
A Rare Case of Polysplenia Syndrome Associated with Severe Cardiac Malformations and Congenital Alveolar Dysplasia in a One-Month-Old Infant: A Complete Macroscopic and Histopathologic Study

 ,
,  , , ,
, , ,
Abstract
1. Introduction
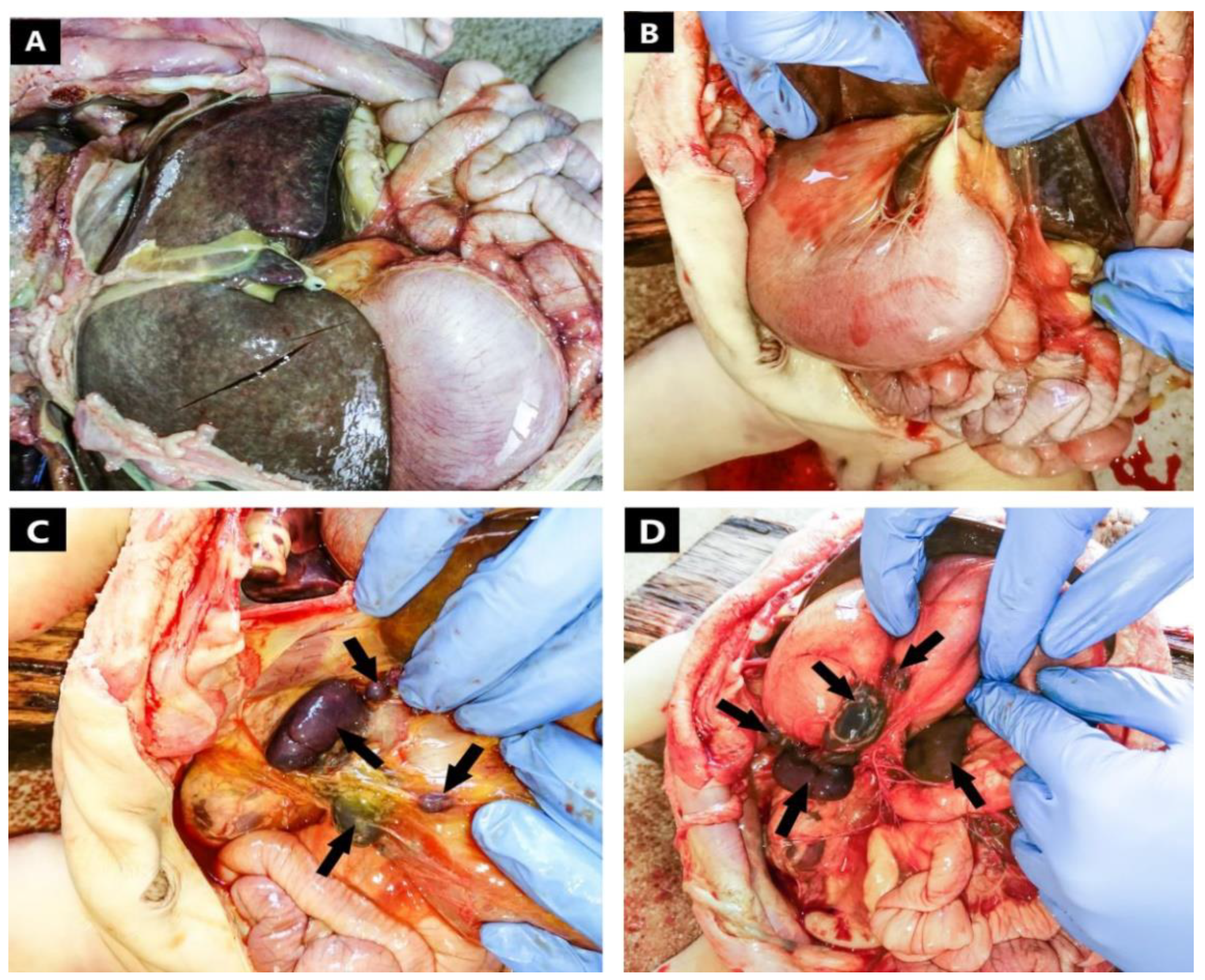
2. Detailed Case Description
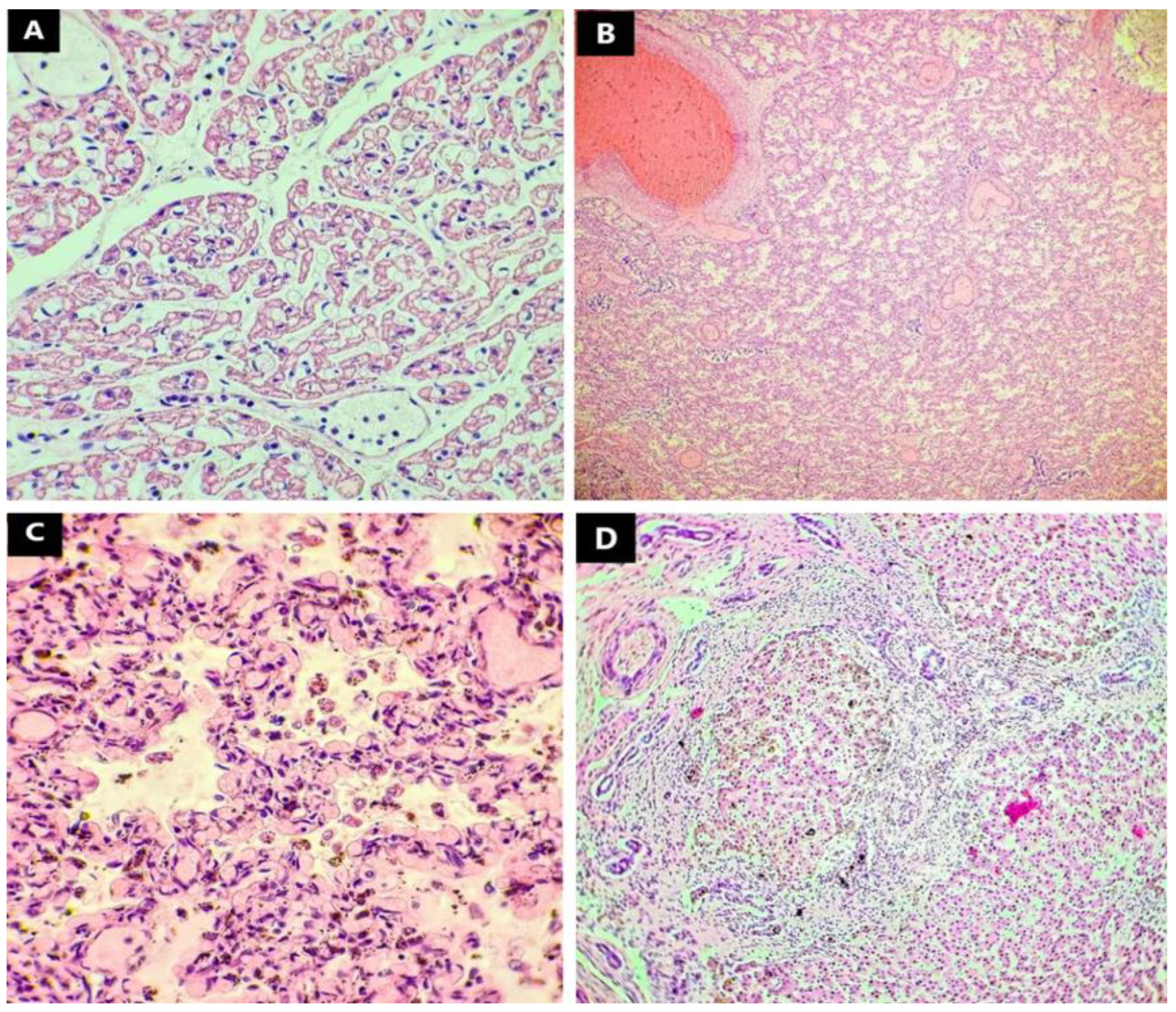
3. Results
4. Discussions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamada, H. Molecular and cellular basis of left-right asymmetry in vertebrates. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2020, 96, 273–296. [Google Scholar] [CrossRef]
- Karakaya, C.; Goktas, S.; Celik, M.; Kowalski, W.J.; Keller, B.B.; Pekkan, K. Asymmetry in Mechanosensitive Gene Expression during Aortic Arch Morphogenesis. Sci. Rep. 2018, 8, 16948. [Google Scholar] [CrossRef]
- Morales, A.V.; Acloque, H.; Ocaña, O.H.; de Frutos, C.A.; Gold, V.; Nieto, M.A. Snail genes at the crossroads of symmetric and asymmetric processes in the developing mesoderm. EMBO Rep. 2007, 8, 104–109. [Google Scholar] [CrossRef]
- Murray, S.A.; Gridley, T. Snail family genes are required for left–right asymmetry determination, but not neural crest formation, in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 10300–10304. [Google Scholar] [CrossRef]
- Blue, G.M.; Mekel, M.; Das, D.; Troup, M.; Rath, E.; Ip, E.; Gudkov, M.; Perumal, G.; Harvey, R.P.; Sholler, G.F.; et al. Whole genome sequencing in transposition of the great arteries and associations with clinically relevant heart, brain and laterality genes. Am. Heart J. 2022, 244, 1–13. [Google Scholar] [CrossRef]
- Armes, J.E.; Mifsud, W.; Ashworth, M. Diffuse lung disease of infancy: A pattern-based, algorithmic approach to histological diagnosis. J. Clin. Pathol. 2015, 68, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Friederich, M.W.; Erdogan, A.J.; Coughlin, C.R.; Elos, M.T.; Jiang, H.; O’Rourke, C.P.; Lovell, M.A.; Wartchow, E.; Gowan, K.; Chatfield, K.C.; et al. Mutations in the accessory subunit NDUFB10 result in isolated complex I deficiency and illustrate the critical role of intermembrane space import for complex I holoenzyme assembly. Hum. Mol. Genet. 2017, 26, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.C.; Scheimberg, I. The Pediatric and Perinatal Autopsy Manual; Cambridge University Press: Cambridge, UK, 2014; ISBN 978-1-107-64148-8. [Google Scholar]
- Berg, C.; Geipel, A.; Smrcek, J.; Krapp, M.; Germer, U.; Kohl, T.; Gembruch, U.; Baschat, A.A. Prenatal diagnosis of cardiosplenic syndromes: A 10-year experience. Ultrasound Obstet. Gynecol. 2003, 22, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Mori, H.; Kodo, K.; Nakanishi, T.; Yamagishi, H. Polysplenia syndrome as a risk factor for early progression of pulmonary hypertension. Circ. J. 2019, 83, 831–836. [Google Scholar] [CrossRef]
- Rose, V.; Izukawa, T.; Moes, C.A.F. Syndromes of asplenia and polysplenia. A review of cardiac and non-cardiac malformations in 60 cases with special reference to diagnosis and prognosis. Br. Heart J. 1975, 37, 840–852. [Google Scholar] [CrossRef]
- Digilio, M.C.; Versacci, P.; Lepri, F.; Baban, A.; Dallapiccola, B.; Marino, B. Atrioventricular canal defectand associated genetic disorders: New insights into polydactyly syndromes. Cardiogenetics 2011, 1, e7. [Google Scholar] [CrossRef]
- Peoples, W.M.; Moller, J.H.; Edwards, J.E. Polysplenia: A review of 146 cases. Pediatr. Cardiol. 1983, 4, 129–137. [Google Scholar] [CrossRef]
- Lim, H.G.; Bacha, E.A.; Marx, G.R.; Marshall, A.; Fynn-Thompson, F.; Mayer, J.E.; Del Nido, P.; Pigula, F.A. Biventricular repair in patients with heterotaxy syndrome. Congenit. Heart Dis. 2009, 137, 371–379. [Google Scholar] [CrossRef]
- Moller, J.H.; Nakib, A.; Anderson, R.C.; Edwards, J.E. Congenital Cardiac Disease Associated with Polysplenia: A Developmental Complex of Bilateral “Left-Sidedness”. Circulation 1967, 36, 789–799. [Google Scholar] [CrossRef]
- Arai, H.; Harada, K.; Tamura, M.; Okamura, T.; Takada, G. Polysplenia syndrome with common atrioventricular canal and persistent truncus arteriosus. Tohoku J. Exp. Med. 1995, 177, 171–177. [Google Scholar] [CrossRef][Green Version]
- Gumbiner, C.H.; McManus, B.M.; Latson, L.A. Associated occurrence of persistent truncus arteriosus and asplenia. Pediatr. Cardiol. 1991, 12, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Bass, J.E.; Redwine, M.D.; Kramer, L.A.; Huynh, P.T.; Harris, J.H. Spectrum of Congenital Anomalies of the Inferior Vena Cava: Cross-Sectional Imaging Findings. Radiographics 2000, 20, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.D.; Martins, I. Congenital systemic venous return anomalies to the right atrium review. Insights Imaging 2019, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Rameshbabu, C.S.; Gupta, K.K.; Qasim, M.; Gupta, O.P. Heterotaxy Polysplenia Syndrome in an Adult with Unique Vascular Anomalies: Case Report with Review of Literature. J. Radiol. Case Rep. 2015, 9, 22–37. [Google Scholar] [CrossRef]
- Jo, D.S.; Jung, S.S.; Joo, C.U. A case of unusual visceral heterotaxy syndrome with isolated levocardia. Korean Circ. J. 2013, 43, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Hartog, H.; Mirza, D.F.; Perera, M.T.P.R. Heterotaxy Syndrome with Malrotation of the Gut and Interrupted Vena Cava Does Not Preclude Safe Procurement of Multivisceral Graft. Am. J. Transplant. 2014, 14, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Grubin, J.; Salvatore, M.; Kaur, S. Case Review: Heterotaxy syndrome with polysplenia. Appl. Radiol. 2015. Available online: https://www.appliedradiology.com/articles/case-review-heterotaxy-syndrome-with-polysplenia (accessed on 29 January 2022).
- El Mortaji, H.; Elatiqi, K.; El Hammaoui, H.; Alj, S. Polysplenia syndrome with situs ambiguous, common mesentery, and IVC interruption discovered incidentally in an adult. Radiol. Case Rep. 2019, 14, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.; Bacha, D. Embryology, Pulmonary; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK544372/ (accessed on 24 January 2021).
- Nandyal, R.; Parham, D.; Yu, Z.; Escobedo, M. Congenital Alveolar Capillary Dysplasia and New Associations: A case series with a report of new associations and literature review. Med. Rep. Case Stud. 2017, 2, 1000129. [Google Scholar]
- Bishop, N.B.; Stankiewicz, P.; Steinhorn, R.H. Alveolar capillary dysplasia. Am. J. Respir. Crit. Care Med. 2011, 184, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Belmont, J.W.; Mohapatra, B.; Towbin, J.A.; Ware, S.M. Molecular genetics of heterotaxy syndromes. Curr. Opin. Cardiol. 2004, 19, 216–220. [Google Scholar] [CrossRef]
- Shiraishi, I.; Ichikawa, H. Human heterotaxy syndrome—From molecular genetics to clinical features, management and prognosis. Circ. J. 2012, 76, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Bamford, R.; Roessler, E.; Burdine, R.; Şaplakoğlu, U.; dela Cruz, J.; Splitt, M.; Towbin, J.; Bowers, P.; Marino, B.; Schier, A.F.; et al. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nat. Genet. 2000, 26, 365–369. [Google Scholar] [CrossRef]
- Loomba, R.S.; Frommelt, P.C.; Anderson, R.H. Genetic Disturbances in Patients with Bodily Isomerism from a Single Center: Clinical Implications of Affected Genes and Potential Impact of Ciliary Dyskinesia. Cardiogenetics 2016, 6, 5818. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Organ/System | Macroscopic Findings | Microscopic Findings |
|---|---|---|
| Cardiovascular system | Common atrium | Infantile lactic acidosis |
| Complete atrio-ventricular canal defect | ||
| Left ventricle hypertrophy | ||
| Right ventricle hypoplasia | ||
| Common arterial trunk | ||
| Superior vena cava duplication | ||
| Pulmonary system | Bilateral left-sideness of the pulmonary system (bilobation of the lungs) | Congenital alveolar dysplasia |
| Hepatobiliary system and pancreas | Midline liver | Hepatic fibrosis |
| Symmetrical hepatic lobes | ||
| Gall bladder agenesis | ||
| Inversion of the pancreas | ||
| Digestive tract and spleen | Right-sided stomach | - |
| Complete inversion of the intestines | ||
| Multiple right-sided spleens | ||
| Uro-genital system | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohor, C.I.; Fleaca, S.R.; Oprinca Muja, A.; Oprinca, G.C.; Roman, M.D.; Chicea, R.; Boicean, A.G.; Dura, H.; Tanasescu, C.; Ion, N.C.I.; et al. A Rare Case of Polysplenia Syndrome Associated with Severe Cardiac Malformations and Congenital Alveolar Dysplasia in a One-Month-Old Infant: A Complete Macroscopic and Histopathologic Study. J. Cardiovasc. Dev. Dis. 2022, 9, 135. https://doi.org/10.3390/jcdd9050135
Mohor CI, Fleaca SR, Oprinca Muja A, Oprinca GC, Roman MD, Chicea R, Boicean AG, Dura H, Tanasescu C, Ion NCI, et al. A Rare Case of Polysplenia Syndrome Associated with Severe Cardiac Malformations and Congenital Alveolar Dysplasia in a One-Month-Old Infant: A Complete Macroscopic and Histopathologic Study. Journal of Cardiovascular Development and Disease. 2022; 9(5):135. https://doi.org/10.3390/jcdd9050135
Chicago/Turabian StyleMohor, Cosmin Ioan, Sorin Radu Fleaca, Alexandra Oprinca Muja, George Calin Oprinca, Mihai Dan Roman, Radu Chicea, Adrian Gheorghe Boicean, Horatiu Dura, Ciprian Tanasescu, Nicolas Catalin Ionut Ion, and et al. 2022. "A Rare Case of Polysplenia Syndrome Associated with Severe Cardiac Malformations and Congenital Alveolar Dysplasia in a One-Month-Old Infant: A Complete Macroscopic and Histopathologic Study" Journal of Cardiovascular Development and Disease 9, no. 5: 135. https://doi.org/10.3390/jcdd9050135
APA StyleMohor, C. I., Fleaca, S. R., Oprinca Muja, A., Oprinca, G. C., Roman, M. D., Chicea, R., Boicean, A. G., Dura, H., Tanasescu, C., Ion, N. C. I., Faur, M., Bacila, C. I., Batar, F., & Mohor, C. I. (2022). A Rare Case of Polysplenia Syndrome Associated with Severe Cardiac Malformations and Congenital Alveolar Dysplasia in a One-Month-Old Infant: A Complete Macroscopic and Histopathologic Study. Journal of Cardiovascular Development and Disease, 9(5), 135. https://doi.org/10.3390/jcdd9050135