

Comparative Genomic Analysis of Colletotrichum lini Strains with Different Virulence on Flax
 , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Material
2.2. DNA Extraction and Purification
2.3. DNA Library Preparation and Sequencing on the Oxford Nanopore Technologies and Illumina Platforms
2.4. Genome Assembly
2.5. Genome Analysis
3. Results
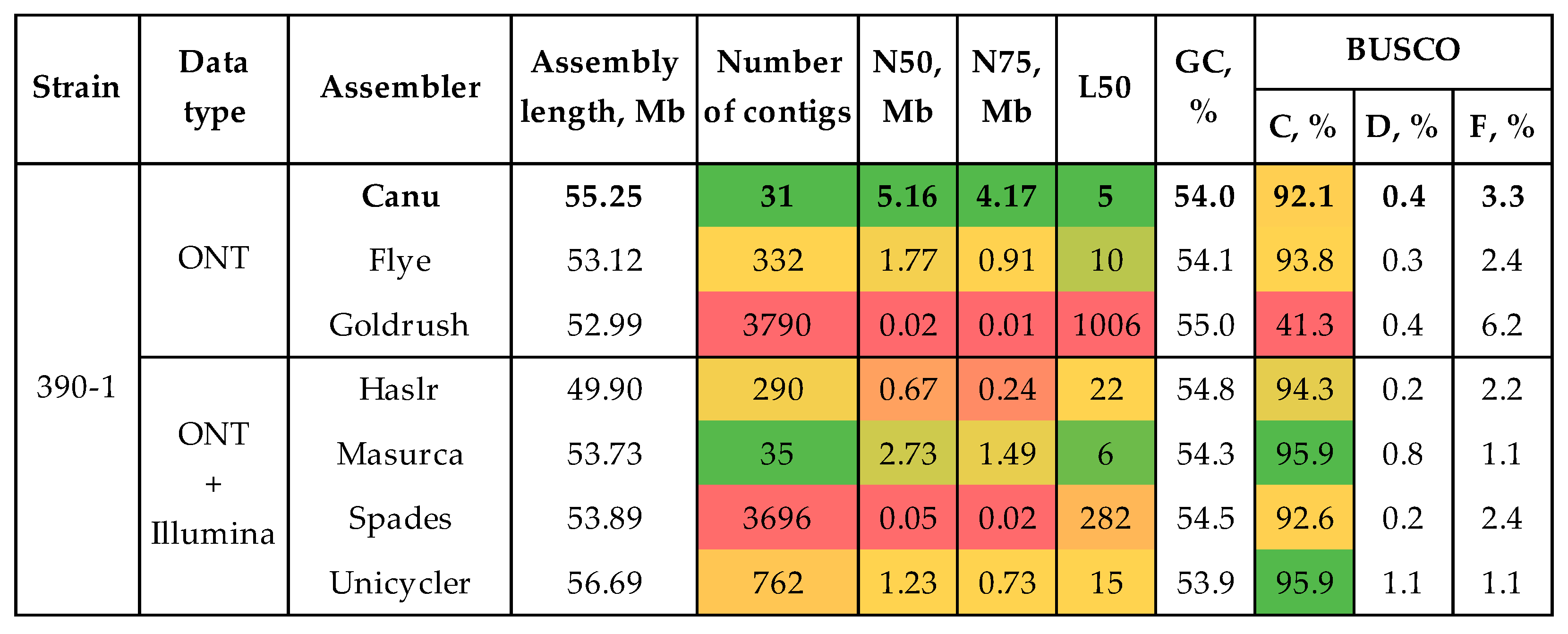
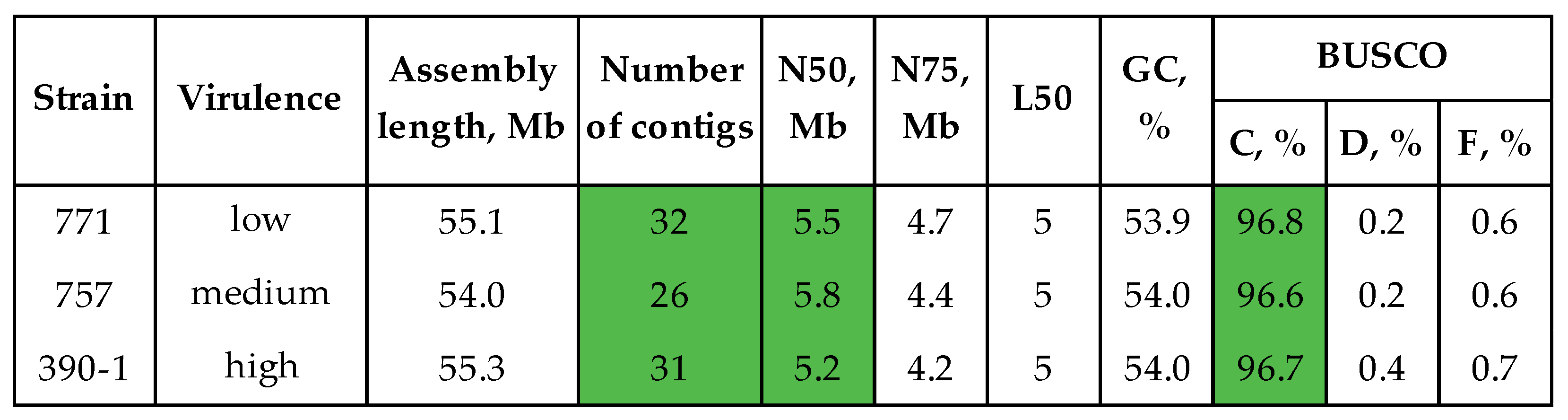
3.1. Genome Assembly and Polishing
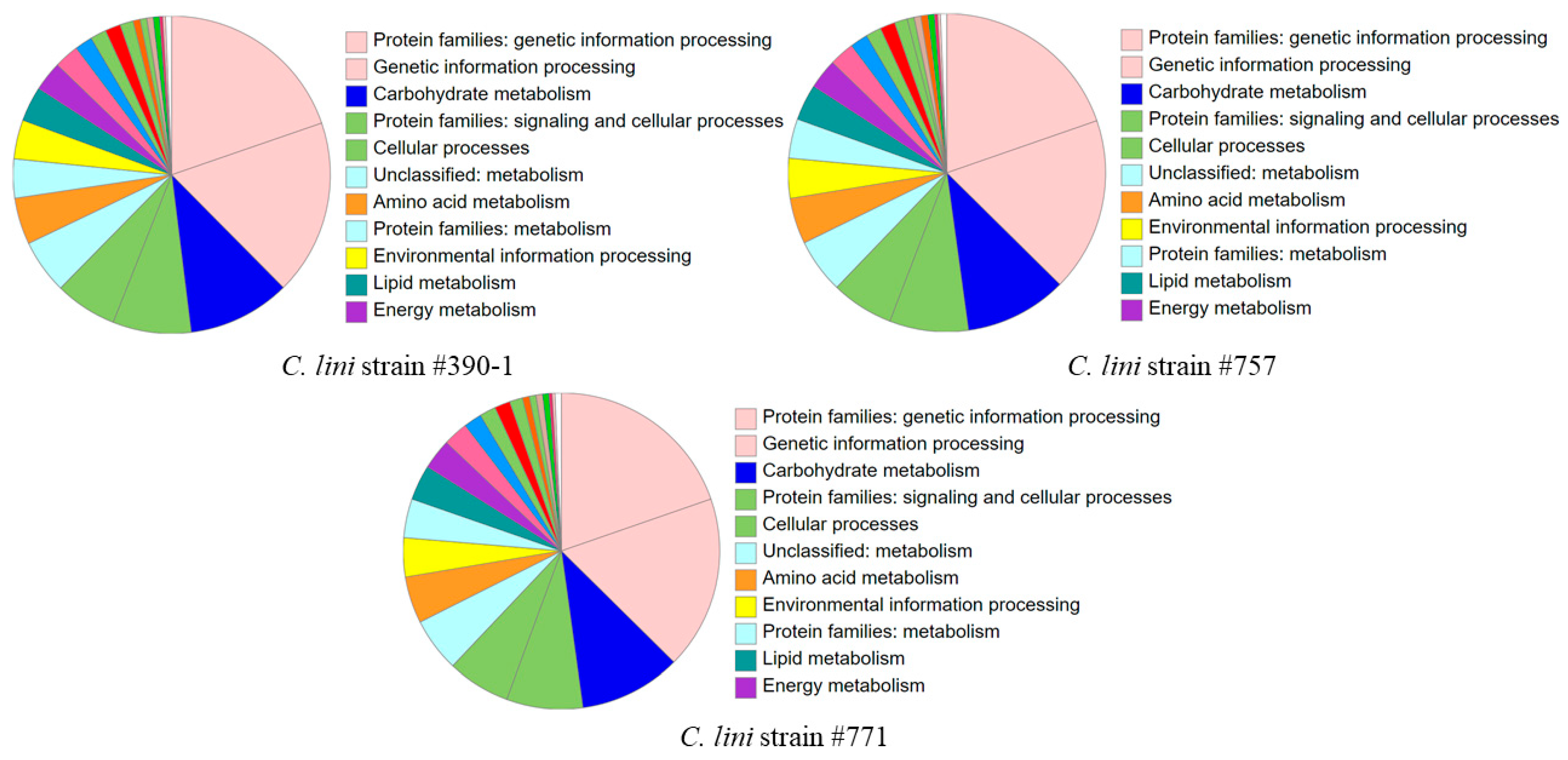
3.2. Genome Annotation and Search for Effector Proteins
3.3. Comparative Genomic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, R.; Kumar, A.; Dhaka, R.K.; Chahar, M.; Malyan, S.K.; Singh, A.P.; Rana, A. Climate change and agriculture: Impact assessment and sustainable alleviation approach using rhizomicrobiome. In Bioinoculants: Biological Option for Mitigating Global Climate Change; Springer: Berlin/Heidelberg, Germany, 2023; pp. 87–114. [Google Scholar]
- Sembiring, H.; Subekti, N.A.; Erythrina; Nugraha, D.; Priatmojo, B.; Stuart, A.M. Yield gap management under seawater intrusion areas of Indonesia to improve rice productivity and resilience to climate change. Agriculture 2020, 10, 1. [Google Scholar] [CrossRef]
- Różewicz, M.; Wyzińska, M.; Grabiński, J. The most important fungal diseases of cereals—Problems and possible solutions. Agronomy 2021, 11, 714. [Google Scholar] [CrossRef]
- Zakaria, L. Diversity of Colletotrichum species associated with anthracnose disease in tropical fruit crops—A review. Agriculture 2021, 11, 297. [Google Scholar] [CrossRef]
- Talhinhas, P.; Loureiro, A.; Oliveira, H. Olive anthracnose: A yield- and oil quality-degrading disease caused by several species of Colletotrichum that differ in virulence, host preference and geographical distribution. Mol. Plant Pathol. 2018, 19, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Prasad, D.; Singh, V.P. Evaluation of fungicides and genotypes against anthracnose disease of mungbean caused by Colletotrichum lindemuthianum. Int. J. Bio-Resour. Stress Manag. 2022, 13, 448–453. [Google Scholar] [CrossRef]
- Gruzdeviene, E.; Dabkevicius, Z. The control of flax anthracnose [Colletotrichum lini [West.] Toch.] by fungicidal seed treatment. J. Plant Prot. Res. 2003, 43, 205–212. [Google Scholar]
- Nyvall, R.F. Diseases of flax. In Field Crop Diseases Handbook; Nyvall, R.F., Ed.; Springer: Boston, MA, USA, 1989; pp. 251–264. [Google Scholar]
- De Silva, D.D.; Crous, P.W.; Ades, P.K.; Hyde, K.D.; Taylor, P.W.J. Life styles of Colletotrichum species and implications for plant biosecurity. Fungal Biol. Rev. 2017, 31, 155–168. [Google Scholar] [CrossRef]
- Münch, S.; Lingner, U.; Floss, D.S.; Ludwig, N.; Sauer, N.; Deising, H.B. The hemibiotrophic lifestyle of Colletotrichum species. J. Plant Physiol. 2008, 165, 41–51. [Google Scholar] [CrossRef]
- da Silva, L.L.; Moreno, H.L.A.; Correia, H.L.N.; Santana, M.F.; de Queiroz, M.V. Colletotrichum: Species complexes, lifestyle, and peculiarities of some sources of genetic variability. Appl. Microbiol. Biotechnol. 2020, 104, 1891–1904. [Google Scholar] [CrossRef] [PubMed]
- Manamgoda, D.S.; Udayanga, D.; Cai, L.; Chukeatirote, E.; Hyde, K.D. Endophytic Colletotrichum from tropical grasses with a new species C. endophytica. Fungal Divers. 2013, 61, 107–115. [Google Scholar] [CrossRef]
- Zheng, H.; Yu, Z.; Jiang, X.; Fang, L.; Qiao, M. Endophytic Colletotrichum species from aquatic plants in southwest China. J. Fungi 2022, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, R.; Padhi, S.; Rai, A.K.; Masi, M.; Evidente, A.; Jha, D.K.; Cimmino, A.; Tayung, K. Isolation and characterization of an endophytic fungus Colletotrichum coccodes producing tyrosol from Houttuynia cordata Thunb. using ITS2 RNA secondary structure and molecular docking study. Front. Bioeng. Biotechnol. 2021, 9, 650247. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Shim, S.H. The fungus Colletotrichum as a source for bioactive secondary metabolites. Arch. Pharmacal Res. 2019, 42, 735–753. [Google Scholar] [CrossRef] [PubMed]
- Vieira, W.; Bezerra, P.A.; Silva, A.C.D.; Veloso, J.S.; Camara, M.P.S.; Doyle, V.P. Optimal markers for the identification of Colletotrichum species. Mol. Phylogenetics Evol. 2020, 143, 106694. [Google Scholar] [CrossRef] [PubMed]
- Bhunjun, C.S.; Phukhamsakda, C.; Jayawardena, R.S.; Jeewon, R.; Promputtha, I.; Hyde, K.D. Investigating species boundaries in Colletotrichum. Fungal Divers. 2021, 107, 107–127. [Google Scholar] [CrossRef]
- Van Hemelrijck, W.; Debode, J.; Heungens, K.; Maes, M.; Creemers, P. Phenotypic and genetic characterization of Colletotrichum isolates from Belgian strawberry fields. Plant Pathol. 2010, 59, 853–861. [Google Scholar] [CrossRef]
- Liu, F.; Ma, Z.Y.; Hou, L.W.; Diao, Y.Z.; Wu, W.P.; Damm, U.; Song, S.; Cai, L. Updating species diversity of Colletotrichum, with a phylogenomic overview. Stud. Mycol. 2022, 101, 1–56. [Google Scholar] [CrossRef]
- Crouch, J.; O’Connell, R.; Gan, P.; Buiate, E.; Torres, M.F.; Beirn, L.; Shirasu, K.; Vaillancourt, L. The genomics of Colletotrichum. In Genomics of Plant-Associated Fungi: Monocot Pathogens; Dean, R.A., Lichens-Park, A., Kole, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 69–102. [Google Scholar]
- Gardiner, D.M.; McDonald, M.C.; Covarelli, L.; Solomon, P.S.; Rusu, A.G.; Marshall, M.; Kazan, K.; Chakraborty, S.; McDonald, B.A.; Manners, J.M. Comparative pathogenomics reveals horizontally acquired novel virulence genes in fungi infecting cereal hosts. PLoS Pathog. 2012, 8, e1002952. [Google Scholar] [CrossRef]
- Lelwala, R.V.; Korhonen, P.K.; Young, N.D.; Scott, J.B.; Ades, P.K.; Gasser, R.B.; Taylor, P.W.J. Comparative genome analysis indicates high evolutionary potential of pathogenicity genes in Colletotrichum tanaceti. PLoS ONE 2019, 14, e0212248. [Google Scholar] [CrossRef]
- Wang, H.; Huang, R.; Ren, J.; Tang, L.; Huang, S.; Chen, X.; Fan, J.; Li, B.; Wang, Q.; Hsiang, T.; et al. The evolution of mini-chromosomes in the fungal genus Colletotrichum. mBio 2023, 14, e00629-23. [Google Scholar] [CrossRef]
- Krasnov, G.S.; Pushkova, E.N.; Novakovskiy, R.O.; Kudryavtseva, L.P.; Rozhmina, T.A.; Dvorianinova, E.M.; Povkhova, L.V.; Kudryavtseva, A.V.; Dmitriev, A.A.; Melnikova, N.V. High-quality genome assembly of Fusarium oxysporum f. sp. lini. Front. Genet. 2020, 11, 959. [Google Scholar] [CrossRef] [PubMed]
- Sigova, E.A.; Pushkova, E.N.; Rozhmina, T.A.; Kudryavtseva, L.P.; Zhuchenko, A.A.; Novakovskiy, R.O.; Zhernova, D.A.; Povkhova, L.V.; Turba, A.A.; Borkhert, E.V.; et al. Assembling quality genomes of flax fungal pathogens from Oxford Nanopore Technologies data. J. Fungi 2023, 9, 301. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Coombe, L.; Nikolić, V.; Zhang, E.; Nip, K.M.; Sidhu, P.; Warren, R.L.; Birol, I. Linear time complexity de novo long read genome assembly with GoldRush. Nat. Commun. 2023, 14, 2906. [Google Scholar] [CrossRef]
- Haghshenas, E.; Asghari, H.; Stoye, J.; Chauve, C.; Hach, F. HASLR: Fast Hybrid Assembly of Long Reads. iScience 2020, 23, 101389. [Google Scholar] [CrossRef]
- Zimin, A.V.; Puiu, D.; Luo, M.C.; Zhu, T.; Koren, S.; Marcais, G.; Yorke, J.A.; Dvorak, J.; Salzberg, S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 2017, 27, 787–792. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Salzberg, S.L. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLoS Comput. Biol. 2020, 16, e1007981. [Google Scholar] [CrossRef]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant-Microbe Interact. MPMI 2022, 35, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.; Hiroyama, R.; Tsushima, A.; Masuda, S.; Shibata, A.; Ueno, A.; Kumakura, N.; Narusaka, M.; Hoat, T.X.; Narusaka, Y.; et al. Telomeres and a repeat-rich chromosome encode effector gene clusters in plant pathogenic Colletotrichum fungi. Environ. Microbiol. 2021, 23, 6004–6018. [Google Scholar] [CrossRef] [PubMed]
- Taga, M.; Tanaka, K.; Kato, S.; Kubo, Y. Cytological analyses of the karyotypes and chromosomes of three Colletotrichum species, C. orbiculare, C. graminicola and C. higginsianum. Fungal Genet. Biol. 2015, 82, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.; Wang, Y.; Hao, Y.; Yao, Y.; Wang, Y.; Zhang, K.; Tan, X.; Li, Z.; Wang, W. Genome sequence resource for Colletotrichum scovillei, the cause of anthracnose disease of chili. Mol. Plant-Microbe Interact. MPMI 2021, 34, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Cao, M.; Li, S.; Kong, Y.; Rollins, J.A.; Zhang, R.; Sun, G. Highly contiguous genome resource of Colletotrichum fructicola generated using long-read sequencing. Mol. Plant-Microbe Interact. MPMI 2020, 33, 790–793. [Google Scholar] [CrossRef]
- O’Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N.; et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012, 44, 1060–1065. [Google Scholar] [CrossRef]
- Gan, P.; Ikeda, K.; Irieda, H.; Narusaka, M.; O’Connell, R.J.; Narusaka, Y.; Takano, Y.; Kubo, Y.; Shirasu, K. Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. New Phytol. 2013, 197, 1236–1249. [Google Scholar] [CrossRef]
- Gan, P.; Narusaka, M.; Kumakura, N.; Tsushima, A.; Takano, Y.; Narusaka, Y.; Shirasu, K. Genus-wide comparative genome analyses of Colletotrichum species reveal specific gene family losses and gains during adaptation to specific infection lifestyles. Genome Biol. Evol. 2016, 8, 1467–1481. [Google Scholar] [CrossRef]
- Gan, P.; Narusaka, M.; Tsushima, A.; Narusaka, Y.; Takano, Y.; Shirasu, K. Draft genome assembly of Colletotrichum chlorophyti, a pathogen of herbaceous plants. Genome Announc. 2017, 5, 10–1128. [Google Scholar] [CrossRef]
- Gan, P.; Tsushima, A.; Narusaka, M.; Narusaka, Y.; Takano, Y.; Kubo, Y.; Shirasu, K. Genome sequence resources for four phytopathogenic fungi from the Colletotrichum orbiculare species complex. Mol. Plant-Microbe Interact. MPMI 2019, 32, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wang, B.; Dong, Q.; Li, L.; Rollins, J.A.; Zhang, R.; Sun, G. Pathogenic adaptations of Colletotrichum fungi revealed by genome wide gene family evolutionary analyses. PLoS ONE 2018, 13, e0196303. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, R.; Bhunjun, C.S.; Gentekaki, E.; Hyde, K.; Promputtha, I. Colletotrichum: Lifestyles, biology, morpho-species, species complexes and accepted species. Mycosphere 2021, 12, 519–669. [Google Scholar] [CrossRef]
- Yan, Y.; Yuan, Q.; Tang, J.; Huang, J.; Hsiang, T.; Wei, Y.; Zheng, L. Colletotrichum higginsianum as a model for understanding host–pathogen interactions: A review. Int. J. Mol. Sci. 2018, 19, 2142. [Google Scholar] [CrossRef]
- Abreha, K.B.; Ortiz, R.; Carlsson, A.S.; Geleta, M. Understanding the sorghum-Colletotrichum sublineola interactions for enhanced host resistance. Front. Plant Sci. 2021, 12, 641969. [Google Scholar] [CrossRef]
- Boufleur, T.R.; Ciampi-Guillardi, M.; Tikami, I.; Rogerio, F.; Thon, M.R.; Sukno, S.A.; Massola Junior, N.S.; Baroncelli, R. Soybean anthracnose caused by Colletotrichum species: Current status and future prospects. Mol. Plant Pathol. 2021, 22, 393–409. [Google Scholar] [CrossRef]
- Dallery, J.F.; Lapalu, N.; Zampounis, A.; Pigne, S.; Luyten, I.; Amselem, J.; Wittenberg, A.H.J.; Zhou, S.; de Queiroz, M.V.; Robin, G.P.; et al. Gapless genome assembly of Colletotrichum higginsianum reveals chromosome structure and association of transposable elements with secondary metabolite gene clusters. BMC Genom. 2017, 18, 667. [Google Scholar] [CrossRef]
- Plaumann, P.L.; Schmidpeter, J.; Dahl, M.; Taher, L.; Koch, C. A dispensable chromosome is required for virulence in the hemibiotrophic plant pathogen Colletotrichum higginsianum. Front. Microbiol. 2018, 9, 1005. [Google Scholar] [CrossRef]
- Bhadauria, V.; MacLachlan, R.; Pozniak, C.; Cohen-Skalie, A.; Li, L.; Halliday, J.; Banniza, S. Genetic map-guided genome assembly reveals a virulence-governing minichromosome in the lentil anthracnose pathogen Colletotrichum lentis. New Phytol. 2019, 221, 431–445. [Google Scholar] [CrossRef]
- Novakovskiy, R.O.; Dvorianinova, E.M.; Rozhmina, T.A.; Kudryavtseva, L.P.; Gryzunov, A.A.; Pushkova, E.N.; Povkhova, L.V.; Snezhkina, A.V.; Krasnov, G.S.; Kudryavtseva, A.V.; et al. Data on genetic polymorphism of flax (Linum usitatissimum L.) pathogenic fungi of Fusarium, Colletotrichum, Aureobasidium, Septoria, and Melampsora genera. Data Brief 2020, 31, 105710. [Google Scholar] [CrossRef]
- Latunde-Dada, A.O.; Lucas, J.A. Localized hemibiotrophy in Colletotrichum: Cytological and molecular taxonomic similarities among C. destructivum, C. linicola and C. truncatum. Plant Pathol. 2007, 56, 437–447. [Google Scholar] [CrossRef]
- Dvorianinova, E.M.; Pushkova, E.N.; Novakovskiy, R.O.; Povkhova, L.V.; Bolsheva, N.L.; Kudryavtseva, L.P.; Rozhmina, T.A.; Melnikova, N.V.; Dmitriev, A.A. Nanopore and Illumina genome sequencing of Fusarium oxysporum f. sp. lini strains of different virulence. Front. Genet. 2021, 12, 662928. [Google Scholar] [CrossRef] [PubMed]
- Becerra, S.; Baroncelli, R.; Boufleur, T.R.; Sukno, S.A.; Thon, M.R. Chromosome-level analysis of the Colletotrichum graminicola genome reveals the unique characteristics of core and minichromosomes. Front. Microbiol. 2023, 14, 1129319. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wang, Y.; McDowell, J. Focus on effector-triggered susceptibility. Mol. Plant-Microbe Interact. 2018, 31, 5. [Google Scholar] [CrossRef]
- Hsieh, D.-K.; Chuang, S.-C.; Chen, C.-Y.; Chao, Y.-T.; Lu, M.-Y.J.; Lee, M.-H.; Shih, M.-C. Comparative genomics of three Colletotrichum scovillei strains and genetic analysis revealed genes involved in fungal growth and virulence on chili pepper. Front. Microbiol. 2022, 13, 818291. [Google Scholar] [CrossRef]
- Cui, H.; Tsuda, K.; Parker, J.E. Effector-triggered immunity: From pathogen perception to robust defense. Annu. Rev. Plant Biol. 2015, 66, 487–511. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dvorianinova, E.M.; Sigova, E.A.; Mollaev, T.D.; Rozhmina, T.A.; Kudryavtseva, L.P.; Novakovskiy, R.O.; Turba, A.A.; Zhernova, D.A.; Borkhert, E.V.; Pushkova, E.N.; et al. Comparative Genomic Analysis of Colletotrichum lini Strains with Different Virulence on Flax. J. Fungi 2024, 10, 32. https://doi.org/10.3390/jof10010032
Dvorianinova EM, Sigova EA, Mollaev TD, Rozhmina TA, Kudryavtseva LP, Novakovskiy RO, Turba AA, Zhernova DA, Borkhert EV, Pushkova EN, et al. Comparative Genomic Analysis of Colletotrichum lini Strains with Different Virulence on Flax. Journal of Fungi. 2024; 10(1):32. https://doi.org/10.3390/jof10010032
Chicago/Turabian StyleDvorianinova, Ekaterina M., Elizaveta A. Sigova, Timur D. Mollaev, Tatiana A. Rozhmina, Ludmila P. Kudryavtseva, Roman O. Novakovskiy, Anastasia A. Turba, Daiana A. Zhernova, Elena V. Borkhert, Elena N. Pushkova, and et al. 2024. "Comparative Genomic Analysis of Colletotrichum lini Strains with Different Virulence on Flax" Journal of Fungi 10, no. 1: 32. https://doi.org/10.3390/jof10010032
APA StyleDvorianinova, E. M., Sigova, E. A., Mollaev, T. D., Rozhmina, T. A., Kudryavtseva, L. P., Novakovskiy, R. O., Turba, A. A., Zhernova, D. A., Borkhert, E. V., Pushkova, E. N., Melnikova, N. V., & Dmitriev, A. A. (2024). Comparative Genomic Analysis of Colletotrichum lini Strains with Different Virulence on Flax. Journal of Fungi, 10(1), 32. https://doi.org/10.3390/jof10010032