
Decoding the Tissue-Specific Profiles of Bioactive Compounds in Helvella leucopus Using Combined Transcriptomic and Metabolomic Approaches



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Scanning Structure and Ultrastructural Spectrometry Analysis
2.3. Determination of Nutrient Composition
2.4. Transcriptomic Analysis
2.5. Metabolomic Analysis
2.6. Statistical Analysis
3. Results
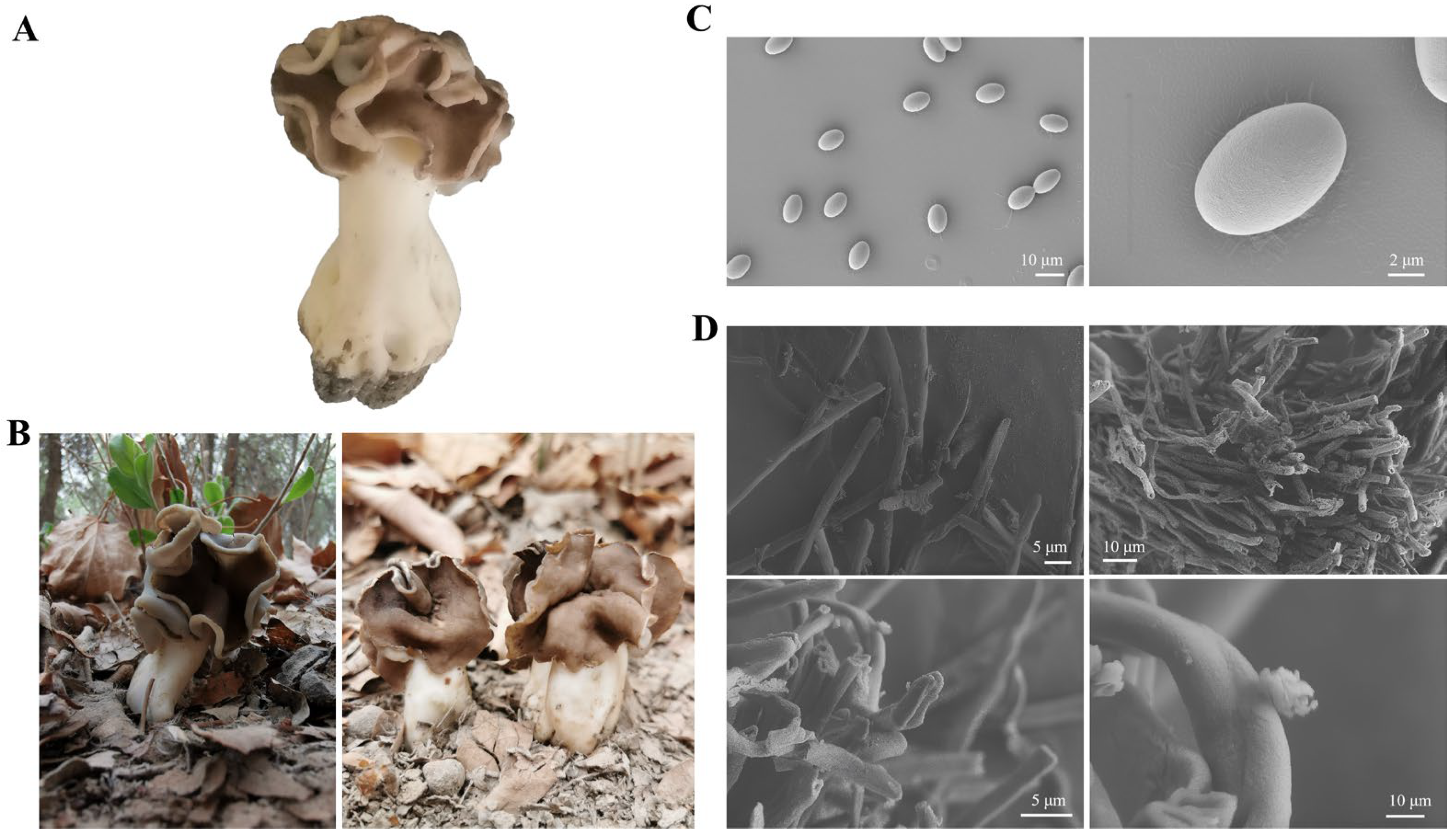
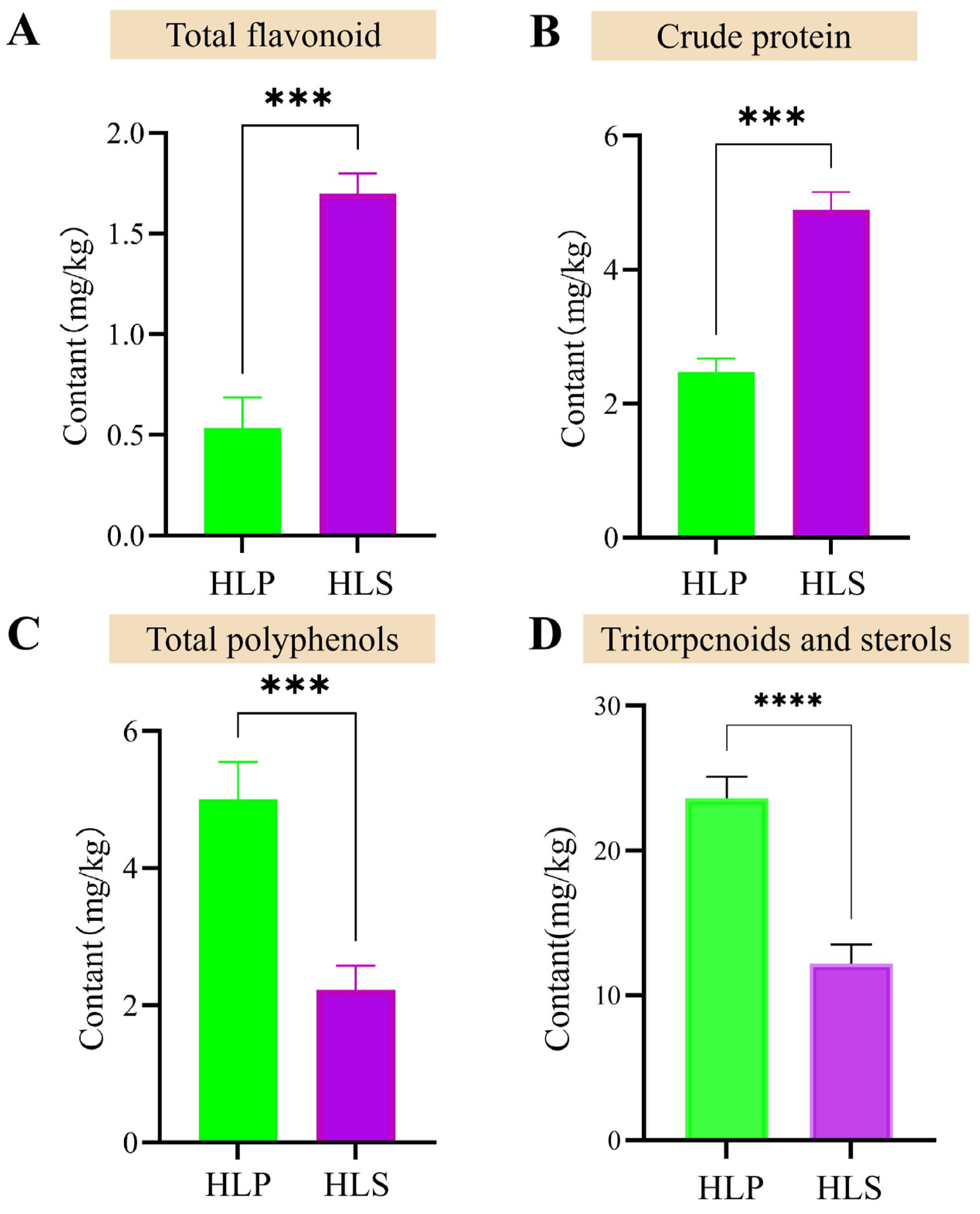
3.1. Changes in Morphological and Bioactive Nutrient of H. leucopus at Stalks and Pileus
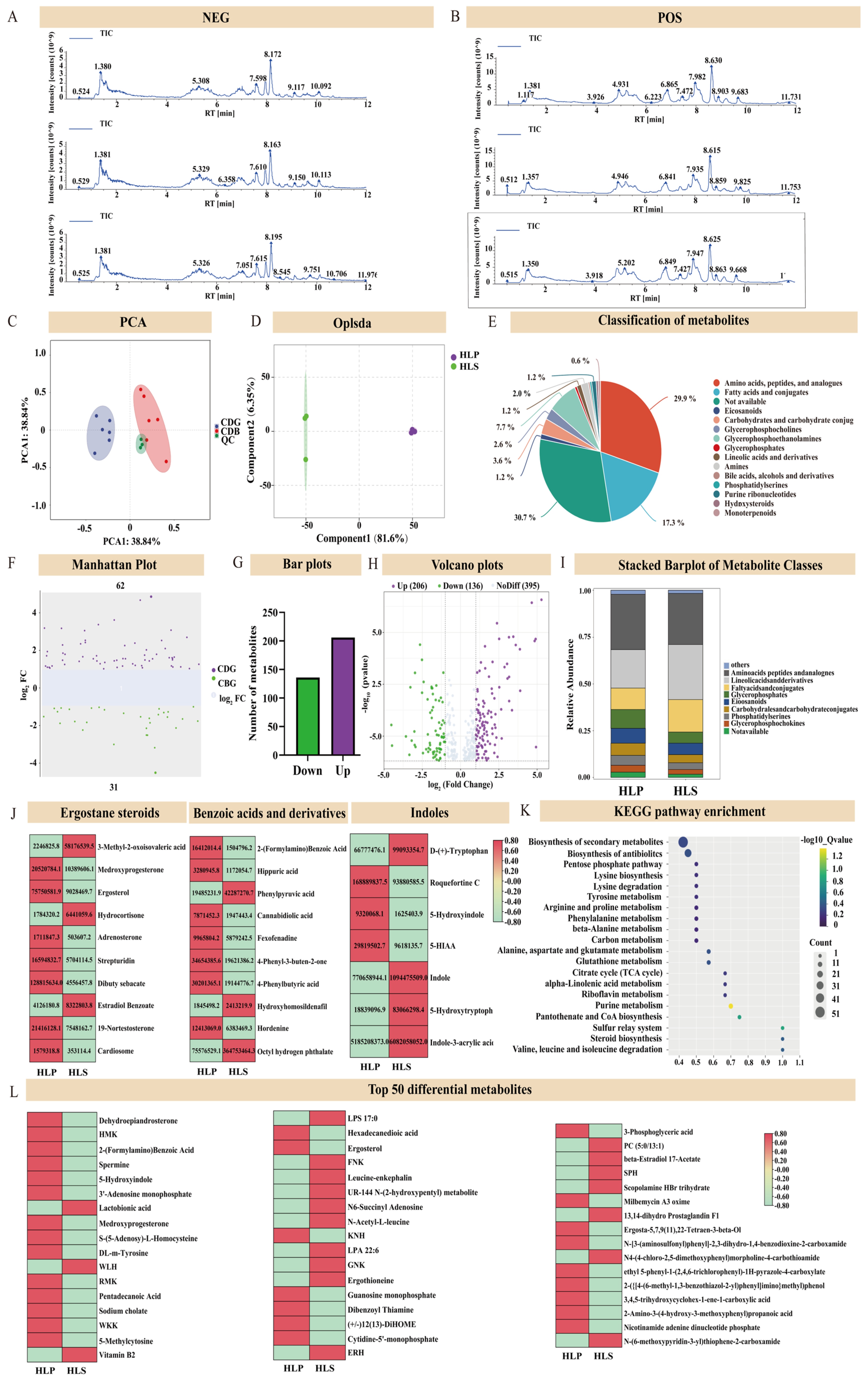
3.2. Metabolomics Analysis of the Stipe and Cap of H. leucopus
3.3. Differential Analysis of the Stipe and Cap of H. leucopus
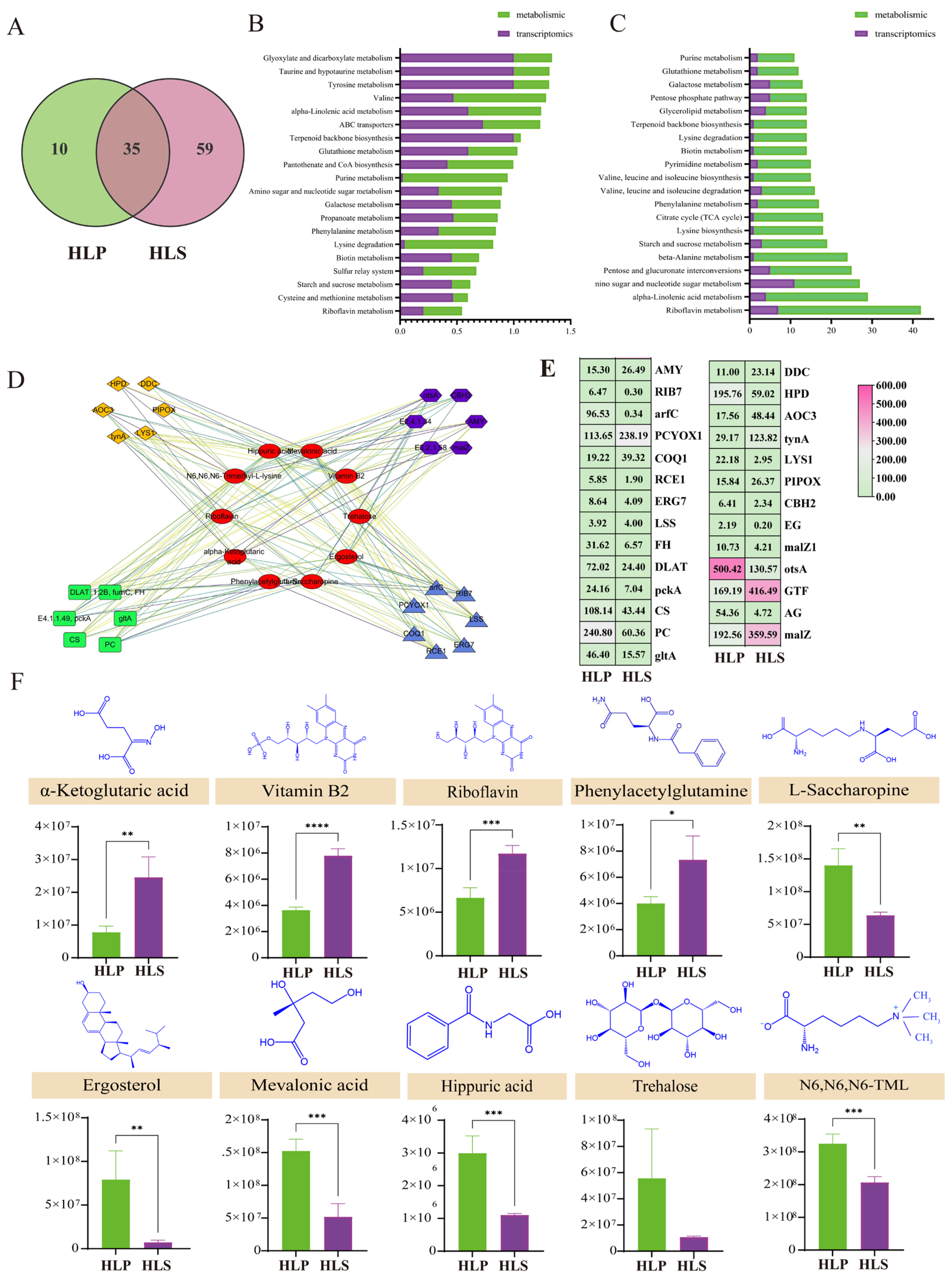
3.4. Transcriptomics Profilin of the Stipe and Cap of H. leucopus
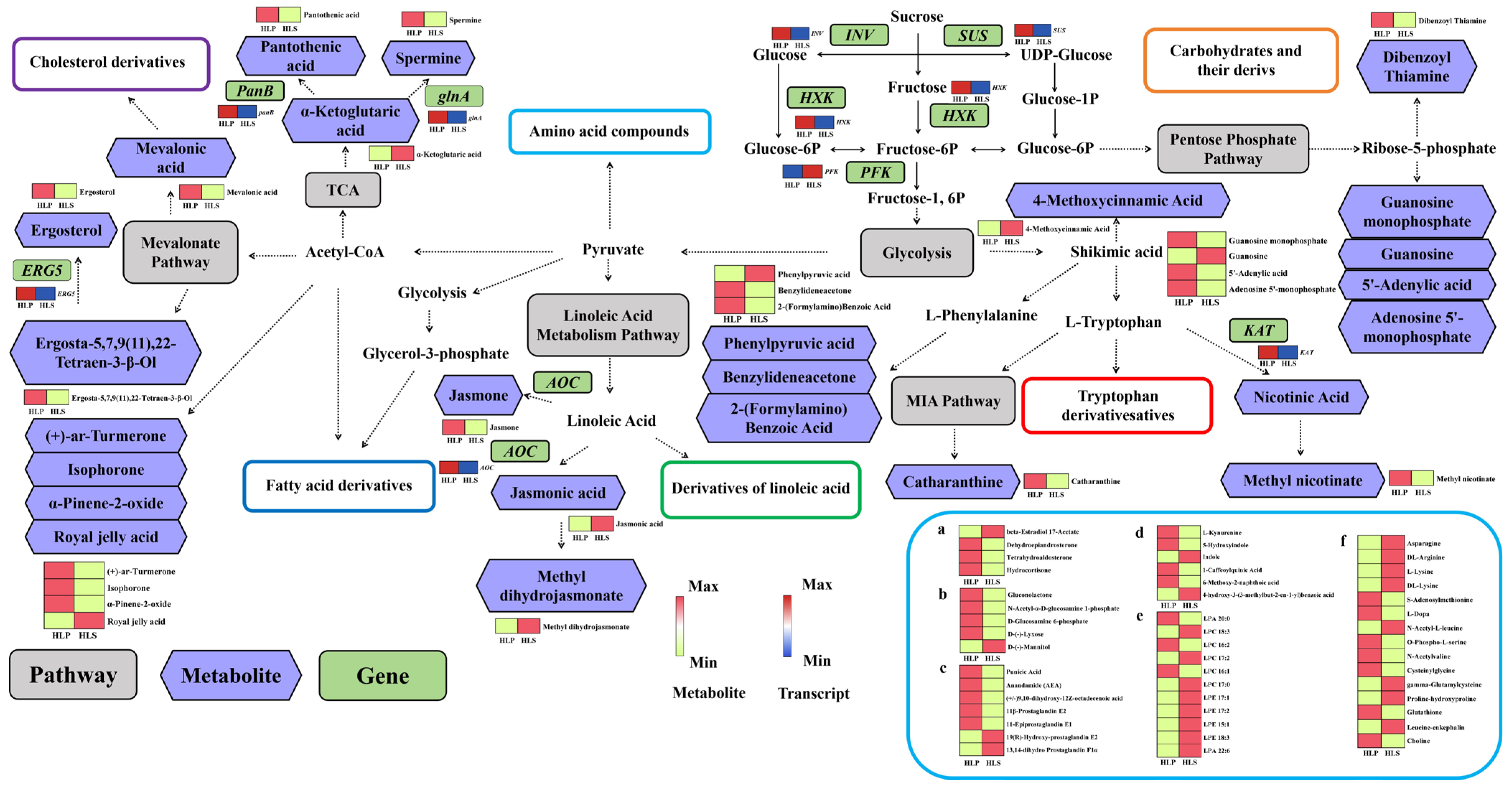
3.5. Integrated Analysis of Metabolomics and Transcriptomics
4. Discussion
4.1. Sterol Synthesis and Defense Mechanism
4.2. Fatty Acid Metabolism and Immune Regulation
4.3. Amino Acid Metabolism and Cell Growth
4.4. Tryptophan Metabolism and Growth Regulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, Q.; Sulayman, M.; Zhu, X.-T.; Zhao, Y.-C.; Yang, Z.-L.; Hyde, K.D. Species clarification of the culinary Bachu mushroom in western China. Mycologia 2016, 108, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Duan, X.; Lin, L.; Wu, W.; Li, X.; Zeng, Z.; Luo, Q.; Liu, Y. A review on the edible mushroom as a source of special flavor: Flavor categories, influencing factors, and challenges. Food Front. 2023, 4, 1561–1577. [Google Scholar] [CrossRef]
- Leuschner, C.; Ellenberg, H. Ecology of Central European Forests: Vegetation Ecology of Central Europe; Springer: Berlin/Heidelberg, Germany, 2017; Volume I, p. 972. [Google Scholar] [CrossRef]
- Thakur, M.P.; Singh, H.K. Mushrooms, their bioactive compounds and medicinal uses: A review. Med. Plants-Int. J. Phytomed. Relat. Ind. 2013, 5, 1–20. [Google Scholar] [CrossRef]
- Muszynska, B.; Grzywacz-Kisielewska, A.; Kala, K.; Gdula-Argasinska, J. Anti-inflammatory properties of edible mushrooms: A review. Food Chem. 2018, 243, 373–381. [Google Scholar] [CrossRef]
- Elsayed, E.A.; El Enshasy, H.; Wadaan, M.A.; Aziz, R. Mushrooms: A potential natural source of anti-inflammatory compounds for medical applications. Mediat. Inflamm. 2014, 2014, 805841. [Google Scholar] [CrossRef]
- Rana, K.L.; Kour, D.; Sheikh, I.; Dhiman, A.; Yadav, N.; Yadav, A.N.; Rastegari, A.A.; Singh, K.; Saxena, A.K. Endophytic Fungi: Biodiversity, Ecological Significance, and Potential Industrial Applications. In Recent Advancement in White Biotechnology Through Fungi; Fungal Biology; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–62. [Google Scholar]
- Allen, E.E.; Banfield, J.F. Community genomics in microbial ecology and evolution. Nat. Rev. Microbiol. 2005, 3, 489–498. [Google Scholar] [CrossRef]
- Yus, E.; Maier, T.; Michalodimitrakis, K.; van Noort, V.; Yamada, T.; Chen, W.H.; Wodke, J.A.; Guell, M.; Martinez, S.; Bourgeois, R.; et al. Impact of genome reduction on bacterial metabolism and its regulation. Science 2009, 326, 1263–1268. [Google Scholar] [CrossRef]
- Liang, C.; Tian, D.; Liu, Y.; Li, H.; Zhu, J.; Li, M.; Xin, M.; Xia, J. Review of the molecular mechanisms of Ganoderma lucidum triterpenoids: Ganoderic acids A, C2, D, F, DM, X and Y. Eur. J. Med. Chem. 2019, 174, 130–141. [Google Scholar] [CrossRef]
- Ge, Y.; Qiu, H.; Zheng, J. Physicochemical characteristics and anti-hyperlipidemic effect of polysaccharide from BaChu mushroom (Helvella leucopus). Food Chem. X 2022, 15, 100443. [Google Scholar] [CrossRef]
- Aziz, W.M.; Hamed, M.A.; Abd-Alla, H.; Ahmed, S.A. Mitigates nephrotoxicity induced by carbon tetrachloride in rats regulation oxidative, inflammatory, tubular and glomerular indices. Biomarkers 2022, 27, 35–43. [Google Scholar] [CrossRef]
- Llamasares-Castillo, A.; Uclusin-Bolibol, R.; Rojsitthisak, P.; Alcantara, K.P. Studies of the therapeutic potential of extracts in osteoarthritis: Targeting oxidation, inflammation, and chondroprotection. J. Ethnopharmacol. 2024, 333, 118446. [Google Scholar] [CrossRef] [PubMed]
- Kadir, F.A.; Othman, F.; Abdulla, M.A.; Hussan, F.; Hassandarvish, P. Effect of Tinospora crispa on thioacetamide-induced liver cirrhosis in rats. Indian J. Pharmacol. 2011, 43, 64–68. [Google Scholar] [CrossRef]
- Tan, K.C.; Ipcho, S.V.; Trengove, R.D.; Oliver, R.P.; Solomon, P.S. Assessing the impact of transcriptomics, proteomics and metabolomics on fungal phytopathology. Mol. Plant Pathol. 2009, 10, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Li, W.; Yu, X.; Zhang, B.; Shang, L.; Xie, Y.; Li, Y.; Ding, A.; Shi, J.; Dou, Y.; et al. Genome-wide analysis, metabolomics, and transcriptomics reveal the molecular basis of ZlRc overexpression in promoting phenolic compound accumulation in rice seeds. Food Front. 2023, 4, 849–866. [Google Scholar] [CrossRef]
- Stitt, M.; Sulpice, R.; Keurentjes, J. Metabolic networks: How to identify key components in the regulation of metabolism and growth. Plant Physiol. 2010, 152, 428–444. [Google Scholar] [CrossRef]
- Chen, X.L.; Sun, M.C.; Chong, S.L.; Si, J.P.; Wu, L.S. Transcriptomic and Metabolomic Approaches Deepen Our Knowledge of Plant-Endophyte Interactions. Front. Plant Sci. 2022, 12, 700200. [Google Scholar] [CrossRef]
- Sardans, J.; Peñuelas, J.; Rivas-Ubach, A. Ecological metabolomics: Overview of current developments and future challenges. Chemoecology 2011, 21, 191–225. [Google Scholar] [CrossRef]
- Kluger, B.; Lehner, S.; Schuhmacher, R. Metabolomics and Secondary Metabolite Profiling of Filamentous Fungi. In Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites; Zeilinger, S., Martín, J.-F., García-Estrada, C., Eds.; Springer: New York, NY, USA, 2015; Volume 2, pp. 81–101. [Google Scholar]
- Wijayawardene, N.N.; Boonyuen, N.; Ranaweera, C.B.; de Zoysa, H.K.S.; Padmathilake, R.E.; Nifla, F.; Dai, D.Q.; Liu, Y.; Suwannarach, N.; Kumla, J.; et al. OMICS and Other Advanced Technologies in Mycological Applications. J. Fungi 2023, 9, 688. [Google Scholar] [CrossRef]
- Arshadi, N.; Nouri, H.; Moghimi, H. Increasing the production of the bioactive compounds in medicinal mushrooms: An omics perspective. Microb. Cell Factories 2023, 22, 11. [Google Scholar] [CrossRef]
- Culibrk, L.; Croft, C.A.; Tebbutt, S.J. Systems Biology Approaches for Host-Fungal Interactions: An Expanding Multi-Omics Frontier. Omics 2016, 20, 127–138. [Google Scholar] [CrossRef]
- Arora, A.; Raghuraman, H.; Chattopadhyay, A. Influence of cholesterol and ergosterol on membrane dynamics: A fluorescence approach. Biochem. Biophys. Res. Commun. 2004, 318, 920–926. [Google Scholar] [CrossRef] [PubMed]
- T/AHFIA 004-2018; Determination of Total Saponins in Food Spectrophotometric Method. Anhui Food Industry Association: Hefei, China, 2018.
- SN/T 4260-2015; Determination of Crude Polysaccharides in Plant Source Foods for Export. Phehol-Sulfuric Acid Colorimetry. General Administration of Quality Supervision, Inspection and Quarantine of PRC: Beijing, China, 2015.
- SN/T 4592-2016; Determination of Total Flavonoids in Export Food. General Administration of Quality Supervision, Inspection and Quarantine of the People’s Republic of China: Beijing, China, 2016.
- T/AHFIA 005-2018; Determination of Total Polyphenol in Plant Extracts and Its Products Spectrophotometric Method. Anhui Food Industry Association: Hefei, China, 2018.
- NY/T 3676-2020; Determination of Total Triterpenes in Ganoderma Lucidum by Spectrophotometry. Ministry of Agriculture and Rural Affairs: Beijing, China, 2020.
- Liu, J.; Zhang, B.; Wang, L.; Li, S.; Long, Q.; Xiao, X. Bioactive components, pharmacological properties and underlying mechanism of Ganoderma lucidum spore oil: A review. Chin. Herb. Med. 2024, 16, 375–391. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Yuan, W.; Zhou, R.; Lv, G.; Sun, M.; Zhao, Y.; Zheng, W. Production of hispidin polyphenols from medicinal mushroom Sanghuangporus vaninii in submerged cultures. Chin. Herb. Med. 2023, 15, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Pott, D.M.; Osorio, S.; Vallarino, J.G. From central to specialized metabolism: An overview of some secondary compounds derived from the primary metabolism for their role in conferring nutritional and organoleptic characteristics to fruit. Front. Plant Sci. 2019, 10, 835. [Google Scholar] [CrossRef]
- Jasinghe, V.J.; Perera, C.O. Distribution of ergosterol in different tissues of mushrooms and its effect on the conversion of ergosterol to vitamin D2 by UV irradiation. Food Chem. 2005, 92, 541–546. [Google Scholar] [CrossRef]
- Guerrero-Ochoa, P.; Rodríguez-Zapater, S.; Anel, A.; Esteban, L.M.; Camón-Fernández, A.; Espilez-Ortiz, R.; Gil-Sanz, M.J.; Borque-Fernando, Á. Prostate Cancer and the Mevalonate Pathway. Int. J. Mol. Sci. 2024, 25, 2152. [Google Scholar] [CrossRef]
- Banni, S. Conjugated linoleic acid metabolism. Curr. Opin. Lipidol. 2002, 13, 261–266. [Google Scholar] [CrossRef]
- Ruan, J.; Zhou, Y.; Zhou, M.; Yan, J.; Khurshid, M.; Weng, W.; Cheng, J.; Zhang, K. Jasmonic acid signaling pathway in plants. Int. J. Mol. Sci. 2019, 20, 2479. [Google Scholar] [CrossRef]
- Ghorbel, M.; Brini, F.; Sharma, A.; Landi, M. Role of jasmonic acid in plants: The molecular point of view. Plant Cell Rep. 2021, 40, 1471–1494. [Google Scholar] [CrossRef]
- Feduraev, P.; Skrypnik, L.; Riabova, A.; Pungin, A.; Tokupova, E.; Maslennikov, P.; Chupakhina, G. Phenylalanine and tyrosine as exogenous precursors of wheat (Triticum aestivum L.) secondary metabolism through PAL-associated pathways. Plants 2020, 9, 476. [Google Scholar] [CrossRef]
- Cleri, F.; Cleri, F. Energy Production and Storage for Life. In The Physics of Living Systems; Springer: Berlin/Heidelberg, Germany, 2016; pp. 113–158. [Google Scholar]
- Zhang, Y.; Swart, C.; Alseekh, S.; Scossa, F.; Jiang, L.; Obata, T.; Graf, A.; Fernie, A.R. The extra-pathway interactome of the TCA cycle: Expected and unexpected metabolic interactions. Plant Physiol. 2018, 177, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.S.; Laxmi, A. Rooted in Communication: Exploring Auxin–Salicylic Acid Nexus in Root Growth and Development. Plant Cell Environ. 2025; early view. [Google Scholar] [CrossRef] [PubMed]
- Ohri, P.; Bhardwaj, R.; Bali, S.; Kaur, R.; Jasrotia, S.; Khajuria, A.; Parihar, R.D. The common molecular players in plant hormone crosstalk and signaling. Curr. Protein Pept. Sci. 2015, 16, 369–388. [Google Scholar] [CrossRef] [PubMed]
- Chand, T.; Savitri, B. Vitamin B3, niacin. In Industrial Biotechnology of Vitamins, Biopigments, and Antioxidants; Wiley: Hoboken, NJ, USA, 2016; pp. 41–65. [Google Scholar]
- Zhou, H.; Shi, B.J. New roles of DNA-binding and forkhead-associated domains of Fkh1 and Fkh2 in cellular functions. Cell Biochem. Funct. 2022, 40, 888–902. [Google Scholar] [CrossRef]
- An, H.-M.; Dai, Y.-F.; Zhu, J.; Liu, W.; Wang, X.-P. MYST family histone acetyltransferases regulate reproductive diapause initiation. Int. J. Biol. Macromol. 2024, 256, 128269. [Google Scholar] [CrossRef]
- Ohm, R.A.; Aerts, D.; Wösten, H.A.; Lugones, L.G. The blue light receptor complex WC-1/2 of S chizophyllum commune is involved in mushroom formation and protection against phototoxicity. Environ. Microbiol. 2013, 15, 943–955. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Gao, X.; Ma, J.; Zhao, H.; Gao, D.; Zhao, H. Decoding the Tissue-Specific Profiles of Bioactive Compounds in Helvella leucopus Using Combined Transcriptomic and Metabolomic Approaches. J. Fungi 2025, 11, 205. https://doi.org/10.3390/jof11030205
Zhou Q, Gao X, Ma J, Zhao H, Gao D, Zhao H. Decoding the Tissue-Specific Profiles of Bioactive Compounds in Helvella leucopus Using Combined Transcriptomic and Metabolomic Approaches. Journal of Fungi. 2025; 11(3):205. https://doi.org/10.3390/jof11030205
Chicago/Turabian StyleZhou, Qian, Xusheng Gao, Junxia Ma, Haoran Zhao, Dan Gao, and Huixin Zhao. 2025. "Decoding the Tissue-Specific Profiles of Bioactive Compounds in Helvella leucopus Using Combined Transcriptomic and Metabolomic Approaches" Journal of Fungi 11, no. 3: 205. https://doi.org/10.3390/jof11030205
APA StyleZhou, Q., Gao, X., Ma, J., Zhao, H., Gao, D., & Zhao, H. (2025). Decoding the Tissue-Specific Profiles of Bioactive Compounds in Helvella leucopus Using Combined Transcriptomic and Metabolomic Approaches. Journal of Fungi, 11(3), 205. https://doi.org/10.3390/jof11030205