Multiplex PCR Based Strategy for Detection of Fungal Pathogen DNA in Patients with Suspected Invasive Fungal Infections

 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Primer Selection and Panel Design
2.2. Strains Tested
2.3. Genomic DNA Extraction
2.4. DNA Extraction from Serum Spiked with DNA
2.5. PCR Amplification Conditions
2.6. PCR Fragment Size Determination
2.7. Clinical Samples
3. Results
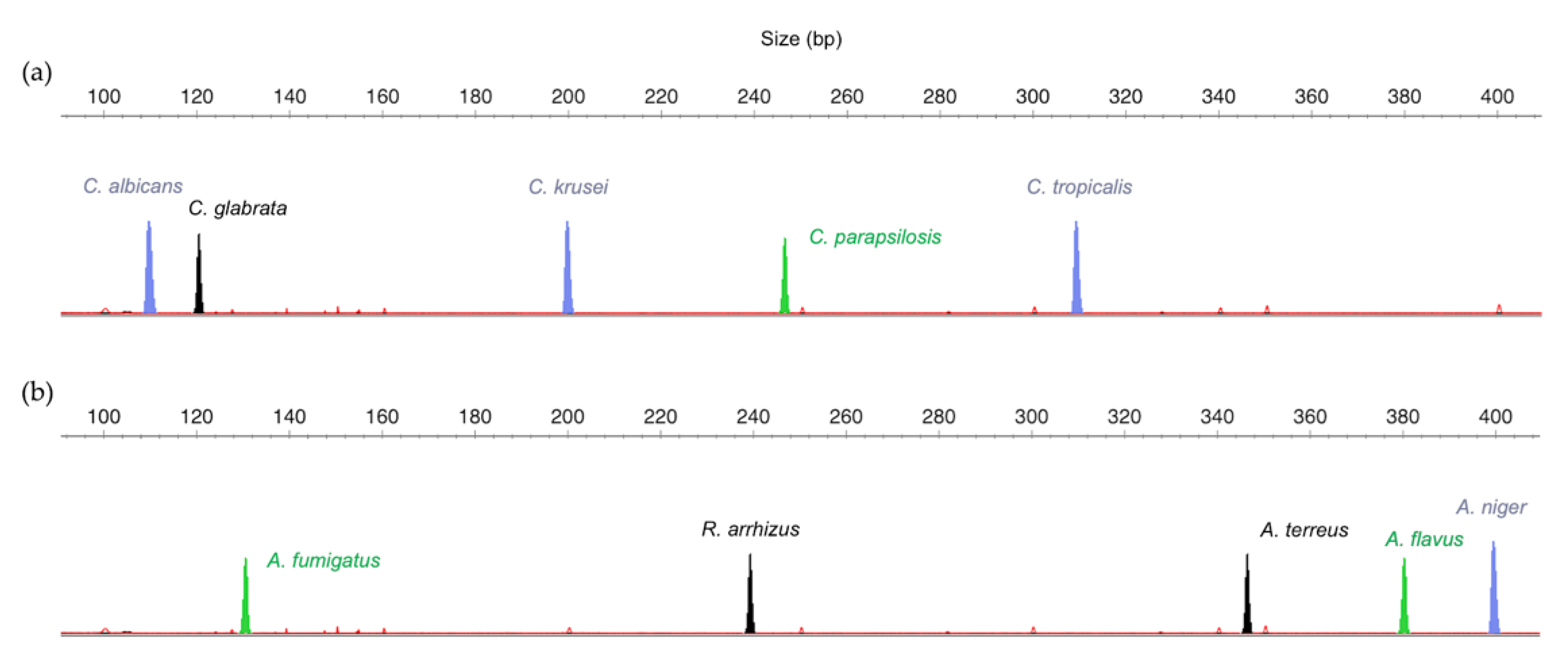
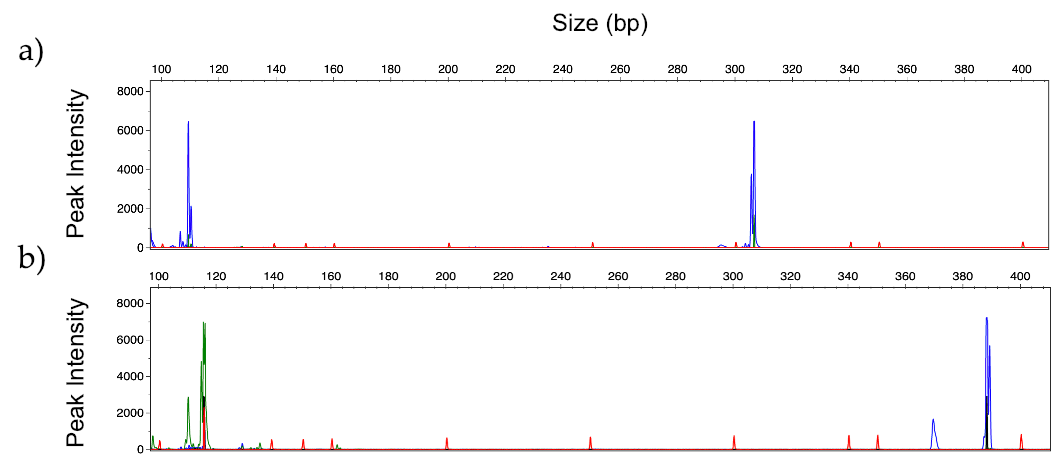
3.1. Design of the Multiplex Strategy
3.2. Optimization of Multiplex Amplification Conditions
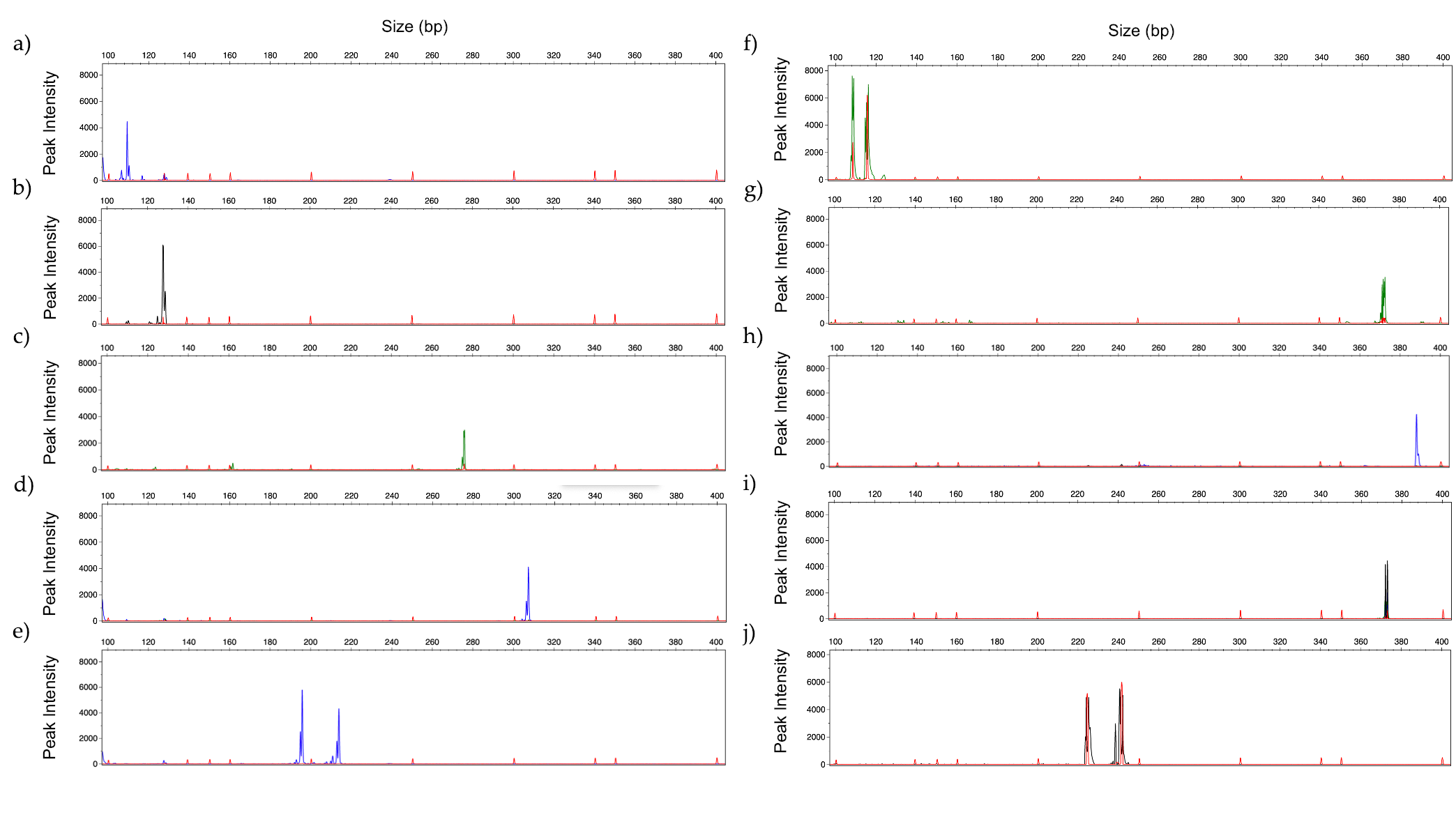
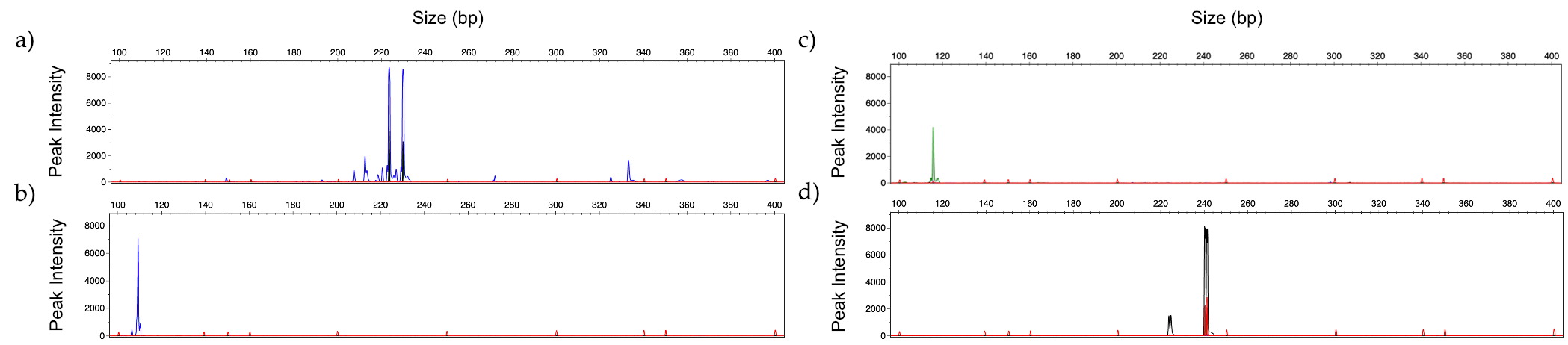
3.3. Limit of Detection
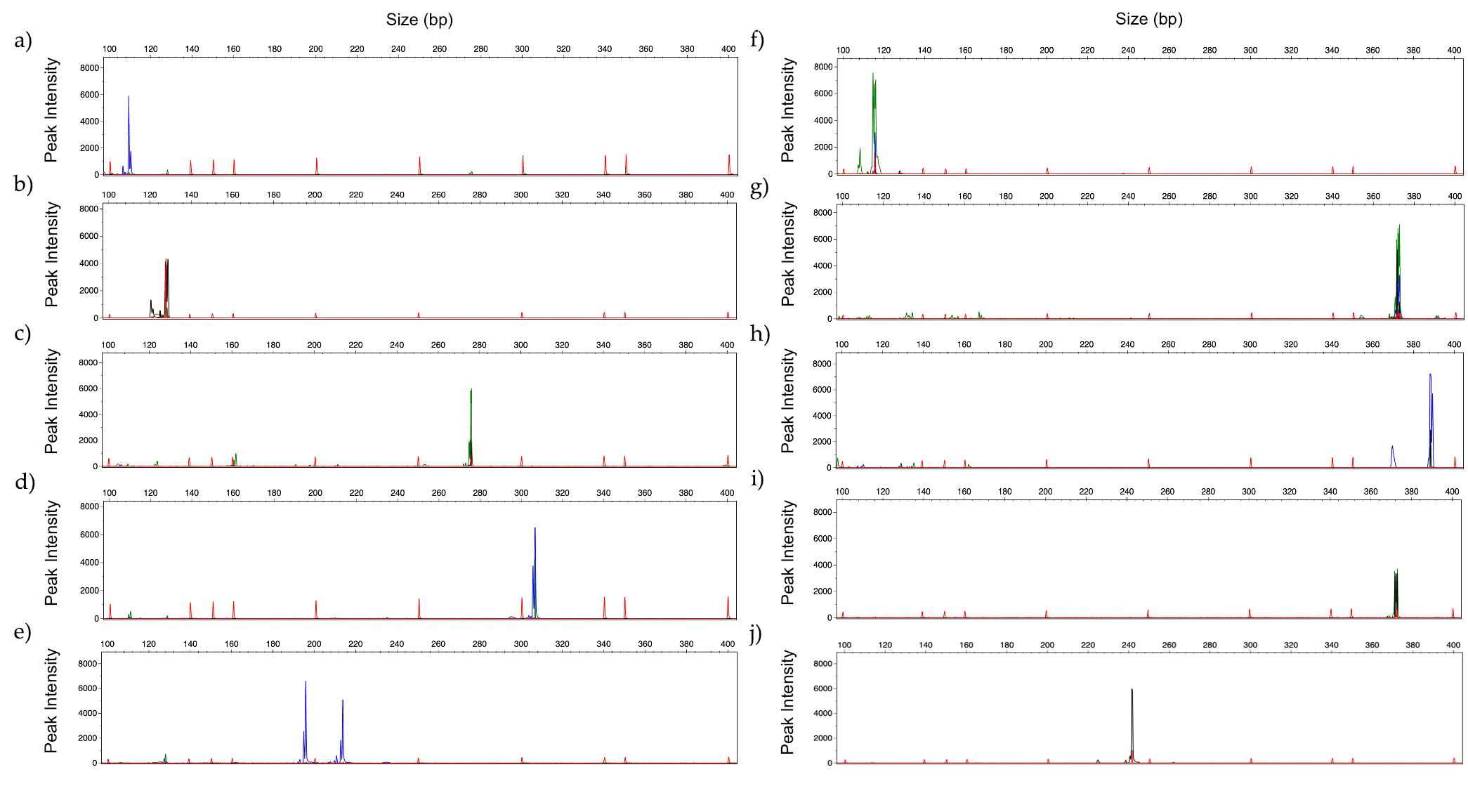
3.4. Identification of Mixed Fungal DNA
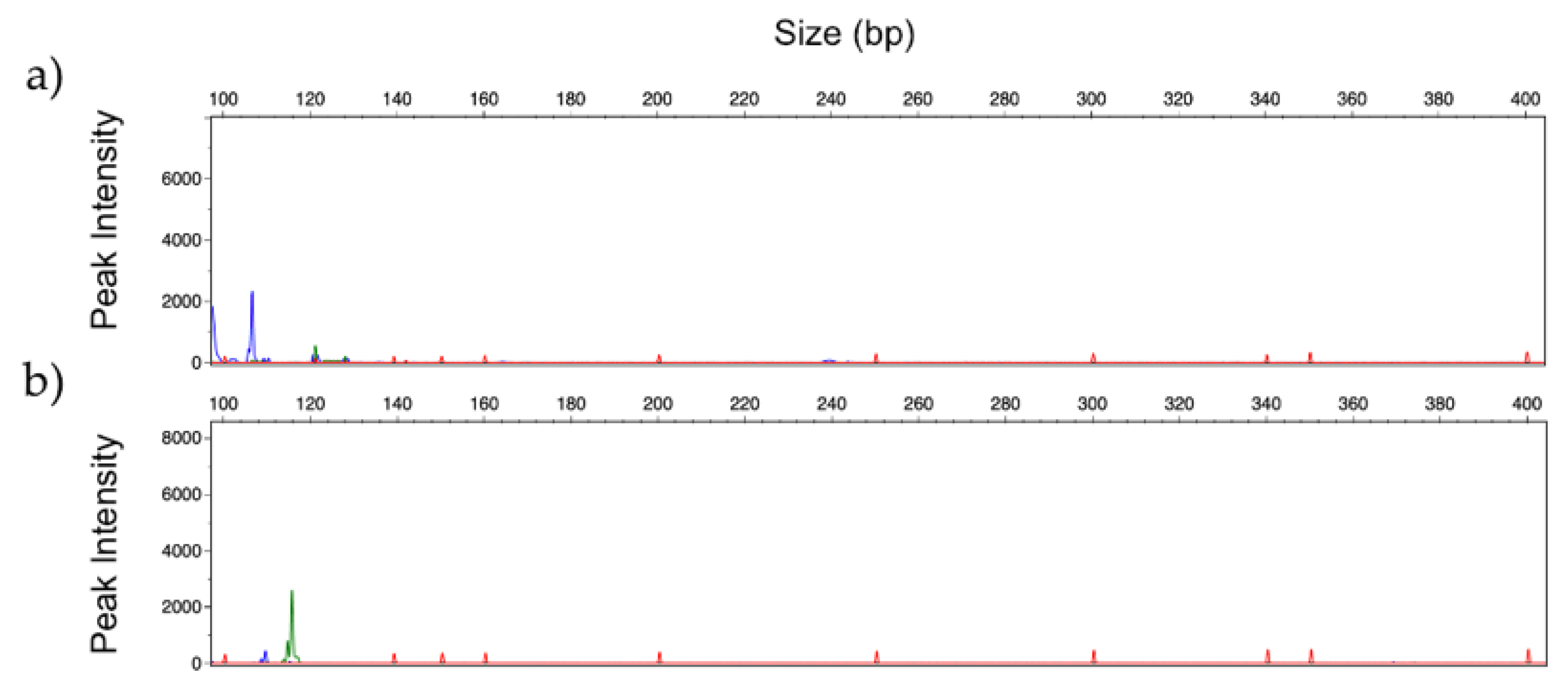
3.5. Human Serum Spiked with Fungal DNA
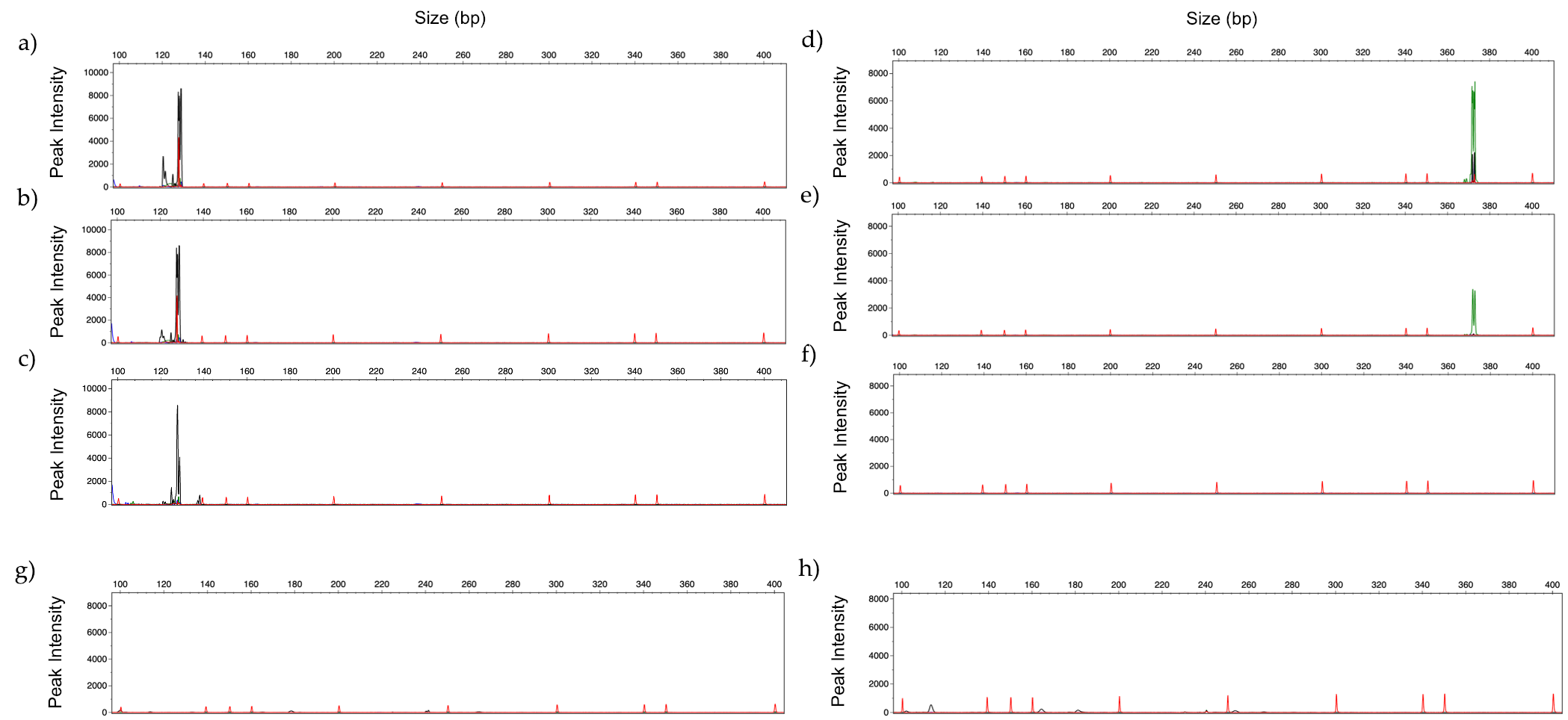
3.6. Identification of Fungal DNA in Clinical Samples
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pappas, P.G.; Alexander, B.D.; Andes, D.R.; Hadley, S.; Kauffman, C.A.; Freifeld, A.; Anaissie, E.J.; Brumble, L.M.; Herwaldt, L.; Lto, J.; et al. Invasive fungal infections among organ transplant recipients: Results of the transplant-associated infection surveillance network (Transnet). Clin. Infect. Dis. 2010. [Google Scholar] [CrossRef]
- Gavaldà, J.; Meije, Y.; Fortún, J.; Roilides, E.; Saliba, F.; Lortholary, O.; Muñoz, P.; Grossi, P.; Cuenca-Estrella, M. Invasive fungal infections in solid organ transplant recipients. Clin. Microbiol. Infect. 2014, 20, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Dignani, M.C. Epidemiology of invasive fungal diseases on the basis of autopsy reports. F1000Prime Rep. 2014, 6, 81. [Google Scholar] [PubMed]
- Pal, M. Morbidity and Mortality Due to Fungal Infections. J. Appl. Microbiol. Biochem. 2018. [Google Scholar] [CrossRef]
- Benedict, K.; Jackson, B.R.; Chiller, T.; Beer, K.D. Estimation of Direct Healthcare Costs of Fungal Diseases in the United States. Clin. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Pana, Z.D.; Roilides, E.; Warris, A.; Groll, A.H.; Zaoutis, T. Epidemiology of Invasive Fungal Disease in Children. J. Pediatric Infect. Dis. Soc. 2017, 6, S3–S11. [Google Scholar]
- Klingspor, L.; Tortorano, A.M.; Peman, J.; Willinger, B.; Hamal, P.; Sendid, B.; Velegraki, A.; Kibbler, C.; Meis, J.F.; Sabino, R.; et al. Invasive Candida infections in surgical patients in intensive care units: A prospective, multicentre survey initiated by the European Confederation of Medical Mycology (ECMM) (2006–2008). Clin. Microbiol. Infect. 2015, 21, 87.e1–87.e10. [Google Scholar] [CrossRef]
- Lortholary, O.; Gangneux, J.P.; Sitbon, K.; Lebeau, B.; de Monbrison, F.; Le Strat, Y.; Coignard, B.; Dromer, F.; Bretagne, S. Epidemiological trends in invasive aspergillosis in France: The SAIF network (2005–2007). Clin. Microbiol. Infect. 2011. [Google Scholar] [CrossRef]
- Garcia-Hermoso, D.; Criscuolo, A.; Lee, S.C.; Legrand, M.; Chaouat, M.; Denis, B.; Lafaurie, M.; Rouveau, M.; Soler, C.; Schaal, J.-V.; et al. Outbreak of Invasive Wound Mucormycosis in a Burn Unit Due to Multiple Strains of Mucor circinelloides f. circinelloides Resolved by Whole-Genome Sequencing. MBio 2018. [Google Scholar] [CrossRef]
- Jeong, W.; Keighley, C.; Wolfe, R.; Lee, W.L.; Slavin, M.A.; Kong, D.C.M.; Chen, S.C.A. The epidemiology and clinical manifestations of mucormycosis: A systematic review and meta-analysis of case reports. Clin. Microbiol. Infect. 2019, 25, 26–34. [Google Scholar] [PubMed]
- Vallés, J.; Rello, J.; Ochagavía, A.; Garnacho, J.; Alcalá, M.A. Community-acquired bloodstream infection in critically III adult patients: Impact of shock and inappropriate antibiotic therapy on survival. Chest 2003. [Google Scholar] [CrossRef]
- De Rosa, F.G.; Trecarichi, E.M.; Montrucchio, C.; Losito, A.R.; Raviolo, S.; Posteraro, B.; Corcione, S.; Di Giambenedetto, S.; Fossati, L.; Sanguinetti, M.; et al. Mortality in patients with early- or late-onset candidaemia. J. Antimicrob. Chemother. 2013. [Google Scholar] [CrossRef]
- Pfaller, M.; Neofytos, D.; Diekema, D.; Azie, N.; Meier-Kriesche, H.U.; Quan, S.P.; Horn, D. Epidemiology and outcomes of candidemia in 3648 patients: Data from the Prospective Antifungal Therapy (PATH Alliance®) registry, 2004–2008. Diagn. Microbiol. Infect. Dis. 2012. [Google Scholar] [CrossRef]
- Cao, J.H.; Li, G.H. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) C. Chin. J. Infect. Chemother. 2008. [Google Scholar] [CrossRef]
- Bunn, T.; Sikarwar, A. Diagnostics: Conventional Versus Modern Methods. J. Adv. Med. Pharm. Sci. 2016. [Google Scholar] [CrossRef]
- Colombo, A.L.; de Almeida Júnior, J.N.; Slavin, M.A.; Chen, S.C.A.; Sorrell, T.C. Candida and invasive mould diseases in non-neutropenic critically ill patients and patients with haematological cancer. Lancet Infect. Dis. 2017, 17, e344–e356. [Google Scholar]
- Terrero-Salcedo, D.; Powers-Fletcher, M.V. Updates in laboratory diagnostics for invasive fungal infections. J. Clin. Microbiol. 2020. [Google Scholar] [CrossRef]
- Levesque, E.; El Anbassi, S.; Sitterle, E.; Foulet, F.; Merle, J.C.; Botterel, F. Contribution of (1,3)-beta-D-glucan to diagnosis of invasive candidiasis after liver transplantation. J. Clin. Microbiol. 2015. [Google Scholar] [CrossRef]
- Theel, E.S.; Jespersen, D.J.; Iqbal, S.; Bestrom, J.E.; Rollins, L.O.; Misner, L.J.; Markley, B.J.; Mandrekar, J.; Baddour, L.M.; Limper, A.H.; et al. Detection of (1,3)-β-d-glucan in bronchoalveolar lavage and serum samples collected from immunocompromised hosts. Mycopathologia 2013. [Google Scholar] [CrossRef]
- Friedrich, R.; Rappold, E.; Bogdan, C.; Held, J. Comparative analysis of the wako Β-glucan test and the fungitell assay for diagnosis of candidemia and Pneumocystis jirovecii pneumonia. J. Clin. Microbiol. 2018. [Google Scholar] [CrossRef]
- Khan, Z.U.; Ahmad, S.; Theyyathel, A.M. Diagnostic value of DNA and (1→3)-β-D-glucan detection in serum and bronchoalveolar lavage of mice experimentally infected with Fusarium oxysporum. J. Med. Microbiol. 2008. [Google Scholar] [CrossRef]
- Sulahian, A.; Porcher, R.; Bergeron, A.; Touratier, S.; Raffoux, E.; Menotti, J.; Derouin, F.; Ribaud, P. Use and limits of (1-3)-β-D-glucan assay (fungitell), compared to galactomannan determination (platelia Aspergillus), for diagnosis of invasive aspergillosis. J. Clin. Microbiol. 2014. [Google Scholar] [CrossRef]
- Patel, R. MALDI-TOF MS for the diagnosis of infectious diseases. Clin. Chem. 2015, 61, 100–111. [Google Scholar]
- Santos, C.; Lima, N.; Sampaio, P.; Pais, C. Matrix-assisted laser desorption/ionization time-of-flight intact cell mass spectrometry to detect emerging pathogenic Candida species. Diagn. Microbiol. Infect. Dis. 2011, 71, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Maubon, D.; Dard, C.; Garnaud, C.; Cornet, M. Profile of GenMark’s ePlex® blood culture identification fungal pathogen panel. Expert Rev. Mol. Diagn. 2018. [Google Scholar] [CrossRef]
- White, P.L.; Perry, M.D.; Moody, A.; Follett, S.A.; Morgan, G.; Barnes, R.A. Evaluation of analytical and preliminary clinical performance of myconostica mycassay aspergillus when testing serum specimens for diagnosis of invasive aspergillosis. J. Clin. Microbiol. 2011. [Google Scholar] [CrossRef]
- Dierkes, C.; Ehrenstein, B.; Siebig, S.; Linde, H.J.; Reischl, U.; Salzberger, B. Clinical impact of a commercially available multiplex PCR system for rapid detection of pathogens in patients with presumed sepsis. BMC Infect. Dis. 2009. [Google Scholar] [CrossRef]
- Schabereiter-Gurtner, C.; Selitsch, B.; Rotter, M.L.; Hirschl, A.M.; Willinger, B. Development of novel real-time PCR assays for detection and differentiation of eleven medically important Aspergillus and Candida species in clinical specimens. J. Clin. Microbiol. 2007. [Google Scholar] [CrossRef]
- Warhurst, G.; Maddi, S.; Dunn, G.; Ghrew, M.; Chadwick, P.; Alexander, P.; Bentley, A.; Moore, J.; Sharman, M.; Carlson, G.L.; et al. Diagnostic accuracy of SeptiFast multi-pathogen real-time PCR in the setting of suspected healthcare-associated bloodstream infection. Intensive Care Med. 2015. [Google Scholar] [CrossRef]
- Kidd, S.E.; Chen, S.C.A.; Meyer, W.; Halliday, C.L. A New Age in Molecular Diagnostics for Invasive Fungal Disease: Are We Ready? Front. Microbiol. 2020, 10, 2903. [Google Scholar]
- Lehmann, L.E.; Hunfeld, K.P.; Emrich, T.; Haberhausen, G.; Wissing, H.; Hoeft, A.; Stüber, F. A multiplex real-time PCR assay for rapid detection and differentiation of 25 bacterial and fungal pathogens from whole blood samples. Med. Microbiol. Immunol. 2008. [Google Scholar] [CrossRef]
- Tziolos, N.; Giamarellos-Bourboulis, E.J. Contemporary approaches to the rapid molecular diagnosis of sepsis. Expert Rev. Mol. Diagn. 2016, 16, 1201–1207. [Google Scholar] [PubMed]
- Marklein, G.; Josten, M.; Klanke, U.; Müller, E.; Horré, R.; Maier, T.; Wenzel, T.; Kostrzewa, M.; Bierbaum, G.; Hoerauf, A.; et al. Matrix-assisted laser desorption ionization-time of flight mass spectrometry for fast and reliable identification of clinical yeast isolates. J. Clin. Microbiol. 2009. [Google Scholar] [CrossRef]
- Vaz, C.; Sampaio, P.; Clemons, K.V.; Huang, Y.C.; Stevens, D.A.; Pais, C. Microsatellite multilocus genotyping clarifies the relationship of candida parapsilosis strains involved in a neonatal intensive care unit outbreak. Diagn. Microbiol. Infect. Dis. 2011, 71, 159–162. [Google Scholar] [CrossRef]
- Loeffler, J.; Mengoli, C.; Springer, J.; Bretagne, S.; Cuenca-Estrella, M.; Klingspor, L.; Lagrou, K.; Melchers, W.J.G.; Morton, C.O.; Barnes, R.A.; et al. Analytical Comparison of In Vitro -Spiked Human Serum and Plasma for PCR-Based Detection of Aspergillus fumigatus DNA: A Study by the European Aspergillus PCR Initiative. J. Clin. Microbiol. 2015. [Google Scholar] [CrossRef]
- Lion, T. Human Fungal Pathogen Identification: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2017; ISBN 9781493965137. [Google Scholar]
- Carvalho-Pereira, J.; Springer, J.; Buítrago, M.J.; Löffler, J.; Pais, C.; Sampaio, P. Multiplex PCR system for fungal pathogen detection. Ann. Med. 2019, 51. [Google Scholar] [CrossRef]
- Arvanitis, M.; Anagnostou, T.; Mylonakis, E. Galactomannan and Polymerase Chain Reaction-Based Screening for Invasive Aspergillosis among High-Risk Hematology Patients: A Diagnostic Meta-analysis. Clin. Infect. Dis. 2015. [Google Scholar] [CrossRef]
- Kim, W.B.; Park, C.; Cho, S.Y.; Chun, H.S.; Lee, D.G. Development of multiplex real-time PCR for rapid identification and quantitative analysis of Aspergillus species. PLoS ONE 2020. [Google Scholar] [CrossRef]
- Chong, G.L.M.; Van De Sande, W.W.J.; Dingemans, G.J.H.; Gaajetaan, G.R.; Vonk, A.G.; Hayette, M.P.; Van Tegelen, D.W.E.; Simons, G.F.M.; Rijnders, B.J.A. Validation of a new Aspergillus real-time PCR assay for direct detection of Aspergillus and azole resistance of Aspergillus fumigatus on bronchoalveolar lavage fluid. J. Clin. Microbiol. 2015. [Google Scholar] [CrossRef]
- Khot, P.D.; Ko, D.L.; Hackman, R.C.; Fredricks, D.N. Development and optimization of quantitative PCR for the diagnosis of invasive aspergillosis with bronchoalveolar lavage fluid. BMC Infect. Dis. 2008. [Google Scholar] [CrossRef]
- Arvanitis, M.; Anagnostou, T.; Fuchs, B.B.; Caliendo, A.M.; Mylonakis, E. Molecular and nonmolecular diagnostic methods for invasive fungal infections. Clin. Microbiol. Rev. 2014. [Google Scholar] [CrossRef]
- Mylonakis, E.; Clancy, C.J.; Ostrosky-Zeichner, L.; Garey, K.W.; Alangaden, G.J.; Vazquez, J.A.; Groeger, J.S.; Judson, M.A.; Vinagre, Y.M.; Heard, S.O.; et al. T2 magnetic resonance assay for the rapid diagnosis of candidemia in whole blood: A clinical trial. Clin. Infect. Dis. 2015. [Google Scholar] [CrossRef]
- Lucignano, B.; Ranno, S.; Liesenfeld, O.; Pizzorno, B.; Putignani, L.; Bernaschi, P.; Menichella, D. Multiplex PCR allows rapid and accurate diagnosis of bloodstream infections in newborns and children with suspected sepsis. J. Clin. Microbiol. 2011. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Candida Panel | Filamentous Fungi Panel | ||||
|---|---|---|---|---|---|
| Species | Primer Sequence (5′ to 3′) | Dye Label | Species | Primer Sequence (5′ to 3′) | Dye Label |
| C. albicans | F-ttggaatcacttcaccagga R-tttccgtggcatcagtatca | FAM | A. fumigatus | F-gccctcttccgttattcctt R-gcgcattgatagctacctcaggc | HEX |
| C. glabrata | F-acacctacgagaaaccaaca R-tagcggtcatccagcatca | NED | A. flavus | F-gggatcgacactcggactt R-ctggtaagagcttgtgggtg | HEX |
| C. krusei | F-acagcagtcgcaggccc R-gtcggagacataaccgc | FAM | A. niger | F-ccctccttccaaacaaacaa R-tccagatcggctacacagaa | FAM |
| C. parapsilosis | F-aaagtgctacacacgcatcg R-ggcttgcaatttcatttcct | HEX | A. terreus | F-gcggatgcaaggtgtaattt R-tactgcgcgttagttgaagc | NED |
| C. tropicalis | F- ccccaccaaaaacatacatacat R-ttacattcagcccgccacag | FAM | R. arrhizus | F-agaagcaaaatcatcgtcgaaag R-cgtaggtccagcgtaaacttg | NED |
| Species | Isolates Tested | No. of Isolates Amplified with Each Primer Pair | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CA | CP | CG | CT | CK | AF | AFL | AN | AT | RA | Absence of Amplification | ||
| Candida albicans | 21 | 21 | - | - | - | - | - | - | - | - | - | - |
| Candida parapsilosis | 14 | - | 14 | - | - | - | - | - | - | - | - | - |
| Candida glabrata | 20 | - | - | 20 | - | - | - | - | - | - | - | - |
| Candida tropicalis | 7 | - | - | - | 7 | - | - | - | - | - | - | - |
| Candida krusei | 5 | - | - | - | 5 | - | - | - | - | - | - | |
| Candida bracarensis | 4 | - | - | - | - | - | - | - | - | - | - | 4 |
| Candida metapsilosis | 5 | - | - | - | - | - | - | - | - | - | - | 5 |
| Candida orthopsilosis | 5 | - | - | - | - | - | - | - | - | - | - | 5 |
| Candida guilliermondii | 1 | - | - | - | - | - | - | - | - | - | - | 1 |
| Candida lusitaniae | 1 | - | - | - | - | - | - | - | - | - | - | 1 |
| Loderomyces elongisporus | 1 | - | - | - | - | - | - | - | - | - | - | 1 |
| Aspergillus fumigatus | 2 | - | - | - | - | - | 2 | - | - | - | - | - |
| Aspergillus flavus | 8 | - | - | - | - | - | - | 8 | - | - | - | - |
| Aspergillus terreus | 4 | - | - | - | - | - | - | - | - | 4 | - | - |
| Aspergillus niger | 6 | - | - | - | - | - | - | - | 6 | - | - | - |
| Aspergillus versicolor | 3 | - | - | - | - | - | - | - | - | - | - | 3 |
| Aspergillus unguis | 1 | - | - | - | - | - | - | - | - | - | - | 1 |
| Aspergillus westerdijkiae | 1 | - | - | - | - | - | - | - | - | - | - | 1 |
| Aspergillus tamarii | 1 | - | - | - | - | - | - | - | - | - | - | 1 |
| Aspergillus wentii | 1 | - | - | - | - | - | - | - | - | - | - | 1 |
| Aspergillus nidulans | 3 | - | - | - | - | - | - | - | - | - | - | 3 |
| Aspergillus tubingensis | 1 | - | - | - | - | - | - | - | - | - | - | 1 |
| Rhizopus arrhizus | 3 | - | - | - | - | - | - | - | - | - | 3 | - |
| Rhizopus microsporus | 1 | - | - | - | - | - | - | - | - | - | - | 1 |
| Lichtheimia corymbifera | 1 | - | - | - | - | - | - | - | - | - | - | 1 |
| Mucor sp. | 1 | - | - | - | - | - | - | - | - | - | - | 1 |
| TOTAL | 121 | 21 | 14 | 20 | 7 | 5 | 2 | 8 | 6 | 4 | 3 | 28 |
| Candida Panel | Filamentous Fungi Panel | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | DNA | Amplification Cycles | Species | DNA | Amplification Cycles | ||||||
| 35 | 40 | 45 | 50 | 35 | 40 | 45 | 50 | ||||
| C. albicans | 1 ng | +++ | +++ | +++ | +++ | A. fumigatus | 1 ng | +++ | +++ | +++ | +++ |
| 100 pg | ++ | ++ | ++ | ++ | 100 pg | ++ | ++ | +++ | +++ | ||
| 10 pg | - | + | ++ | ++ | 10 pg | - | + | ++ | ++ | ||
| 1 pg | - | - | + | + | 1 pg | - | + | + | + | ||
| C. glabrata | 1 ng | +++ | +++ | +++ | +++ | A. flavus | 1 ng | +++ | +++ | +++ | +++ |
| 100 pg | +++ | +++ | +++ | +++ | 100 pg | +++ | +++ | +++ | +++ | ||
| 10 pg | - | ++ | +++ | +++ | 10 pg | - | + | + | + | ||
| 1 pg | - | + | ++ | ++ | 1 pg | - | - | - | - | ||
| C. parapsilosis | 1 ng | ++ | ++ | ++ | ++ | A. niger | 1 ng | +++ | +++ | +++ | +++ |
| 100 pg | ++ | ++ | ++ | ++ | 100 pg | +++ | +++ | +++ | +++ | ||
| 10 pg | - | + | + | + | 10 pg | - | + | ++ | ++ | ||
| 1 pg | - | - | - | - | 1 pg | - | - | + | + | ||
| C. tropicalis | 1 ng | ++ | ++ | ++ | ++ | A. terreus | 1 ng | +++ | +++ | +++ | +++ |
| 100 pg | ++ | ++ | ++ | ++ | 100 pg | ++ | +++ | +++ | +++ | ||
| 10 pg | - | + | + | + | 10 pg | - | - | + | + | ||
| 1 pg | - | - | - | - | 1 pg | - | - | - | - | ||
| C. krusei | 1 ng | +++ | +++ | +++ | +++ | R. arrhizus | 1 ng | +++ | +++ | +++ | +++ |
| 100 pg | +++ | +++ | +++ | +++ | 100 pg | +++ | +++ | +++ | +++ | ||
| 10 pg | - | + | ++ | ++ | 10 pg | - | + | ++ | ++ | ||
| 1 pg | - | + | ++ | ++ | 1 pg | - | - | - | - | ||
| Samples | EORTC/MSG Classification | Previous Identification (qPCR) | Multiplex PCR Results | ||
|---|---|---|---|---|---|
| Code | Origin | Candida Panel | Filamentous Fungi Panel | ||
| CNM1 | Vitreous humor | Probable | C. albicans | C. albicans | - |
| CNM2 | CSF | Probable | C. albicans | C. albicans | - |
| CNM3 | Broncho-aspirate | Probable | A. fumigatus | - | - |
| CNM4 | Liver biopsy | Probable | C. parapsilosis | C. parapsilosis | - |
| CNM5 | Liver biopsy | Probable | C. albicans | C. albicans | - |
| CNM6 | BAL | Probable | C. albicans | C. albicans | - |
| CNM7 | CSF | Proven | C. albicans | C. albicans | - |
| CNM8 | Biopsy | Proven | C. albicans | C. albicans | - |
| CNM9 | BAL | Probable | A. fumigatus | - | A. fumigatus |
| CNM10 | BAL | Probable | A. fumigatus | - | - |
| CNM11 | BAL | Probable | C. krusei | C. krusei | - |
| CNM12 | Biopsy | Probable | C. tropicalis | C. tropicalis | - |
| CNM13 | Biopsy | Probable | C. glabrata | - | - |
| CNM14 | BAL | Probable | C. parapsilosis | - | - |
| UHW1 | BAL | NA | A. fumigatus | - | Inconclusive |
| UHW2 | BAL | NA | A. fumigatus | - | Inconclusive |
| UHW3 | BAL | NA | A. fumigatus | - | Inconclusive |
| UHW4 | BAL | NA | A. fumigatus | - | Inconclusive |
| UHW5 | BAL | NA | A. fumigatus | - | Inconclusive |
| Controls | |||||
| CNM15 | Pulmonary biopsy | NA | C. guilliermondii | - | - |
| CNM16 | CSF | NA | T. asahii | - | - |
| CNM17 | Biopsy | NA | L. corymbifera | - | - |
| CNM18 | Intestinal biopsy | NA | Rhizopus sp. | - | - |
| CNM19 | BAL | NA | P. jiroveccii | - | - |
| CNM20 | BAL | Not classified | Neg. controls | - | - |
| CNM21 | Broncho-aspirate | Not classified | Neg. controls | - | - |
| CNM22 | Pulmonary biopsy | Not classified | Neg. controls | - | - |
| CNM23 | Whole blood | Not classified | Neg. controls | - | - |
| CNM24 | Hepatic biopsy | Not classified | Neg. controls | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvalho-Pereira, J.; Fernandes, F.; Araújo, R.; Springer, J.; Loeffler, J.; Buitrago, M.J.; Pais, C.; Sampaio, P. Multiplex PCR Based Strategy for Detection of Fungal Pathogen DNA in Patients with Suspected Invasive Fungal Infections. J. Fungi 2020, 6, 308. https://doi.org/10.3390/jof6040308
Carvalho-Pereira J, Fernandes F, Araújo R, Springer J, Loeffler J, Buitrago MJ, Pais C, Sampaio P. Multiplex PCR Based Strategy for Detection of Fungal Pathogen DNA in Patients with Suspected Invasive Fungal Infections. Journal of Fungi. 2020; 6(4):308. https://doi.org/10.3390/jof6040308
Chicago/Turabian StyleCarvalho-Pereira, Joana, Filipa Fernandes, Ricardo Araújo, Jan Springer, Juergen Loeffler, María José Buitrago, Célia Pais, and Paula Sampaio. 2020. "Multiplex PCR Based Strategy for Detection of Fungal Pathogen DNA in Patients with Suspected Invasive Fungal Infections" Journal of Fungi 6, no. 4: 308. https://doi.org/10.3390/jof6040308
APA StyleCarvalho-Pereira, J., Fernandes, F., Araújo, R., Springer, J., Loeffler, J., Buitrago, M. J., Pais, C., & Sampaio, P. (2020). Multiplex PCR Based Strategy for Detection of Fungal Pathogen DNA in Patients with Suspected Invasive Fungal Infections. Journal of Fungi, 6(4), 308. https://doi.org/10.3390/jof6040308