Dorado, J.; Almendros, G.; Camarero, S.; Martínez, A.T.; Vares, T.; Hatakka, A. Transformation of wheat straw in the course of solid-state fermentation by four ligninolytic basidiomycetes. EMT 1999, 25, 605–612. [Google Scholar] [CrossRef]
- Eichlerova, I.; Ruel, K.; Homolka, L.; Joseleau, J.P.; Nerud, F. Ligninolytic characteristics of Pleurotus ostreatus strain F6 and its monokaryotic protoplast derivative P19. Can. J. Microbiol. 2000, 46, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Valmaseda, M.; Almendros, G.; Martínez, A.T. Substrate-dependent degradation patterns in the decay of wheat straw and beech wood by ligninolytic fungi. AMB 1990, 33, 481–484. [Google Scholar] [CrossRef]
- Valmaseda, M.; Martínez, M.J.; Martínez, A.T. Kinetics of wheat straw solid-state fermentation with Trametes versicolor and Pleurotus ostreatus-Lignin and polysaccharide alteration and production of related enzymatic activities. AMB 1991, 35, 817–823. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña, A.; Babiker, R.; Chaduli, D.; Lipzen, A.; Wang, M.; Chovatia, M.; Rencoret, J.; Marques, G.; Sánchez-Ruiz, M.I.; Kijpornyongpan, T.; et al. A Multiomic Approach to Understand How Pleurotus eryngii Transforms Non-Woody Lignocellulosic Material. J. Fungi 2021, 7, 426. https://doi.org/10.3390/jof7060426
Peña A, Babiker R, Chaduli D, Lipzen A, Wang M, Chovatia M, Rencoret J, Marques G, Sánchez-Ruiz MI, Kijpornyongpan T, et al. A Multiomic Approach to Understand How Pleurotus eryngii Transforms Non-Woody Lignocellulosic Material. Journal of Fungi. 2021; 7(6):426. https://doi.org/10.3390/jof7060426
Chicago/Turabian StylePeña, Ander, Rashid Babiker, Delphine Chaduli, Anna Lipzen, Mei Wang, Mansi Chovatia, Jorge Rencoret, Gisela Marques, María Isabel Sánchez-Ruiz, Teeratas Kijpornyongpan, and et al. 2021. "A Multiomic Approach to Understand How Pleurotus eryngii Transforms Non-Woody Lignocellulosic Material" Journal of Fungi 7, no. 6: 426. https://doi.org/10.3390/jof7060426
APA StylePeña, A., Babiker, R., Chaduli, D., Lipzen, A., Wang, M., Chovatia, M., Rencoret, J., Marques, G., Sánchez-Ruiz, M. I., Kijpornyongpan, T., Salvachúa, D., Camarero, S., Ng, V., Gutiérrez, A., Grigoriev, I. V., Rosso, M.-N., Martínez, A. T., & Ruiz-Dueñas, F. J. (2021). A Multiomic Approach to Understand How Pleurotus eryngii Transforms Non-Woody Lignocellulosic Material. Journal of Fungi, 7(6), 426. https://doi.org/10.3390/jof7060426