
Allergic Bronchopulmonary Aspergillosis


Abstract
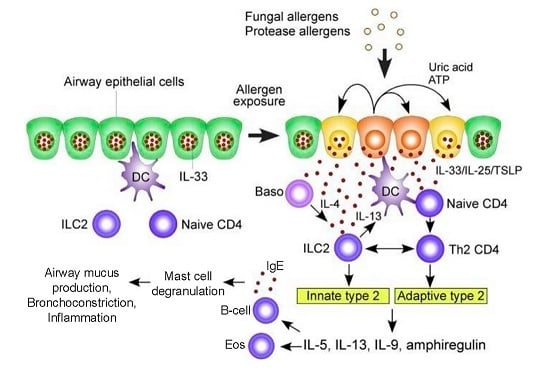
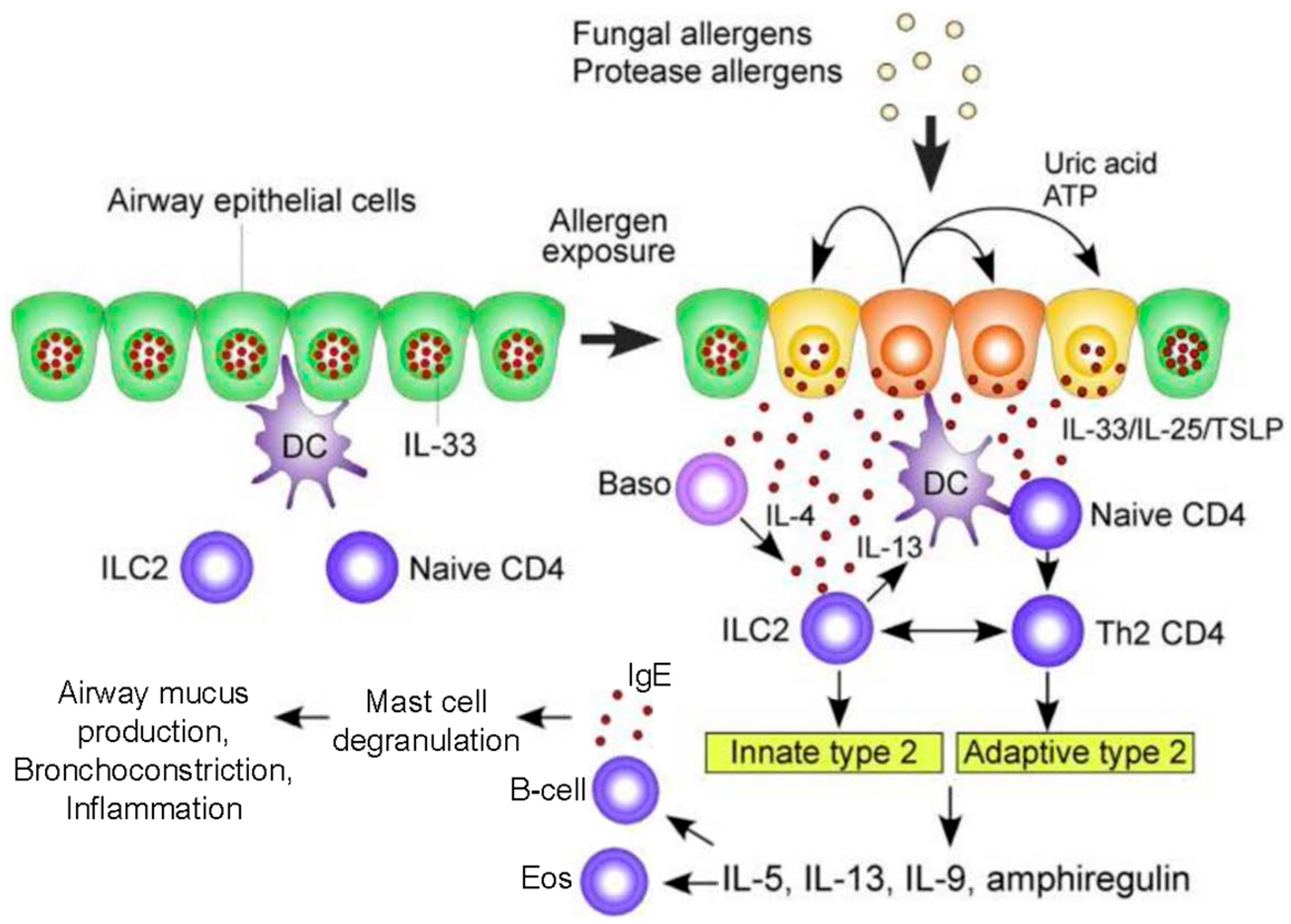
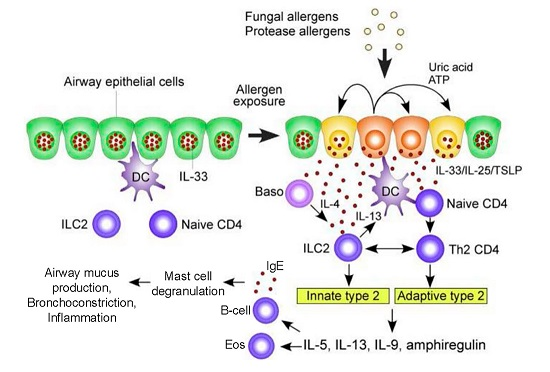
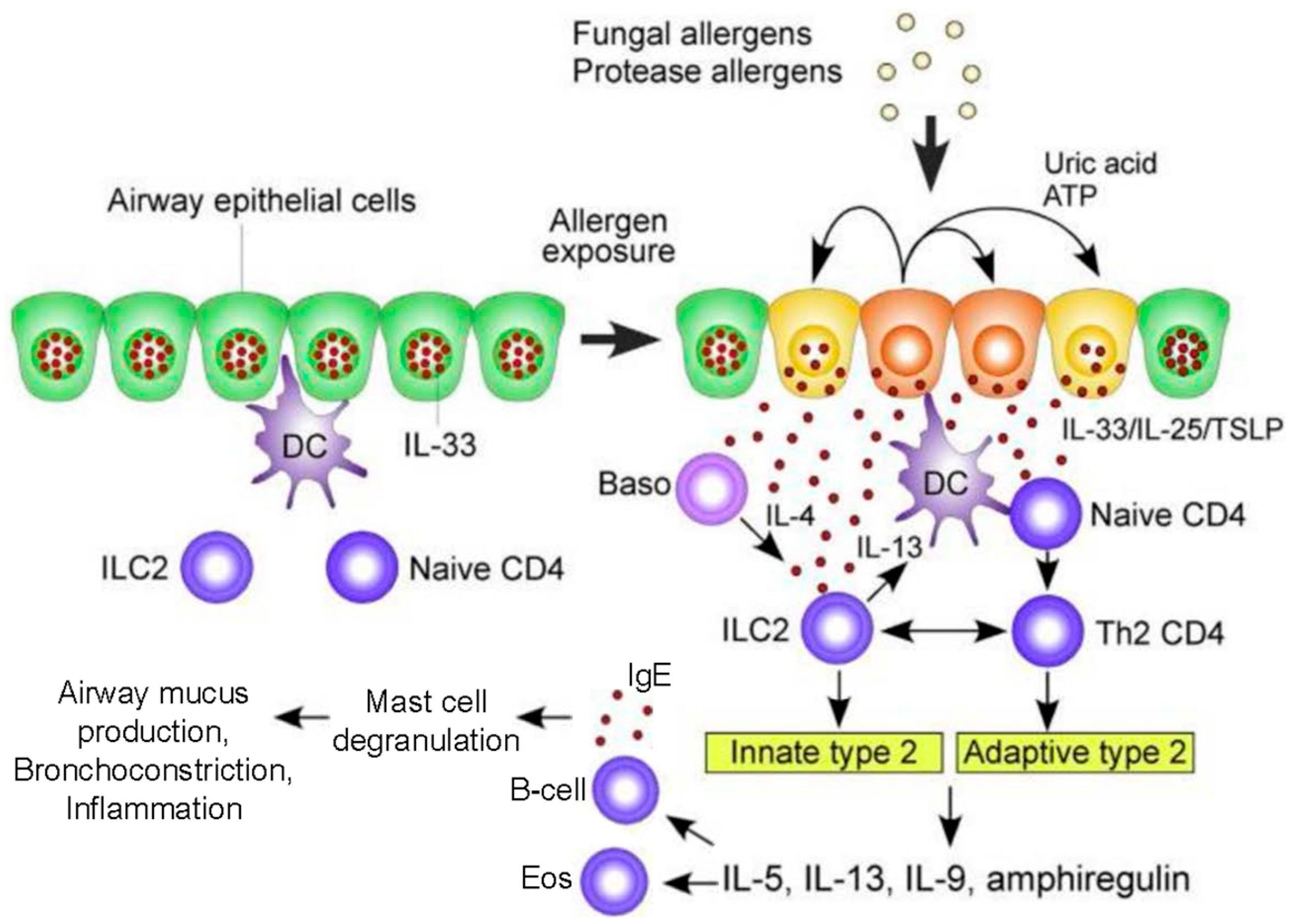
:
1. Introduction
1.1. Pathogenesis of ABPA
1.2. Diagnosis of ABPA
2. Treatment of ABPA
2.1. Glucocorticosteroids
2.2. Antifungal Agents
2.3. Immunotherapy
3. Conclusions
Conflicts of Interest
References
- Denning, D.W.; Pleuvry, A.; Cole, D.C. Global burden of allergic bronchopulmonary aspergillosis with asthma and its complication chronic pulmonary aspergillosis in adults. Med. Mycol. 2013, 51, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Armstead, J.; Morris, J.; Denning, D.W. Multi-country estimate of different manifestations of aspergillosis in cystic fibrosis. PLoS ONE 2014, 9, e98502. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.W.; O’Driscoll, B.R.; Hogaboam, C.M.; Bowyer, P.; Niven, R.M. The link between fungi and asthma: A summary of the evidence. Eur. Respir. J. 2006, 27, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R. Severe asthma with fungal sensitization. Curr. Allergy Asthma Rep. 2011, 11, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Knutsen, A.P.; Bush, R.K.; Demain, J.G.; Denning, D.W.; Dixit, A.; Fairs, A.; Greenberger, P.A.; Kariuki, B.; Kita, H.; Kurup, V.P.; et al. Fungi and allergic lower respiratory diseases. J. Allergy Clin. Immunol. 2012, 129, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Knutsen, A.P.; Slavin, R.G. Allergic bronchopulmonary aspergillosis in asthma and cystic fibrosis. Clin. Dev. Immunol. 2011, 2011, 843763. [Google Scholar] [CrossRef] [PubMed]
- Kwon-Chung, K.J.; Sugui, J.A. Aspergillus fumigatus—What makes the species a ubiquitous human fungal pathogen? PLoS Pathog. 2013, 9, e1003743. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Datta, K.; Askin, F.B.; Staab, J.F.; Marr, K.A. Cystic fibrosis transmembrane conductance regulator regulates epithelial cell response to Aspergillus and resultant pulmonary inflammation. Am. J. Respir. Crit. Care Med. 2012, 185, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Aggarwal, A.N.; Gupta, D.; Jindal, S.K. Aspergillus hypersensitivity and allergic bronchopulmonary aspergillosis in patients with bronchial asthma: Systematic review and meta-analysis. Int. J. Tuberc. Lung Dis. 2009, 13, 936–944. [Google Scholar] [PubMed]
- Maturu, V.N.; Agarwal, R. Prevalence of Aspergillus sensitization and allergic bronchopulmonary aspergillosis in cystic fibrosis: Systematic review and meta-analysis. Clin. Exp. Allergy 2015, 45, 1765–1778. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Kala, J.; Sahay, S.; Panjabi, C. Frequency of familial occurrence in 164 patients with allergic bronchopulmonary aspergillosis. Ann. Allergy Asthma Immunol. 2008, 101, 363–369. [Google Scholar] [CrossRef]
- Pana, Z.-D.; Farmaki, E.; Roilides, E. Host genetics and opportunistic fungal infections. Clin. Microbiol. Infect. 2014, 20, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Khan, A.; Aggarwal, A.N.; Gupta, D. Link between CFTR mutations and ABPA: A systematic review and meta-analysis. Mycoses 2012, 55, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Muro, M.; Mondejar-López, P.; Moya-Quiles, M.R.; Salgado, G.; Pastor-Vivero, M.D.; Lopez-Hernandez, R.; Boix, F.; Campillo, J.A.; Minguela, A.; Garcia-Alonso, A.; et al. HLA-DRB1 and HLA-DQB1 genes on susceptibility to and protection from allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Microbiol. Immunol. 2013, 57, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Corvol, H.; Blackman, S.M.; Boëlle, P.-Y.; Gallins, P.J.; Pace, R.G.; Stonebraker, J.R.; Accurso, F.J.; Clement, A.; Collaco, J.M.; Dang, H.; et al. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Aimanianda, V.; Bayry, J.; Bozza, S.; Kniemeyer, O.; Perruccio, K.; Elluru, S.R.; Clavaud, C.; Paris, S.; Brakhage, A.A.; Kaveri, S.V.; et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature 2009, 460, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Eickmeier, O.; Rieber, N.; Eckrich, J.; Hector, A.; Graeppler-Mainka, U.; Hartl, D. Immune response, diagnosis and treatment of allergic bronchopulmonary aspergillosis in cystic fibrosis lung disease. Curr. Pharm. Des. 2013, 19, 3669–3678. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, A.; Fontaine, T.; Aimanianda, V.; Latgé, J.-P. Aspergillus cell wall and biofilm. Mycopathologia 2014, 178, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Gresnigt, M.S.; Netea, M.G.; van de Veerdonk, F.L. Pattern recognition receptors and their role in invasive aspergillosis. Ann. N. Y. Acad. Sci. 2012, 1273, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.; Anand, R.; Tiwari, S.; Singh, A.P.; Tiwary, B.N.; Shankar, J. Cytokines induce effector T-helper cells during invasive aspergillosis; what we have learned about T-helper cells? Front. Microbiol. 2015, 6, 429. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.Y.A.; van de Veerdonk, F.; Marijnissen, R.J.; Cheng, S.-C.; Khoo, A.L.; Hectors, M.; Lagrou, K.; Vonk, A.G.; Maertens, J.; Joosten, L.A.B.; et al. Anti- human host defence relies on type 1 T helper (Th1), rather than type 17 T helper (Th17), cellular immunity. Immunology 2010, 130, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Kita, H. ILC2s and fungal allergy. Allergol. Int. 2015, 64, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, B.; Homma, T.; Norton, J.E.; Sha, Q.; Siebert, J.; Gupta, D.S.; Schroeder, J.W.; Schleimer, R.P. Suppression of epithelial signal transducer and activator of transcription 1 activation by extracts of Aspergillus fumigatus. Am. J. Respir. Cell Mol. Biol. 2015, 53, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Kato, A.; Bhushan, B.; Norton, J.E.; Suh, L.A.; Carter, R.G.; Gupta, D.S.; Schleimer, R.P. Role of Aspergillus fumigatus in triggering protease-activated receptor-2 in airway epithelial cells and skewing the cells toward a T-helper 2 bias. Am. J. Respir. Cell Mol. Biol. 2016, 54, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.L.; Gresnigt, M.S.; Smeekens, S.P.; Jacobs, C.W.; Magis-Escurra, C.; Jaeger, M.; Wang, X.; Lubbers, R.; Oosting, M.; Joosten, L.A.B.; et al. Pattern recognition pathways leading to a Th2 cytokine bias in allergic bronchopulmonary aspergillosis patients. Clin. Exp. Allergy 2015, 45, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Romani, L. Immunity to fungal infections. Nat. Rev. Immunol. 2011, 11, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Carevic, M.; Singh, A.; Rieber, N.; Eickmeier, O.; Griese, M.; Hector, A.; Hartl, D. CXCR4+ granulocytes reflect fungal cystic fibrosis lung disease. Eur. Respir. J. 2015, 46, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B. Treatment options in severe fungal asthma and allergic bronchopulmonary aspergillosis. Eur. Respir. J. 2014, 43, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Licona-Limón, P.; Kim, L.K.; Palm, N.W.; Flavell, R.A. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B. Fungi in CF and non-CF bronchiectasis. Semin. Resp. Crit. Care Med. 2015, 36, 207–216. [Google Scholar]
- Rosenberg, M.; Patterson, R.; Mintzer, R.; Cooper, B.J.; Roberts, M.; Harris, K.E. Clinical and immunologic criteria for the diagnosis of allergic bronchopulmonary aspergillosis. Ann. Intern. Med. 1977, 86, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Chakrabarti, A.; Shah, A.; Gupta, D.; Meis, J.F.; Guleria, R.; Moss, R.; Denning, D.W.; ABPA complicating asthma ISHAM working group. Allergic bronchopulmonary aspergillosis: Review of literature and proposal of new diagnostic and classification criteria. Clin. Exp. Allergy 2013, 43, 850–873. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Maskey, D.; Aggarwal, A.N.; Saikia, B.; Garg, M.; Gupta, D.; Chakrabarti, A. Diagnostic performance of various tests and criteria employed in allergic bronchopulmonary aspergillosis: A latent class analysis. PLoS ONE 2013, 8, e61105. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.S.; Moss, R.; Kurup, V.P.; Knutsen, A.P.; Greenberger, P.; Judson, M.A.; Denning, D.W.; Crameri, R.; Brody, A.; Light, M.; et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis. Clin. Infect. Dis. 2003, 37, S225–S264. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, A.J.; Woolnough, K.; Pashley, C.H. Lassoing a chimera: The semantics of allergic fungal airway disease. Clin. Exp. Allergy 2015, 45, 1746–1749. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Aggarwal, A.N.; Garg, M.; Saikia, B.; Chakrabarti, A. Cutoff values of serum IgE (total and A. fumigatus-specific) and eosinophil count in differentiating allergic bronchopulmonary aspergillosis from asthma. Mycoses 2014, 57, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Page, I.D.; Richardson, M.; Denning, D.W. Antibody testing in aspergillosis—Quo vadis? Med. Mycol. 2015, 53, 417–439. [Google Scholar] [CrossRef] [PubMed]
- Page, I.D.; Richardson, M.D.; Denning, D.W. Comparison of six Aspergillus-specific IgG assays for the diagnosis of chronic pulmonary aspergillosis. J. Infect. 2016, 72, 240–249. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, B.R.; Powell, G.; Chew, F.; Niven, R.M.; Miles, J.F.; Vyas, A.; Denning, D.W. Comparison of skin prick tests with specific serum immunoglobulin E in the diagnosis of fungal sensitization in patients with severe asthma. Clin. Exp. Allergy 2009, 39, 1677–1683. [Google Scholar] [CrossRef] [PubMed]
- Kurup, V.P.; Knutsen, A.P.; Moss, R.B.; Bansal, N.K. Specific antibodies to recombinant allergens of Aspergillus fumigatus in cystic fibrosis patients with ABPA. Clin. Mol. Allergy 2006, 4. [Google Scholar] [CrossRef] [PubMed]
- Delhaes, L.; Frealle, E.; Pinel, C. Serum markers for allergic bronchopulmonary aspergillosis in cystic fibrosis: State of the art and further challenges. Med. Mycol. 2010, 48, S77–S87. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, S.J.; Hansell, D.M. Aspergillus in the lung: Diverse and coincident forms. Eur. Radiol. 2003, 13, 1786–1800. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Gupta, D.; Aggarwal, A.N.; Saxena, A.K.; Chakrabarti, A.; Jindal, S.K. Clinical significance of hyperattenuating mucoid impaction in allergic bronchopulmonary aspergillosis: An analysis of 155 patients. Chest 2007, 132, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Phuyal, S.; Garg, M.K.; Agarwal, R.; Gupta, P.; Chakrabarti, A.; Sandhu, M.S.; Khandelwal, N. High-attenuation mucus impaction in patients with allergic bronchopulmonary aspergillosis: Objective criteria on high-resolution computed tomography and correlation with serologic parameters. Curr. Probl. Diagn. Radiol. 2016, 45, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, P.A.; Miller, T.P.; Roberts, M.; Smith, L.L. Allergic bronchopulmonary aspergillosis in patients with and without evidence of bronchiectasis. Ann. Allergy 1993, 70, 333–338. [Google Scholar] [PubMed]
- Gernez, Y.; Walters, J.; Mirković, B.; Lavelle, G.M.; Colleen, D.E.; Davies, Z.A.; Everson, C.; Tirouvanziam, R.; Silver, E.; Wallenstein, S.; et al. Blood basophil activation is a reliable biomarker of allergic bronchopulmonary aspergillosis in cystic fibrosis. Eur. Respir. J. 2016, 47, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Gernez, Y.; Dunn, C.E.; Everson, C.; Mitsunaga, E.; Gudiputi, L.; Krasinska, K.; Davies, Z.A.; Herzenberg, L.A.; Tirouvanziam, R.; Moss, R.B. Blood basophils from cystic fibrosis patients with allergic bronchopulmonary aspergillosis are primed and hyper-responsive to stimulation by Aspergillus allergens. J. Cyst. Fibros. 2012, 11, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Mirković, B.; Lavelle, G.M.; Azim, A.A.; Helma, K.; Gargoum, F.S.; Molloy, K.; Gernez, Y.; Dunne, K.; Renwick, J.; Murphy, P.; et al. The Basophil surface marker CD203C identifies Aspergillus species sensitization in patients with cystic fibrosis. J. Allergy Clin. Immunol. 2016, 137, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, P.A.; Bush, R.K.; Demain, J.G.; Luong, A.; Slavin, R.G.; Knutsen, A.P. Allergic bronchopulmonary aspergillosis. J. Allergy Clin. Immunol. Pract. 2014, 2, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.F.; Patterson, R.; Roberts, M.; Ghory, A.C. The management of allergic bronchopulmonary aspergillosis. Am. Rev. Respir. Dis. 1979, 120, 87–92. [Google Scholar] [PubMed]
- Agarwal, R. Allergic bronchopulmonary aspergillosis. Chest 2009, 135, 805–826. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, P.A. Allergic bronchopulmonary aspergillosis. J. Allergy Clin. Immunol. 2002, 110, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Mahdavinia, M.; Grammer, L.C. Management of allergic bronchopulmonary aspergillosis: A review and update. Ther. Adv. Respir. Dis. 2012, 6, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Tillie-Leblond, I.; Tonnel, A.B. Allergic bronchopulmonary aspergillosis. Allergy 2005, 60, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Gupta, D.; Aggarwal, A.N.; Behera, D.; Jindal, S.K. Allergic bronchopulmonary aspergillosis: Lessons from 126 patients attending a chest clinic in north India. Chest 2006, 130, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Aggarwal, A.N.; Dhooria, S.; Singh Sehgal, I.; Garg, M.; Saikia, B.; Behera, D.; Chakrabarti, A. A randomised trial of glucocorticoids in acute-stage allergic bronchopulmonary aspergillosis complicating asthma. Eur. Respir. J. 2016, 47, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Khan, A.; Aggarwal, A.N.; Saikia, B.; Gupta, D.; Chakrabarti, A. Role of inhaled corticosteroids in the management of serological allergic bronchopulmonary aspergillosis (ABPA). Intern. Med. 2011, 50, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Aggarwal, A.N.; Sehgal, I.S.; Dhooria, S.; Behera, D.; Chakrabarti, A. Utility of IgE (total and Aspergillus fumigatus specific) in monitoring for response and exacerbations in allergic bronchopulmonary aspergillosis. Mycoses 2016, 59, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Singh Sehgal, I.; Agarwal, R. Pulse methylprednisolone in allergic bronchopulmonary aspergillosis exacerbations. Eur. Respir. Rev. 2014, 23, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Cymberknoh, M.; Blau, H.; Shoseyov, D.; Mei-Zahav, M.; Efrati, O.; Armoni, S.; Kerem, E. Intravenous monthly pulse methylprednisolone treatment for ABPA in patients with cystic fibrosis. J. Cyst. Fibros. 2009, 8, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.M.; Wesley, A.; Byrnes, C.A.; Nixon, G.M. Pulse intravenous methylprednisolone for resistant allergic bronchopulmonary aspergillosis in cystic fibrosis. Pediatr. Pulmonol. 2006, 41, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Skov, M.; Pressler, T. High-dose IV-pulse methylprednisolone (HDIVPM) successful treatment of allergic bronchopulmonary aspergillosis (ABPA). Pediatr. Pulmonol. 2010, 45, 1. [Google Scholar]
- British Thoracic Association. Inhaled beclomethasone dipropionate in allergic bronchopulmonary aspergillosis. Report to the Research Committee of the British Thoracic Association. Br. J. Dis. Chest 1979, 73, 349–356. [Google Scholar]
- Imbeault, B.; Cormier, Y. Usefulness of inhaled high-dose corticosteroids in allergic bronchopulmonary aspergillosis. Chest 1993, 103, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.S.; Silva, D.; Ferreira, A.R.; Delgado, L. Antifungal treatment in allergic bronchopulmonary aspergillosis with and without cystic fibrosis: A systematic review. Clin. Exp. Allergy 2014, 44, 1210–1227. [Google Scholar] [CrossRef] [PubMed]
- Stark, J.E. Allergic pulmonary aspergillosis successfully treated with inhalations of nystatin. Report of a case. Dis. Chest 1967, 51, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Wark, P.A.; Gibson, P.G.; Wilson, A.J. Azoles for allergic bronchopulmonary aspergillosis associated with asthma. Cochrane Database Syst. Rev. 2004, 3. [Google Scholar] [CrossRef]
- Shale, D.J.; Faux, J.A.; Lane, D.J. Trial of ketoconazole in non-invasive pulmonary aspergillosis. Thorax 1987, 42, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.A.; Schwartz, H.J.; Lee, J.Y.; Moskovitz, B.L.; Jerome, D.C.; Catanzaro, A.; Bamberger, D.M.; Weinmann, A.J.; Tuazon, C.U.; Judson, M.A.; et al. A randomized trial of itraconazole in allergic bronchopulmonary aspergillosis. N. Engl. J. Med. 2000, 342, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Wark, P.A.B.; Hensley, M.J.; Saltos, N.; Boye, M.J.; Toneguzzi, R.C.; Epid, G.D.; Simpson, J.L.; McElduff, P.; Gibson, P.G. Anti-inflammatory effect of itraconazole in stable allergic bronchopulmonary aspergillosis: A randomized controlled trial. J. Allergy Clin. Immunol. 2003, 111, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Wark, P. Pathogenesis of allergic bronchopulmonary aspergillosis and an evidence-based review of azoles in treatment. Respir. Med. 2004, 98, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.A.; Kan, V.L.; Judson, M.A.; Morrison, V.A.; Dummer, S.; Denning, D.W.; Bennett, J.E.; Walsh, T.J.; Patterson, T.F.; Pankey, G.A. Practice guidelines for diseases caused by Aspergillus. Clin. Infect. Dis. 2000, 30, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Chishimba, L.; Niven, R.M.; Cooley, J.; Denning, D.W. Voriconazole and posaconazole improve asthma severity in allergic bronchopulmonary aspergillosis and severe asthma with fungal sensitization. J. Asthma 2012, 49, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Lebrun-Vignes, B.; Archer, V.C.; Diquet, B.; Levron, J.C.; Chosidow, O.; Puech, A.J.; Warot, D. Effect of itraconazole on the pharmacokinetics of prednisolone and methylprednisolone and cortisol secretion in healthy subjects. Br. J. Clin. Pharmacol. 2001, 51, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.P.; Paquette, K.; Lands, L.C.; Ovetchkine, P.; Theoret, Y.; Quach, C. Voriconazole inhibition of vitamin a metabolism: Are adverse events increased in cystic fibrosis patients? Pediatr. Pulmonol. 2010, 45, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Clancy, C.J.; Nguyen, M.H. Long-term voriconazole and skin cancer: Is there cause for concern? Curr. Infect. Dis. Rep. 2011, 13, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Dolton, M.J.; McLachlan, A.J. Voriconazole pharmacokinetics and exposure-response relationships: Assessing the links between exposure, efficacy and toxicity. Int. J. Antimicrob. Agents 2014, 44, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Luong, M.-L.; Al-Dabbagh, M.; Groll, A.H.; Racil, Z.; Nannya, Y.; Mitsani, D.; Husain, S. Utility of voriconazole therapeutic drug monitoring: A meta-analysis. J. Antimicrob. Chemother. 2016. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Mansh, M.; Chin-Hong, P.; Singer, J.; Arron, S.T. Voriconazole-associated cutaneous malignancy: A literature review on photocarcinogenesis in organ transplant recipients. Clin. Infect. Dis. 2014, 58, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.P.; Boker, A.; Metchnikoff, C.; Binstock, M.; Boettger, R.; Golden, J.A.; Glidden, D.V.; Arron, S.T. High cumulative dose exposure to voriconazole is associated with cutaneous squamous cell carcinoma in lung transplant recipients. J. Heart Lung Trans. 2012, 31, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.J.; Ronan, N.J.; Khan, K.A.; O’Callaghan, G.; Murphy, D.M.; Plant, B.J. Ivacaftor therapy in siblings with cystic fibrosis-the potential implications of Itraconazole in dosage and efficacy. Pulm. Pharmacol. Ther. 2015, 31, 49–50. [Google Scholar] [CrossRef] [PubMed]
- Conway, S.P.; Etherington, C.; Peckham, D.G.; Brownlee, K.G.; Whitehead, A.; Cunliffe, H. Pharmacokinetics and safety of itraconazole in patients with cystic fibrosis. J. Antimicrob. Chemo. 2004, 53, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Berge, M.; Guillemain, R.; Boussaud, V.; Pham, M.H.; Chevalier, P.; Batisse, A.; Amrein, C.; Dannaoui, E.; Loriot, M.A.; Lillo-Le Louet, A.; et al. Voriconazole pharmacokinetic variability in cystic fibrosis lung transplant patients. Transpl. Infect. Dis. 2009, 11, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Clifton, I.J.; Whitaker, P.; Metcalfe, R.; Phillip, M.; Shaw, N.; Conway, S.P.; Peckham, D.G. Pharmacokinetics of oral voriconazole in patients with cystic fibrosis. J. Antimicrob. Chemo. 2011, 66, 2438–2440. [Google Scholar] [CrossRef] [PubMed]
- Warris, A. Azole-resistant aspergillosis. J. Infect. 2015, 71, S121–S125. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, I.S.; Agarwal, R. Role of inhaled amphotericin in allergic bronchopulmonary aspergillosis. J. Postgrad. Med. 2014, 60, 41–45. [Google Scholar] [PubMed]
- Diot, P.; Rivoire, B.; Le Pape, A.; Lemarie, E.; Dire, D.; Furet, Y.; Breteau, M.; Smaldone, G.C. Deposition of amphotericin B aerosols in pulmonary aspergilloma. Eur. Respir. J. 1995, 8, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Proesmans, M.; Vermeulen, F.; Vreys, M.; De Boeck, K. Use of nebulized amphotericin B in the treatment of allergic bronchopulmonary aspergillosis in cystic fibrosis. Int. J. Pediatr. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Ram, B.; Aggarwal, A.N.; Dhooria, S.; Sehgal, I.S.; Garg, M.; Behera, D.; Chakrabarti, A.; Agarwal, R. A pilot randomized trial of nebulized amphotericin in patients with allergic bronchopulmonary aspergillosis. J. Asthma 2016, 53, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Casciaro, R.; Naselli, A.; Cresta, F.; Ros, M.; Castagnola, E.; Minicucci, L. Role of nebulized amphotericin B in the management of allergic bronchopulmonary aspergillosis in cystic fibrosis: Case report and review of literature. J. Chemother. 2015, 27, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Busse, W.W.; Morgan, W.J.; Gergen, P.J.; Mitchell, H.E.; Gern, J.E.; Liu, A.H.; Gruchalla, R.S.; Kattan, M.; Teach, S.J.; Pongracic, J.A.; et al. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N. Engl. J. Med. 2011, 364, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Busse, W.; Hanania, N.A.; Lowe, P.J.; Canvin, J.; Erpenbeck, V.J.; Holgate, S. Omalizumab in asthma: An update on recent developments. J. Allergy Clin. Immunol. Pract. 2014, 2, 525 e1–536 e1. [Google Scholar] [CrossRef] [PubMed]
- Normansell, R.; Walker, S.; Milan, S.J.; Walters, E.H.; Nair, P. Omalizumab for asthma in adults and children. Cochrane Database System. Rev. 2014. [Google Scholar] [CrossRef]
- Pérez-de-Llano, L.A.; Vennera, M.C.; Parra, A.; Guallar, J.; Marin, M.; Asensio, O.; Ausin, P.; Borderías, L.; Fernández, C.; Granel, C.; et al. Effects of omalizumab in Aspergillus-associated airway disease. Thorax 2011, 66, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Tillie-Leblond, I.; Germaud, P.; Leroyer, C.; Tétu, L.; Girard, F.; Devouassoux, G.; Grignet, J.P.; Prudhomme, A.; Dusser, D.; Wallaert, B. Allergic bronchopulmonary aspergillosis and omalizumab. Allergy 2011, 66, 1254–1256. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.; Devos, G.; Hudes, G.; Rosenstreich, D. Allergic bronchopulmonary aspergillosis treated successfully for one year with omalizumab. J. Asthma Allergy 2012, 5, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.; Wong, M.; Robinson, P.D.; Fitzgerald, D.A. Omalizumab in the management of steroid dependent allergic bronchopulmonary aspergillosis (ABPA) complicating cystic fibrosis. Paediatr. Respir. Rev. 2013, 14, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Tanou, K.; Zintzaras, E.; Kaditis, A.G. Omalizumab therapy for allergic bronchopulmonary aspergillosis in children with cystic fibrosis: A synthesis of published evidence. Pediatr. Pulmonol. 2014, 49, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Pfannenstiel, C.; Friedrichs, F.; Kroger, K.; Wagner, N.; Tenbrock, K. Omalizumab: A new treatment option for allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Ther. Adv. Respir. Dis. 2014, 8, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Ö.; Sözener, Z.Ç.; Soyyiğit, Ş.; Kendirlinan, R.; Gençtürk, Z.; Mısırlıgil, Z.; Mungan, D.; Sin, B.A.; Demirel, Y.S.; Çelik, G.E.; et al. Omalizumab in the treatment of allergic bronchopulmonary aspergillosis: One center’s experience with 14 cases. Allergy Asthma Proceed. 2015, 36, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Emiralioglu, N.; Dogru, D.; Tugcu, G.D.; Yalcin, E.; Kiper, N.; Ozcelik, U. Omalizumab treatment for allergic bronchopulmonary aspergillosis in cystic fibrosis. Ann. Pharmacother. 2016, 50, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Cusack, R.P.; Sahadevan, A.; Lane, S.J. Qualitative effects of omalizumab on concomitant IgE-mediated disease in a severe asthmatic population—A real life observational study. QJM 2016. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.G. Monitoring allergic patients on omalizumab with free and total serum IgE measurements. J. Allergy Clin. Immunol. Pract. 2016, 4, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Jat, K.R.; Walia, D.K.; Khairwa, A. Anti-igE therapy for allergic bronchopulmonary aspergillosis in people with cystic fibrosis. Cochrane Database System. Rev. 2015, 9. [Google Scholar] [CrossRef]
- Voskamp, A.L.; Gillman, A.; Symons, K.; Sandrini, A.; Rolland, J.M.; O’Hehir, R.E.; Douglass, J.A. Clinical efficacy and immunologic effects of omalizumab in allergic bronchopulmonary aspergillosis. J. Allergy Clin. Immunol. Pract. 2015, 3, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.W.; Pashley, C.; Hartl, D.; Wardlaw, A.; Godet, C.; Del Giacco, S.; Delhaes, L.; Sergejeva, S. Fungal allergy in asthma-state of the art and research needs. Clin. Transl. Allergy 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Hogan, C.; Denning, D.W. Allergic bronchopulmonary aspergillosis and related allergic syndromes. Semin. Respir. Crit. Care Med. 2011, 32, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.J.; McSharry, C.; Chaudhuri, R.; Farrow, S.; Thomson, N.C. Long-term macrolide treatment of chronic inflammatory airway diseases: Risks, benefits and future developments. Clin. Exp. Allergy Br. Soc. Allergy Clin. Immunol. 2012, 42, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Hebisawa, A.; Kurashima, A.; Kawabe, Y.; Machida, K.; Yotsumoto, H.; Mori, M. The use of bronchofiberscopy for diagnosis of allergic bronchopulmonary aspergillosis. Intern. Med. 1997, 36, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Khalil, K.F. Therapeutic bronchoalveolar lavage with conventional treatment in allergic bronchopulmonary aspergillosis. J. Coll. Phys. Surg. Pak. 2015, 25, 359–362. [Google Scholar]
- Coughlan, C.A.; Chotirmall, S.H.; Renwick, J.; Hassan, T.; Low, T.B.; Bergsson, G.; Eshwika, A.; Bennett, K.; Dunne, K.; Greene, C.M.; et al. The effect of Aspergillus fumigatus infection on vitamin D receptor expression in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.L.; Pilewski, J.M.; Celedón, J.C.; Mandalapu, S.; Blanchard, M.L.; DeRicco, A.; Hartigan, E.; Alcorn, J.F.; Kolls, J.K. Vitamin D supplementation decreases Aspergillus fumigatus specific Th2 responses in CF patients with Aspergillus sensitization: A phase one open-label study. Asthma Res. Pract. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B. The use of biological agents for the treatment of fungal asthma and allergic bronchopulmonary aspergillosis. Ann. N. Y. Acad. Sci. 2012, 1272, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Capewell, S.; Chapman, B.J.; Alexander, F.; Greening, A.P.; Crompton, G.K. Corticosteroid treatment and prognosis in pulmonary eosinophilia. Thorax 1989, 44, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Garg, M.; Aggarwal, A.N.; Saikia, B.; Gupta, D.; Chakrabarti, A. Serologic bronchopulmonary aspegillosis: Long-term outcomes. Respir. Med. 2012, 106, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Greenberger, P.A.; Patterson, R.; Roberts, M.; Liotta, J.L. Stage V (fibrotic) allergic bronchopulmonary aspergillosis. A review of 17 cases followed from diagnosis. Arch. Intern. Med. 1987, 147, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Ye, L.; Ye, M.; Yang, D.; Jin, M. A retrospective study of patients with a delayed diagnosis of allergic bronchopulmonary aspergillosis/allergic bronchopulmonary mycosis. Allergy Asthma Proc. 2014, 35, e21–e26. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
|
| Secondary (supportive) criteria |
|
|
|
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tracy, M.C.; Okorie, C.U.A.; Foley, E.A.; Moss, R.B. Allergic Bronchopulmonary Aspergillosis. J. Fungi 2016, 2, 17. https://doi.org/10.3390/jof2020017
Tracy MC, Okorie CUA, Foley EA, Moss RB. Allergic Bronchopulmonary Aspergillosis. Journal of Fungi. 2016; 2(2):17. https://doi.org/10.3390/jof2020017
Chicago/Turabian StyleTracy, Michael C., Caroline U. A. Okorie, Elizabeth A. Foley, and Richard B. Moss. 2016. "Allergic Bronchopulmonary Aspergillosis" Journal of Fungi 2, no. 2: 17. https://doi.org/10.3390/jof2020017
APA StyleTracy, M. C., Okorie, C. U. A., Foley, E. A., & Moss, R. B. (2016). Allergic Bronchopulmonary Aspergillosis. Journal of Fungi, 2(2), 17. https://doi.org/10.3390/jof2020017