
Myocardial Afterload Is a Key Biomechanical Regulator of Atrioventricular Myocyte Differentiation in Zebrafish


 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Zebrafish Husbandry and Vasopressin Application
2.2. Live Imaging
2.3. Quantification of Cardiac Functional Parameters
2.4. Immunohistochemistry
2.5. Reverse Transcriptase PCR
3. Results
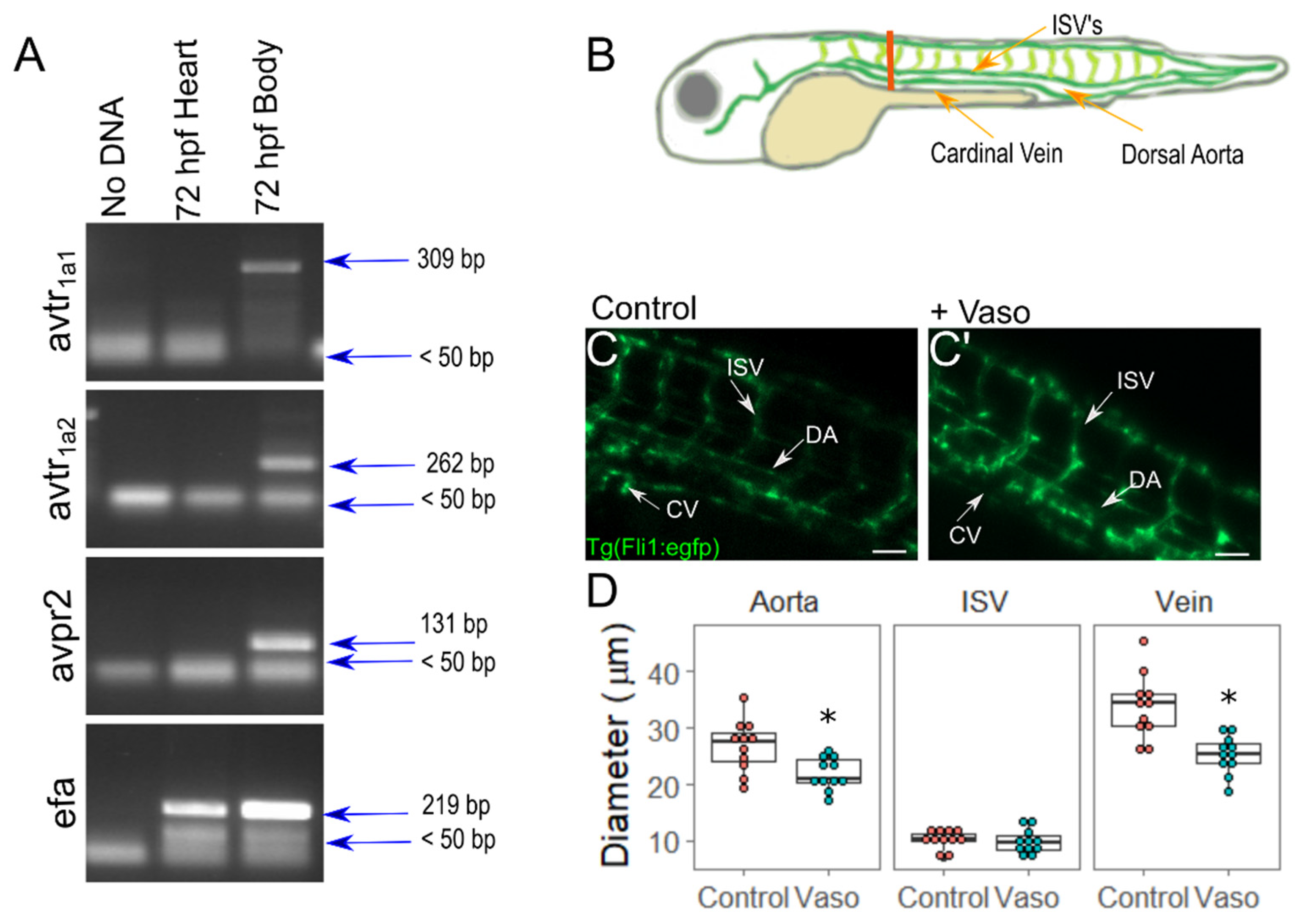
3.1. Vasopressin Actively Elicits Vasoconstriction in Zebrafish Embryos
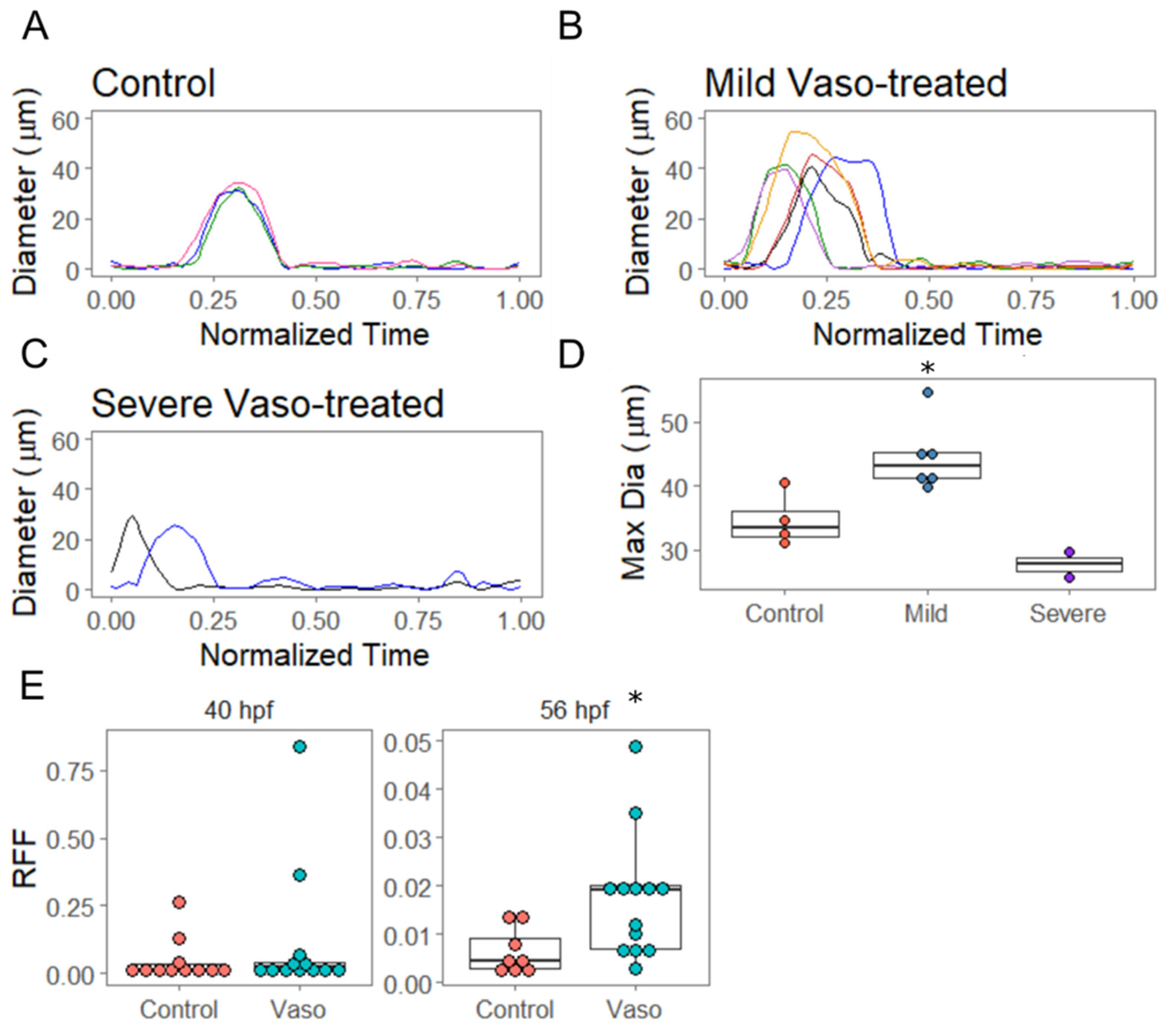
3.2. Increased Afterload Alters Cardiac Function and Growth
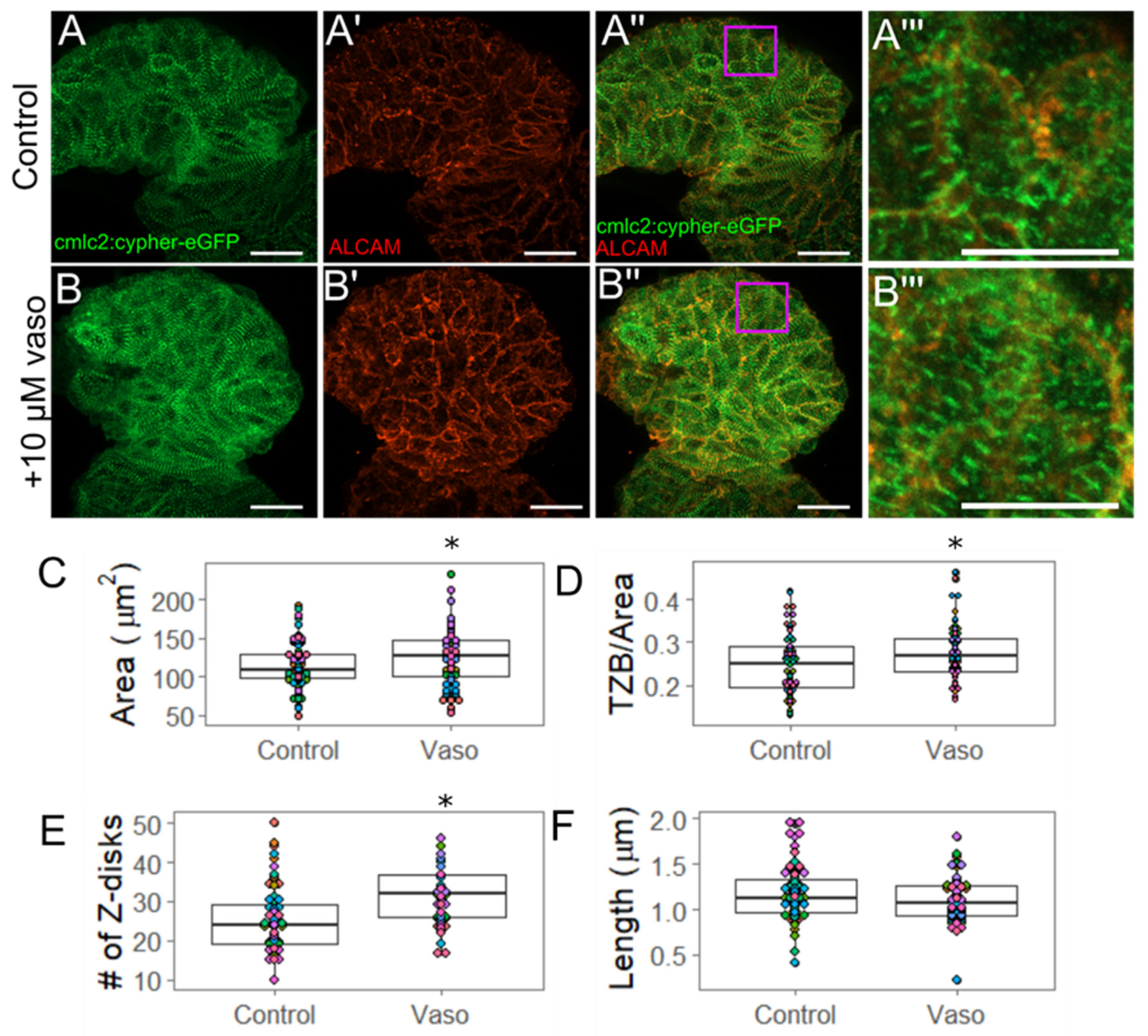
3.3. Increased Afterload Induces Cardiac Hypertrophy and Alters Sarcomere Abundance
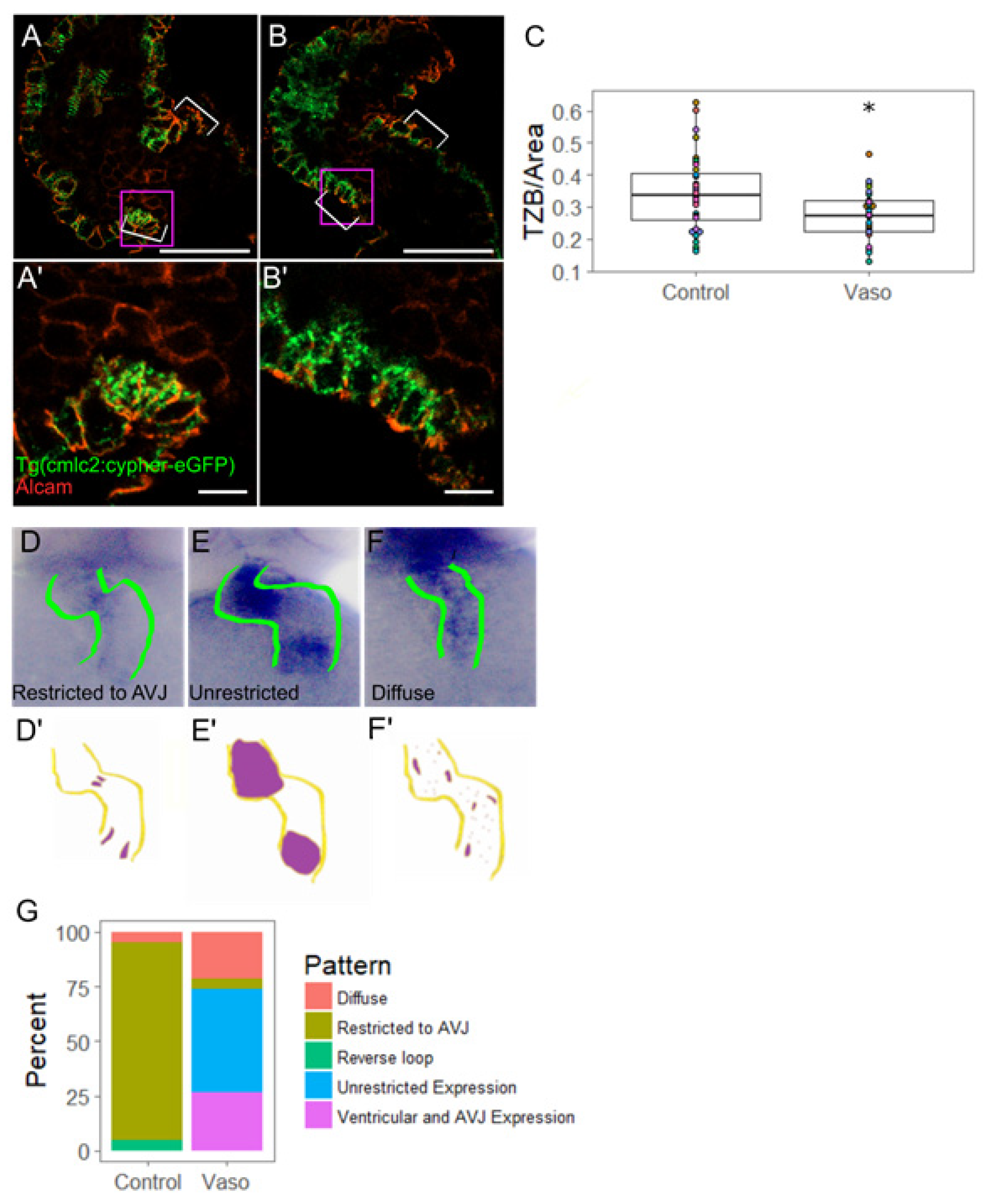
3.4. Increased Afterload Induces Retrograde Flow and Alters AVJ Mechanics
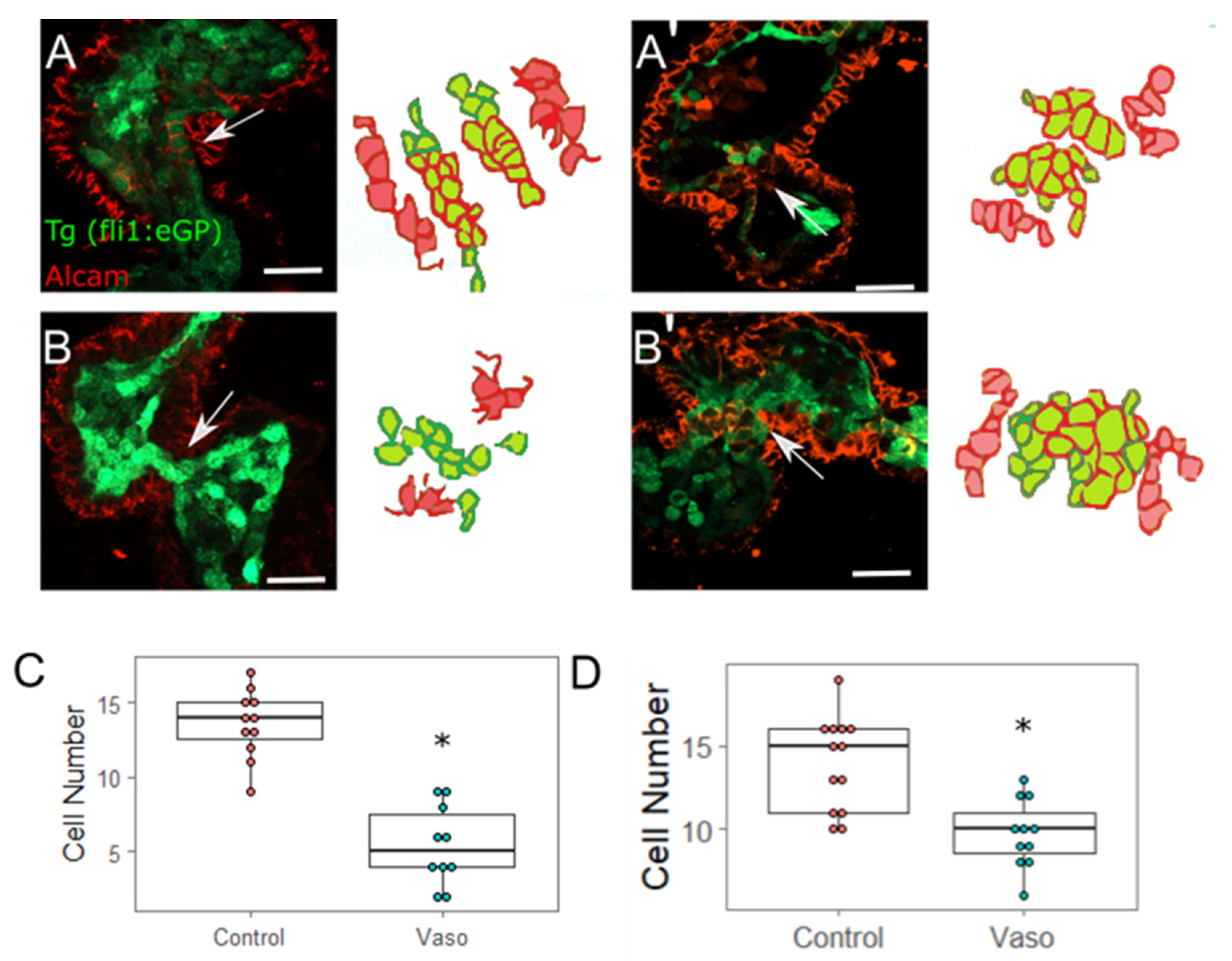
3.5. Increased Afterload Alters Myocardial AVJ Cell Specification
3.6. Increased Afterload Alters Endocardial AVJ Cell Specification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Der Linde, D.; Konings, E.; Slager, M.; Witsenburg, M.; Helbing, W.; Takkenberg, J.; Roos-Hesselinkm, J. Birth prevalence of congenital heart disease worldwide: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.; Kaplan, S. The Incidence of congential heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef]
- Blue, G.M.; Kirk, E.P.; Sholler, G.F.; Harvey, R.P.; Winlaw, D.S. Congenital heart disease: Current knowledge about causes and inheritance. Med. J. Aust. 2012, 197, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Buja, L.M.; Butany, J. Cardiovascular Pathology; Academic Press: San Diego, CA USA, 2015. [Google Scholar]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of Congenital Heart Disease: The Glass Half Empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef]
- Boselli, F.; Freund, J.B.; Vermot, J. Blood flow mechanics in cardiovascular development. Cell. Mol. Life Sci. 2015, 72, 2545–2559. [Google Scholar] [CrossRef]
- Haack, T.; Abdelilah-Seyfried, S. The force within: Endocardial development, mechanotransduction and signalling during cardiac morphogenesis. Development 2016, 143, 373–386. [Google Scholar] [CrossRef]
- Mongkoldhumrongkul, N.; Latif, N.; Yacoub, M.H.; Chester, A.H. Effect of Side-Specific Valvular Shear Stress on the Content of Extracellular Matrix in Aortic Valves. Cardiovasc. Eng. Technol. 2016, 9, 151–157. [Google Scholar] [CrossRef]
- Pappano, A.J.; Wier, W.G. 10—Control of Cardiac Output: Coupling of Heart and Blood Vessels. In Cardiovascular Physiology, 10th ed.; Elsevier: Philadelphia, PA, USA, 2013; pp. 195–222. [Google Scholar]
- Davey, B.; Szwast, A.; Rychik, J. Diagnosis and management of heart failure in the fetus. Minerva Pediatr. 2012, 64, 471–492. [Google Scholar] [PubMed]
- Verburg, B.O.; Jaddoe, V.W.; Wladimiroff, J.W.; Hofman, A.; Witteman, J.C.; Steegers, E.A. Fetal hemodynamic adaptive changes related to intrauterine growth: The Generation R Study. Circulation 2008, 117, 649–659. [Google Scholar] [CrossRef]
- Endo, H.; Shiraishi, H.; Yanagisawa, M. Afterload reduction by hydralazine in children with a ventricular septal defect as determined by aortic input impedance. Cardiovasc. Drugs Ther. 1994, 8, 161–166. [Google Scholar] [CrossRef]
- Beekman, R.H.; Rocchini, A.P.; Dick, M., 2nd; Crowley, D.C.; Rosenthal, A. Vasodilator Therapy in Children: Acute and Chronic Effects in Children with Left Ventricular Dysfunction or Mitral Regurgitation. Pediatrics 1984, 73, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Davila, C.D.; Forfia, P.R. Management of Severe Pulmonary Hypertension in Patients Undergoing Mitral Valve Surgery. Curr. Treat. Options Cardiovasc. Med. 2015, 17, 382. [Google Scholar] [CrossRef]
- Hirt, M.N.; Sörensen, N.A.; Bartholdt, L.M.; Boeddinghaus, J.; Schaaf, S.; Eder, A.; Eschenhagen, T. Increased afterload induces pathological cardiac hypertrophy: A new in vitro model. Basic Res. Cardiol. 2012, 107, 307. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Larson, M.G.; Vasan, R.S.; Kannel, W.B.; Ho, K.K. The progression from hypertension to congestive heart failure. JAMA 1996, 275, 1557–1562. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef]
- Midgett, M.; Rugonyi, S. Congenital heart malformations induced by hemodynamic altering surgical interventions. Front. Physiol. 2014, 5, 287. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.L.; Parnall, M.; Loughna, S. Effect of altered haemodynamics on the developing mitral valve in chick embryonic heart. J. Mol. Cell. Cardiol. 2017, 108, 114–126. [Google Scholar] [CrossRef]
- Slough, J.; Cooney, L.; Brueckner, M. Monocilia in the embryonic mouse heart imply a direct role for cilia in cardiac morphogenesis. Dev. Dyn. 2008, 237, 2304–2314. [Google Scholar] [CrossRef]
- Heckel, E.; Boselli, F.; Roth, S.; Krudewig, A.; Belting, H.-G.; Charvin, G.; Vermot, J. Oscillatory Flow Modulates Mechanosensitive klf2a Expression through trpv4 and trpp2 during Heart Valve Development. Curr. Biol. 2015, 25, 1354–1361. [Google Scholar] [CrossRef]
- McCain, M.L.; Parker, K.K. Mechanotransduction: The role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function. Eur. J. Physiol. 2011, 462, 89–104. [Google Scholar] [CrossRef]
- Dietrich, A.C.; Lombardo, V.A.; Veerkamp, J.; Priller, F.; Abdelilah-Seyfried, S. Blood flow and Bmp signaling control endocardial chamber morphogenesis. Dev. Cell 2014, 30, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K.; Taguchi, M.; Bonkowsky, J.L.; Kuwada, J.Y. Expression of Arginine Vasotocin Receptors in the Developing Zebrafish CNS. GEP 2013, 13, 335–342. [Google Scholar] [CrossRef][Green Version]
- Daza, D.O.; Lewicka, M.; Larhammar, D. The Oxytocin/vasopressin Receptor Family Has at Least Five Members in the Gnathostome Lineage, Inclucing Two Distinct V2 Subtypes. Gen. Comp. Endocrinol. 2012, 175, 135–143. [Google Scholar] [CrossRef]
- Holmes, C.L.; Landry, D.W.; Granton, J.T. Science Review: Vasopressin and the cardiovascular system part 2—Clinical physiology. Crit. Care 2004, 8, 15–23. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; Dezonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hartjes, K.; Nelson, T.; Xu, X. Cessation of contraction induces cardiomyocyte remodeling during zebrafish cardiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H382–H395. [Google Scholar] [CrossRef]
- Berndt, C.; Poschmann, G.; Stühler, K.; Holmgren, A.; Bräutigam, L. Zebrafish heart development is regulated via glutaredoxin 2 dependent migration and survival of neural crest cells. Redox. Biol. 2014, 2, 673–678. [Google Scholar] [CrossRef]
- Johnson, B.; Garrity, D.; Dasi, P. The transitional cardiac pumping mechanics in the embryonic heart. Cardiovasc. Eng. Technol. 2013, 4, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, X. Immunostaining of Dissected Zebrafish Embryonic Heart. J. Vis. Exp. 2012, 59, e3510. [Google Scholar] [CrossRef]
- Bulk, A.; Bark, D.; Johnson, B.; Garrity, D.; Dasi, L. Mechanisms influencing retrograde flow in the atrioventricular canal during early embryonic cardiogenesis. J. Biomech. 2016, 49, 3162–3167. [Google Scholar] [CrossRef]
- Maarman, G.; Lecour, S.; Butrous, G.; Thienemann, F.; Sliwa, K.; Sliwa, K. A Comprehensive Review: The Evolution of Animal Models in Pulmonary Hypertension Research; 3.4, Are We There Yet? Pulm. Circ. 2013, 3, 739–756. [Google Scholar] [CrossRef]
- Clark, E.B.; Hu, N.; Frommelt, P.; Vandekieft, G.K.; Dummett, J.L.; Tomanek, R.J. Effect of increased pressure on ventricular growth in stage 21 chick embryos. Am. J. Physiol. Heart Circ. Physiol. 1989, 257, H55–H61. [Google Scholar] [CrossRef]
- Huang, C.J.; Tu, C.T.; Hsiao, C.D.; Hsieh, F.J.; Tsai, H.J. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev. Dyn. 2003, 228, 30–40. [Google Scholar] [CrossRef]
- Lin, Y.F.; Swinburne, I.; Yelon, D. Multiple influences of blood flow on cardiomyocyte hypertrophy in the embryonic zebrafish heart. Dev. Biol. 2012, 362, 242–253. [Google Scholar] [CrossRef]
- Chi, N.C.; Shaw, R.M.; De Val, S.; Kang, G.; Jan, L.Y.; Black, B.L.; Stainier, D.Y. Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev. 2008, 22, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Patra, C.; Diehl, F.; Ferrazzi, F.; van Amerongen, M.J.; Novoyatleva, T.; Schaefer, L.; Mühlfeld, C.; Jungblut, B.; Engel, F.B. Nephronectin regulates atrioventricular canal differentiation via Bmp4-Has2 signaling in zebrafish. Development 2011, 138, 4499–4509. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.N.; van Eeden, F.; Warren, K.; Chin, A.; Nusslein-Volhard, C.; Haffter, P.; Fishman, M. Left-right pattern of cardiac BMP4 may drive asymmetry of the heart in zebrafish. Development 1997, 124, 4373–4382. [Google Scholar] [CrossRef] [PubMed]
- Combs, M.D.; Yutzey, K.E. Heart valve development: Regulatory networks in development and disease. Circ. Res. 2009, 105, 408–421. [Google Scholar] [CrossRef]
- Scherz, P.J.; Huisken, J.; Sahai-Hernandez, P.; Stainier, D.Y. High-speed imaging of developing heart valves reveals interplay of morphogenesis and function. Development 2008, 135, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Steed, E.; Faggianelli, N.; Roth, S.; Ramspacher, C.; Concordet, J.-P.; Vermot, J. Klf2a Couples Mechanotransduction and Zebrafish Valve Morphogenesis through Fibronectin Synthesis. Nat. Commun. 2016, 7, 11646. [Google Scholar] [CrossRef]
- Beis, D.; Bartman, T.; Jin, S.-W.; Scott, I.; D’Amico, L.A.; Ober, E.A.; Verkade, H.; Frantsve, J.; Field, H.A.; Wehman, A.M.; et al. Genetic and Cellular Analyses of Zebrafish Atrioventricular Cushion and Valve Development. Development 2005, 132, 4193–4204. [Google Scholar] [CrossRef]
- Tulloch, N.; Muskheli, V.; Razumova, M.V.; Korte, F.S.; Regnier, M.; Hauch, K.D.; Pabon, L.; Reinecke, H.; Murry, C.E. Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ. Res. 2011, 109, 47–59. [Google Scholar] [CrossRef]
- Tarazi, R.C.; Levy, M.N. Cardiac responses to increased afterload. State-of-the-art review. Hypertension 1982, 4 Pt 2, 8–18. [Google Scholar]
- Machackova, J.; Barta, J.; Dhalla, N.S. Myofibrillar remodeling in cardiac hypertrophy, heart failure and cardiomyopathies. Can. J. Cardiol. 2006, 22, 953–968. [Google Scholar] [CrossRef]
- Vermot, J.; Forouhar, A.; Liebling, M.; Wu, D.; Plummer, D.; Gharib, M.; Fraser, S.E. Reversing Blood Flows Act through klf2a to Ensure Normal Valvulogenesis in the Developing Heart. PLoS Biol. 2009, 7, e1000246. [Google Scholar] [CrossRef] [PubMed]
- Hove, J.R.; Köster, R.W.; Forouhar, A.S.; Acevedo-Bolton, G.; Fraser, S.E.; Gharib, M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 2003, 421, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Boselli, F.; Steed, E.; Freund, J.B.; Vermot, J. Anisotropic shear stress patterns predict the orientation of convergent tissue movements in the embryonic heart. Development 2017, 144, 4322–4327. [Google Scholar] [CrossRef]
- Verhoeven, M.C.; Haase, C.; Christoffels, V.M.; Weidinger, G.; Bakkers, J. Wnt signaling regulates atrioventricular canal formation upstream of BMP and Tbx2. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Hurlstone, A.F.; Haramis, A.-P.G.; Wienholds, E.; Begthel, H.; Korving, J.; Van Eeden, F.; Cuppen, E.; Zivkovic, D.; Plasterk, R.H.A.; Clevers, H. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature 2003, 425, 633–637. [Google Scholar] [CrossRef]
- Frisk, M.; Ruud, M.; Espe, E.K.S.; Aronsen, J.M.; Røe, T.; Zhang, L.; Norseng, P.A.; Sejersted, O.M.; Christensen, G.A.; Sjaastad, I.; et al. Elevated ventricular wall stress disrupts cardiomyocyte t-tubule structure and calcium homeostasis. Cardiovasc. Res. 2016, 112, 443–451. [Google Scholar] [CrossRef]
- Swapna, I.; Borodinsky, L.N. Interplay between electrical activity and bone morphogenetic protein signaling regulates spinal neuron differentiation. Proc. Natl. Acad. Sci. USA 2012, 109, 16336–16341. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahuja, N.; Ostwald, P.; Gendernalik, A.; Guzzolino, E.; Pitto, L.; Bark, D.; Garrity, D.M. Myocardial Afterload Is a Key Biomechanical Regulator of Atrioventricular Myocyte Differentiation in Zebrafish. J. Cardiovasc. Dev. Dis. 2022, 9, 22. https://doi.org/10.3390/jcdd9010022
Ahuja N, Ostwald P, Gendernalik A, Guzzolino E, Pitto L, Bark D, Garrity DM. Myocardial Afterload Is a Key Biomechanical Regulator of Atrioventricular Myocyte Differentiation in Zebrafish. Journal of Cardiovascular Development and Disease. 2022; 9(1):22. https://doi.org/10.3390/jcdd9010022
Chicago/Turabian StyleAhuja, Neha, Paige Ostwald, Alex Gendernalik, Elena Guzzolino, Letizia Pitto, David Bark, and Deborah M. Garrity. 2022. "Myocardial Afterload Is a Key Biomechanical Regulator of Atrioventricular Myocyte Differentiation in Zebrafish" Journal of Cardiovascular Development and Disease 9, no. 1: 22. https://doi.org/10.3390/jcdd9010022
APA StyleAhuja, N., Ostwald, P., Gendernalik, A., Guzzolino, E., Pitto, L., Bark, D., & Garrity, D. M. (2022). Myocardial Afterload Is a Key Biomechanical Regulator of Atrioventricular Myocyte Differentiation in Zebrafish. Journal of Cardiovascular Development and Disease, 9(1), 22. https://doi.org/10.3390/jcdd9010022