
Genome Analyses of the Less Aggressive Rhizoctonia solani AG1-IB Isolates 1/2/21 and O8/2 Compared to the Reference AG1-IB Isolate 7/3/14

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Pathogen Inoculum
2.2. Pathogenicity Test
2.3. Genome Sequencing and Assembly
2.4. Genome Analysis, Comparative Genomics and Phylogenetics
3. Results & Discussion
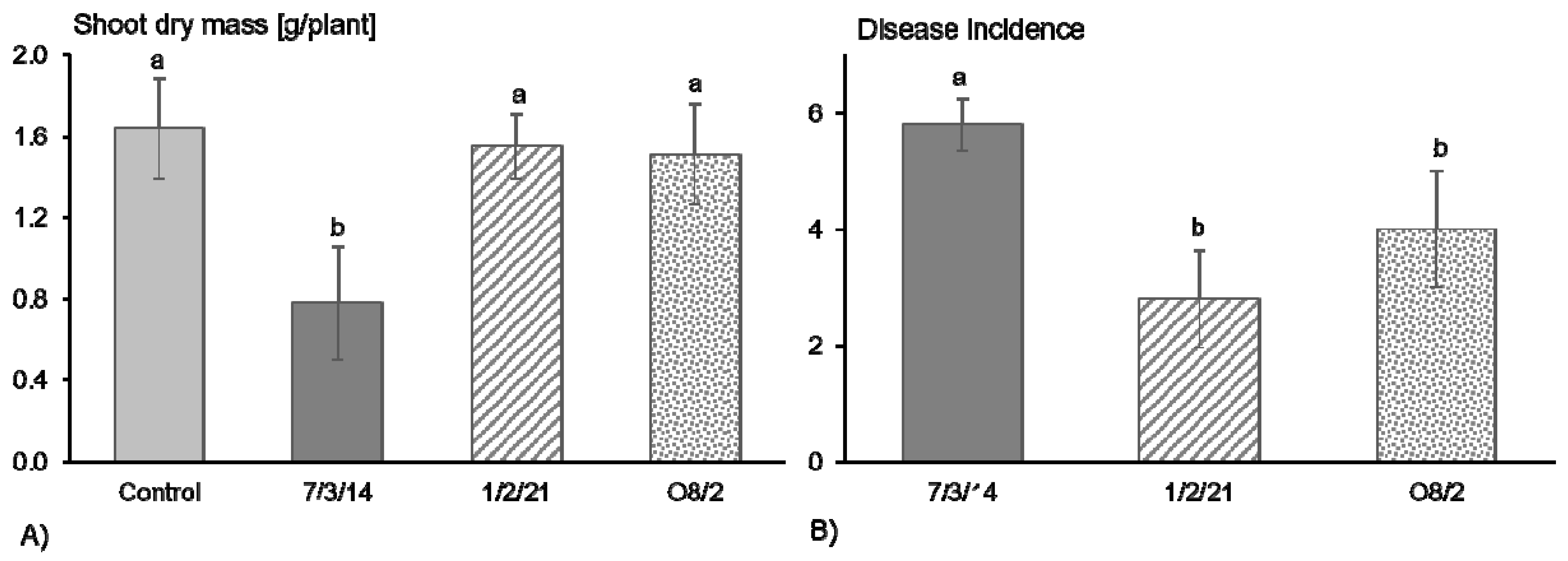
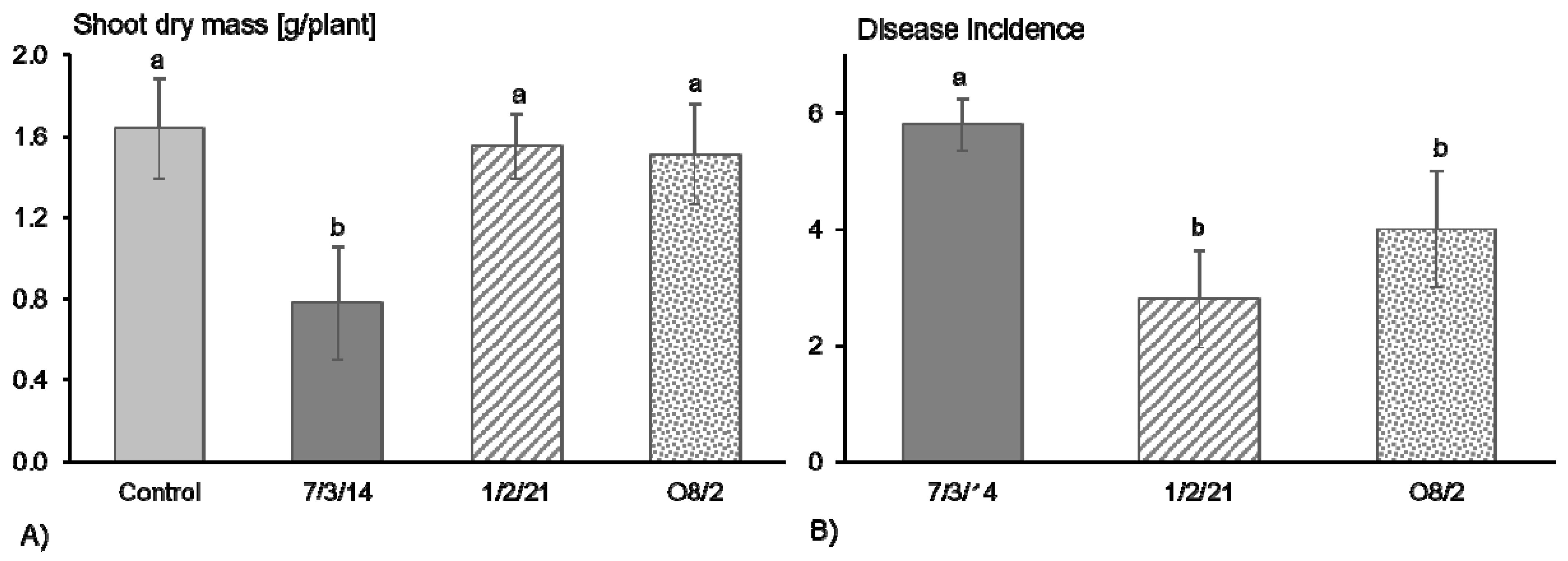
3.1. R. solani AG1-IB Isolates 1/2/21 and O8/2 Appeared to Be Significantly Less Aggressive in Pathogenicity Tests in Comparison to AG1-IB 7/3/14
3.2. Genome Sequencing of the Less Aggressive R. solani AG1-IB Isolates 1/2/21 and O8/2
3.3. Differentiation of R. solani AG1-IB Isolates 1/2/21 and O8/2 Based on Contig-Length vs. Read-Count Analyses
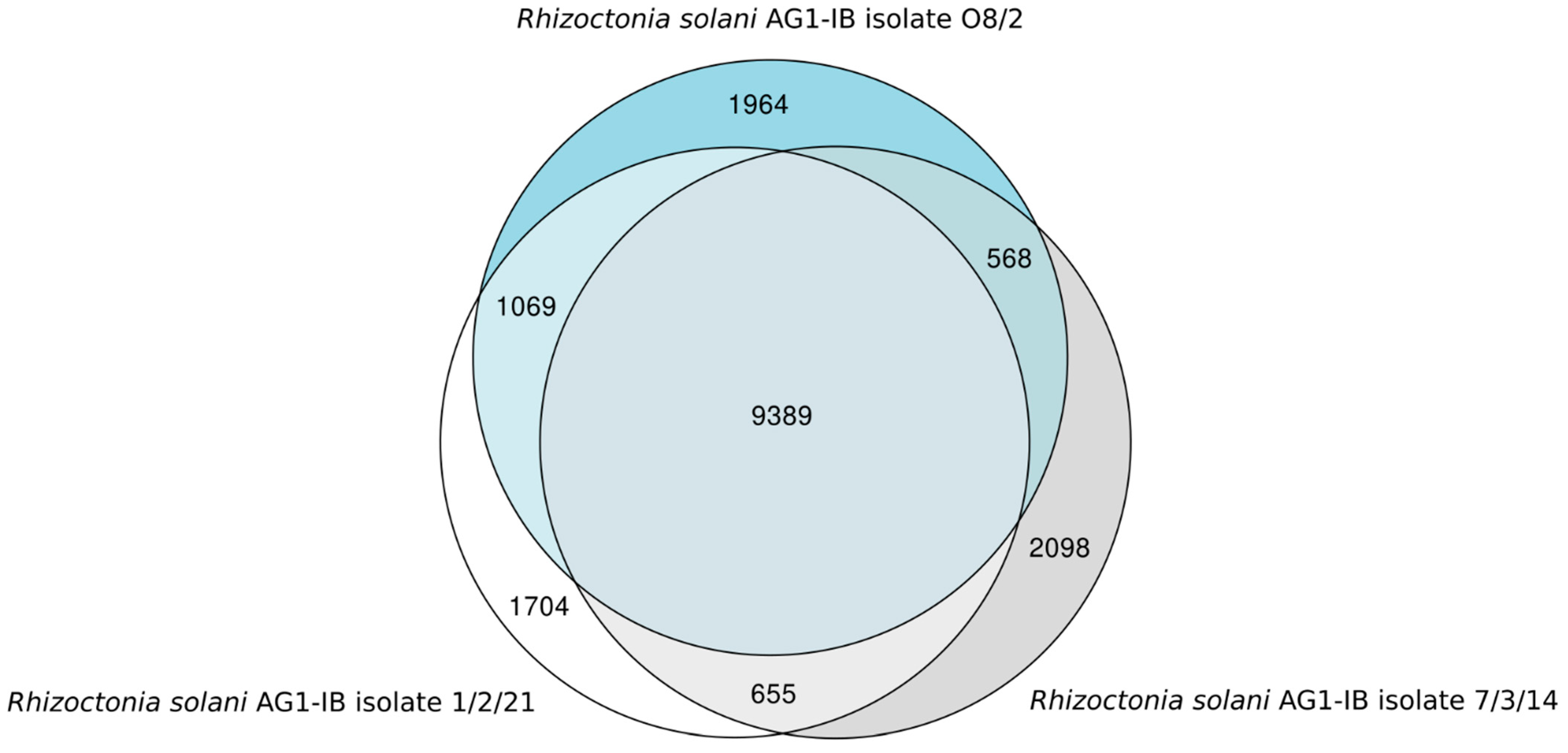
3.4. Synteny Analysis, Gene Prediction, Annotation and Comparative Genomics Provide First Insights into the R. solani AG1-IB Pan-Genome
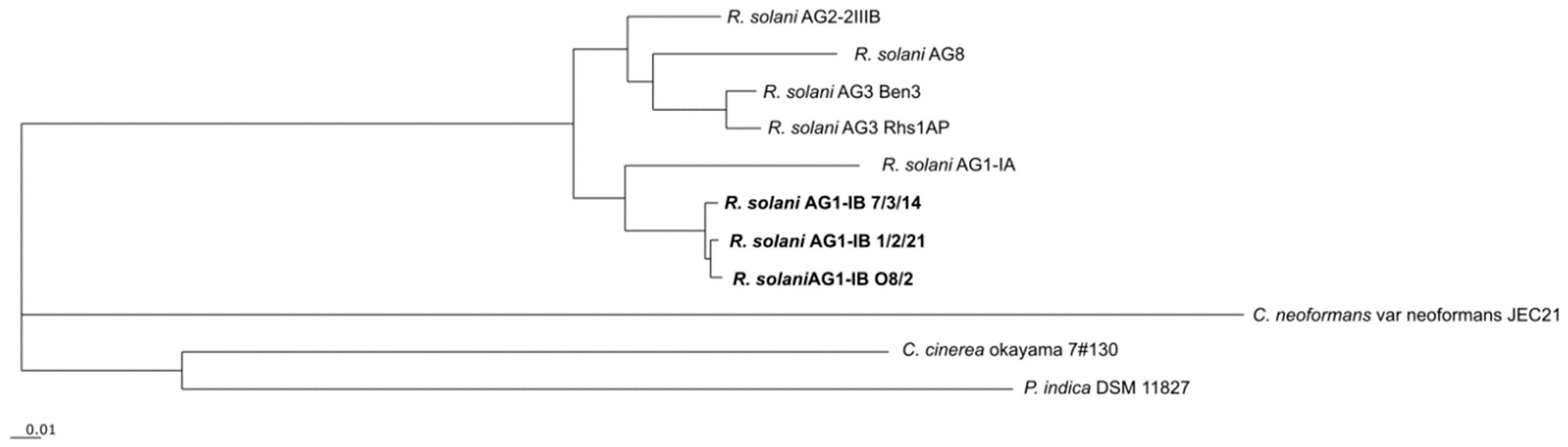
3.5. Phylogenetic Differentiation of R. solani AG1-IB Isolates Based on Shared Core Genes
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- González García, V.; Portal Onco, M.A.; Rubio Susan, V. Biology and Systematics of the form genus Rhizoctonia. Spanish J. Agric. Res. 2006, 4, 55–79. [Google Scholar] [CrossRef] [Green Version]
- Ogoshi, A. Ecology and Pathogenicity of Anastomosis and Intraspecific Groups of Rhizoctonia Solani Kuhn. Annu. Rev. Phytopathol. 1987, 25, 125–143. [Google Scholar] [CrossRef]
- Carling, D.E.; Baird, R.E.; Gitaitis, R.D.; Brainard, K.A.; Kuninaga, S. Characterization of AG-13, a Newly Reported Anastomosis Group of Rhizoctonia solani. Phytopathology 2002, 92, 893–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taheri, P.; Tarighi, S. A survey on basal resistance and riboflavin-induced defense responses of sugar beet against Rhizoctonia solani. J. Plant Physiol. 2011, 168, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Priyatmojo, A.; Escopalao, V.E.; Tangonan, N.G.; Pascual, C.B.; Suga, H.; Kageyama, K.; Hyakumachi, M. Characterization of a New Subgroup of Rhizoctonia solani Anastomosis Group 1 (AG-1-ID), Causal Agent of a Necrotic Leaf Spot on Coffee. Phytopathology 2001, 91, 1054–1061. [Google Scholar] [CrossRef]
- Yang, G.; Li, C. General Description of Rhizoctonia Species Complex. In Plant Pathology; IntechOpen: London, UK, 2012; ISBN 978-953-51-0489-6. [Google Scholar]
- Wibberg, D.; Rupp, O.; Blom, J.; Jelonek, L.; Kröber, M.; Verwaaijen, B.; Goesmann, A.; Albaum, S.; Grosch, R.; Pühler, A.; et al. Development of a Rhizoctonia solani AG1-IB Specific Gene Model Enables Comparative Genome Analyses between Phytopathogenic R. solani AG1-IA, AG1-IB, AG3 and AG8 Isolates. PLoS ONE 2015, 10, e0144769. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, N.; Wang, Y.; Lin, R.; Liang, Y.; Liu, Y.; Zhu, J.; Wang, L.; Wang, S.; Liu, H.; Deng, Q.; et al. Integrative transcriptome analysis discloses the molecular basis of a heterogeneous fungal phytopathogen complex, Rhizoctonia solani AG-1 subgroups. Sci. Rep. 2019, 9, 19626. [Google Scholar] [CrossRef] [Green Version]
- Wibberg, D.; Jelonek, L.; Rupp, O.; Kröber, M.; Goesmann, A.; Grosch, R.; Pühler, A.; Schlüter, A. Transcriptome analysis of the phytopathogenic fungus Rhizoctonia solani AG1-IB 7/3/14 applying high-throughput sequencing of expressed sequence tags (ESTs). Fungal Biol. 2014, 118, 800–813. [Google Scholar] [CrossRef]
- Zrenner, R.; Genzel, F.; Verwaaijen, B.; Wibberg, D.; Grosch, R. Necrotrophic lifestyle of Rhizoctonia solani AG3-PT during interaction with its host plant potato as revealed by transcriptome analysis. Sci. Rep. 2020, 10, 12574. [Google Scholar] [CrossRef]
- Brooks, S.A. Sensitivity to a phytotoxin from Rhizoctonia solani correlates with sheath blight susceptibility in rice. Phytopathology 2007, 97, 1207–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Pan, X.; Li, F.; Dong, W. UPLC-QTOF-MS metabolomics analysis revealed the contributions of metabolites to the pathogenesis of Rhizoctonia solani strain AG-1-IA. PLoS ONE 2018, 13, e0192486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verwaaijen, B.; Wibberg, D.; Kröber, M.; Winkler, A.; Zrenner, R.; Bednarz, H.; Niehaus, K.; Grosch, R.; Pühler, A.; Schlüter, A. The Rhizoctonia solani AG1-IB (isolate 7/3/14) transcriptome during interaction with the host plant lettuce (Lactuca sativa L.). PLoS ONE 2017, 12, e0177278. [Google Scholar] [CrossRef] [PubMed]
- Copley, T.R.; Duggavathi, R.; Jabaji, S. The transcriptional landscape of Rhizoctonia solani AG1-IA during infection of soybean as defined by RNA-seq. PLoS ONE 2017, 12, e0184095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wibberg, D.; Jelonek, L.; Rupp, O.; Hennig, M.; Eikmeyer, F.; Goesmann, A.; Hartmann, A.; Borriss, R.; Grosch, R.; Pühler, A.; et al. Establishment and interpretation of the genome sequence of the phytopathogenic fungus Rhizoctonia solani AG1-IB isolate 7/3/14. J. Biotechnol. 2013, 167, 142–155. [Google Scholar] [CrossRef]
- Wibberg, D.; Rupp, O.; Jelonek, L.; Kröber, M.; Verwaaijen, B.; Blom, J.; Winkler, A.; Goesmann, A.; Grosch, R.; Pühler, A.; et al. Improved genome sequence of the phytopathogenic fungus Rhizoctonia solani AG1-IB 7/3/14 as established by deep mate-pair sequencing on the MiSeq (Illumina) system. J. Biotechnol. 2015, 203, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Grosch, R.; Schneider, J.H.M.; Kofoet, A. Characterisation of Rhizoctonia solani Anastomosis Groups Causing Bottom Rot in Field-Grown Lettuce in Germany. Eur. J. Plant Pathol. 2004, 110, 53–62. [Google Scholar] [CrossRef]
- Laine, A.L.; Barrès, B. Epidemiological and evolutionary consequences of life-history trade-offs in pathogens. Plant Pathol. 2013, 62, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Pariaud, B.; Robert, C.; Goyeau, H.; Lannou, C. Aggressiveness components and adaptation to a host cultivar in wheat leaf rust. Phytopathology 2009, 99, 869–878. [Google Scholar] [CrossRef] [Green Version]
- Grosch, R.; Schneider, J.H.M.; Peth, A.; Waschke, A.; Franken, P.; Kofoet, A.; Jabaji-Hare, S.H. Development of a specific PCR assay for the detection of Rhizoctonia solani AG 1-IB using SCAR primers. J. Appl. Microbiol. 2007, 102, 806–819. [Google Scholar] [CrossRef]
- Schneider, J.H.M.; Schilder, M.T.; Dijst, G. Characterization of Rhizoctonia solani AG 2 isolates causing bare patch in field grown tulips in the Netherlands. Eur. J. Plant Pathol. 1997, 103, 265–279. [Google Scholar] [CrossRef]
- Chowdhury, S.P.; Dietel, K.; Rändler, M.; Schmid, M.; Junge, H.; Borriss, R.; Hartmann, A.; Grosch, R. Effects of Bacillus amyloliquefaciens FZB42 on Lettuce Growth and Health under Pathogen Pressure and Its Impact on the Rhizosphere Bacterial Community. PLoS ONE 2013, 8, e68818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wibberg, D.; Genzel, F.; Verwaaijen, B.; Blom, J.; Rupp, O.; Goesmann, A.; Zrenner, R.; Grosch, R.; Pühler, A.; Schlüter, A. Draft genome sequence of the potato pathogen Rhizoctonia solani AG3-PT isolate Ben3. Arch. Microbiol. 2017, 199, 1065–1068. [Google Scholar] [CrossRef]
- Lee, S.B.; Taylor, J.W. Isolation of DNA from fungal mycelium and single spores. In PCR Protocols: A Guide to Methods and Applications; Academic Press: San Diego, CA, USA, 1990; pp. 282–288. [Google Scholar]
- Wibberg, D.; Andersson, L.; Rupp, O.; Goesmann, A.; Pühler, A.; Varrelmann, M.; Dixelius, C.; Schlüter, A. Genome analysis of the sugar beet pathogen Rhizoctonia solani AG2-2IIIB revealed high numbers in secreted proteins and cell wall degrading enzymes. BMC Genom. 2016, 17, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Mora, J.A.; Campos-Sánchez, R.; Rodríguez, C.; Shi, L.; García, F. High quality 3C de novo assembly and annotation of a multidrug resistant ST-111 Pseudomonas aeruginosa genome: Benchmark of hybrid and non-hybrid assemblers. Sci. Rep. 2020, 10, 1392. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Langmead and Steven L Salzberg Bowtie2. Nat. Methods 2013, 18, 151–154. [CrossRef]
- Husemann, P.; Stoye, J. r2cat: Synteny plots and comparative assembly. Bioinformatics 2010, 26, 570–571. [Google Scholar] [CrossRef]
- Stanke, M.; Stanke, M.; Waack, S.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19 (Suppl. 2), ii215–ii225. [Google Scholar] [CrossRef] [Green Version]
- Meyer, F.; Goesmann, A.; McHardy, A.C.; Bartels, D.; Bekel, T.; Clausen, J.; Kalinowski, J.; Linke, B.; Rupp, O.; Giegerich, R.; et al. GenDB—An open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003, 31, 2187–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, O.; Becker, J.; Brinkrolf, K.; Timmermann, C.; Borth, N.; Pühler, A.; Noll, T.; Goesmann, A. Construction of a public CHO cell line transcript database using versatile bioinformatics analysis pipelines. PLoS ONE 2014, 9, e85568. [Google Scholar] [CrossRef] [PubMed]
- Winnenburg, R.; Baldwin, T.K.; Urban, M.; Rawlings, C.; Köhler, J.; Hammond-Kosack, K.E. PHI-base: A new database for pathogen host interactions. Nucleic Acids Res. 2006, 34, D459–D464. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Blom, J.; Kreis, J.; Spänig, S.; Juhre, T.; Bertelli, C.; Ernst, C.; Goesmann, A. EDGAR 2.0: An enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 2016, 44, W22–W28. [Google Scholar] [CrossRef] [Green Version]
- Blom, J.; Albaum, S.P.; Doppmeier, D.; Pühler, A.; Vorhölter, F.-J.; Zakrzewski, M.; Goesmann, A. EDGAR: A software framework for the comparative analysis of prokaryotic genomes. BMC Bioinform. 2009, 10, 154. [Google Scholar] [CrossRef] [Green Version]
- Dieckmann, M.A.; Beyvers, S.; Nkouamedjo-Fankep, R.C.; Hanel, P.H.G.; Jelonek, L.; Blom, J.; Goesmann, A. EDGAR3.0: Comparative genomics and phylogenomics on a scalable infrastructure. Nucleic Acids Res. 2021, 49, W185–W192. [Google Scholar] [CrossRef]
- Stajich, J.E.; Wilke, S.K.; Ahrén, D.; Au, C.H.; Birren, B.W.; Borodovsky, M.; Burns, C.; Canbäck, B.; Casselton, L.A.; Cheng, C.K. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc. Natl. Acad. Sci. USA 2010, 107, 11889–11894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccaro, A.; Lahrmann, U.; Güldener, U.; Langen, G.; Pfiffi, S.; Biedenkopf, D.; Wong, P.; Samans, B.; Grimm, C.; Basiewicz, M.; et al. Endophytic Life Strategies Decoded by Genome and Transcriptome Analyses of the Mutualistic Root Symbiont Piriformospora indica. PLoS Pathog. 2011, 7, e1002290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loftus, B.J.; Fung, E.; Roncaglia, P.; Rowley, D.; Amedeo, P.; Bruno, D.; Vamathevan, J.; Miranda, M.; Anderson, I.J.; Fraser, J.A.; et al. The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science 2005, 307, 1321–1324. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wibberg, D.; Stadler, M.; Lambert, C.; Bunk, B.; Spröer, C.; Rückert, C.; Kalinowski, J.; Cox, R.J.; Kuhnert, E. High quality genome sequences of thirteen Hypoxylaceae (Ascomycota) strengthen the phylogenetic family backbone and enable the discovery of new taxa. Fungal Divers. 2021, 106, 7–28. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J. eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses. Bachelor’s Thesis, Lund University, Lund, Sweden, 2021. [Google Scholar]
- Cabanettes, F.; Klopp, C. D-GENIES: Dot plot large genomes in an interactive, efficient and simple way. PeerJ 2018, 6, e4958. [Google Scholar] [CrossRef]
- Wibberg, D.; Andersson, L.; Rupp, O.; Goesmann, A.; Pühler, A.; Varrelmann, M.; Dixelius, C.; Schlüter, A. Draft genome sequence of the sugar beet pathogen Rhizoctonia solani AG2-2IIIB strain BBA69670. J. Biotechnol. 2016, 222, 11–12. [Google Scholar] [CrossRef]
- Qin, Q.L.; Xie, B.B.; Zhang, X.Y.; Chen, X.L.; Zhou, B.C.; Zhou, J.; Oren, A.; Zhang, Y.Z. A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 2014, 196, 2210–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Selected Groups | 1/2/21 | O8/2 | 7/3/14 |
|---|---|---|---|
| Celluloytic enzymes 1 | 35 | 32 | 46 |
| Tyrosinases 1 | 0 | 1 | 3 |
| Drug resistance proteins 1 | 0 | 0 | 1 |
| Laccases 1 | 0 | 0 | 1 |
| Cytochrome P450 1 | 7 | 11 | 10 |
| Secondary Metabolite Regions | 1/2/21 | O8/2 | 7/3/14 |
|---|---|---|---|
| All | 17 | 15 | 16 |
| Non-ribosomal peptide synthesis regions | 6 | 6 | 6 |
| Alkaloid/terpene synthesis regions | 10 | 9 | 9 |
| Siderophore regions | 1 | 0 | 1 |
| ANI | 1/2/21 | O8/2 | 7/3/14 |
|---|---|---|---|
| 1/2/21 | 100.00 | 99.12 | 98.92 |
| O8/2 | 99.02 | 100.00 | 99.02 |
| 7/3/14 | 98.92 | 99.06 | 100.00 |
| AAI | 1/2/21 | O8/2 | 7/3/14 |
|---|---|---|---|
| 1/2/21 | 100.00 | 99.46 | 99.40 |
| O8/2 | 99.46 | 100.00 | 99.46 |
| 7/3/14 | 99.40 | 99.46 | 100.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wibberg, D.; Genzel, F.; Verwaaijen, B.; Blom, J.; Rupp, O.; Goesmann, A.; Zrenner, R.; Grosch, R.; Pühler, A.; Schlüter, A. Genome Analyses of the Less Aggressive Rhizoctonia solani AG1-IB Isolates 1/2/21 and O8/2 Compared to the Reference AG1-IB Isolate 7/3/14. J. Fungi 2021, 7, 832. https://doi.org/10.3390/jof7100832
Wibberg D, Genzel F, Verwaaijen B, Blom J, Rupp O, Goesmann A, Zrenner R, Grosch R, Pühler A, Schlüter A. Genome Analyses of the Less Aggressive Rhizoctonia solani AG1-IB Isolates 1/2/21 and O8/2 Compared to the Reference AG1-IB Isolate 7/3/14. Journal of Fungi. 2021; 7(10):832. https://doi.org/10.3390/jof7100832
Chicago/Turabian StyleWibberg, Daniel, Franziska Genzel, Bart Verwaaijen, Jochen Blom, Oliver Rupp, Alexander Goesmann, Rita Zrenner, Rita Grosch, Alfred Pühler, and Andreas Schlüter. 2021. "Genome Analyses of the Less Aggressive Rhizoctonia solani AG1-IB Isolates 1/2/21 and O8/2 Compared to the Reference AG1-IB Isolate 7/3/14" Journal of Fungi 7, no. 10: 832. https://doi.org/10.3390/jof7100832
APA StyleWibberg, D., Genzel, F., Verwaaijen, B., Blom, J., Rupp, O., Goesmann, A., Zrenner, R., Grosch, R., Pühler, A., & Schlüter, A. (2021). Genome Analyses of the Less Aggressive Rhizoctonia solani AG1-IB Isolates 1/2/21 and O8/2 Compared to the Reference AG1-IB Isolate 7/3/14. Journal of Fungi, 7(10), 832. https://doi.org/10.3390/jof7100832