Abstract
Background: Left ventricular noncompaction (LVNC) is a cardiomyopathy that can lead to arrhythmias, embolic events and heart failure. Despite our current knowledge of cardiac development, the mechanisms underlying noncompaction of the ventricular myocardium are still poorly understood. The small GTPase Rac1 acts as a crucial regulator of numerous developmental events. The present study aimed to investigate the cardiomyocyte specific role of Rac1 in embryonic heart development. Methods and Results: The Nkx2.5-Cre transgenic mice were crossed with Rac1f/f mice to generate mice with a cardiomyocyte specific deletion of Rac1 (Rac1Nkx2.5) during heart development. Embryonic Rac1Nkx2.5 hearts at E12.5–E18.5 were collected for histological analysis. Overall, Rac1Nkx2.5 hearts displayed a bifid apex, along with hypertrabeculation and a thin compact myocardium. Rac1Nkx2.5 hearts also exhibited ventricular septal defects (VSDs) and double outlet right ventricle (DORV) or overriding aorta. Cardiomyocytes had a rounded morphology and were highly disorganized, and the myocardial expression of Scrib, a planar cell polarity protein, was reduced in Rac1Nkx2.5 hearts. In addition, cell proliferation rate was significantly decreased in the Rac1Nkx2.5 ventricular myocardium at E9.5. Conclusions: Rac1 deficiency in the myocardium impairs cardiomyocyte elongation and organization, and proliferative growth of the heart. A spectrum of CHDs arises in Rac1Nkx2.5 hearts, implicating Rac1 signaling in the ventricular myocardium as a crucial regulator of OFT alignment, along with compact myocardium growth and development.
1. Introduction
Congenital heart defects (CHDs) are the most common human birth defects, affecting up to 5% of live births [1,2]. The severity of these types of defects ranges from simple defects with no symptoms at birth, such as a small atrial septal defect (ASD), to complicated defects that are life-threatening and require intervention, such as Tetralogy of Fallot (TOF) [3,4]. One particular defect known as left ventricular noncompaction (LVNC) or “spongy myocardium” has no treatment at present, with the major therapy being heart failure and anticoagulation medications [5]. LVNC is classified as a rare genetic cardiomyopathy, occurring in 0.01% to 0.27% of the population, characterized by arrest of normal myocardium development, leading to a thin compact myocardial layer and an extensive non-compacted trabecular network [6,7]. Noncompaction of the ventricular myocardium can increase the risk of cardiac embolism, atrial fibrillation, ventricular arrhythmia and heart failure [5,7]. Although this defect occurs mostly in the LV, right ventricular (RV) noncompaction has also been reported in less than one-half of LVNC patients [8,9].
The ventricles grow and mature through a process of proliferation and differentiation to form trabeculation and compact myocardium [10]. In the mouse, trabecular formation begins at approximately E9.5 where cardiomyocytes grow to form protrusions of muscular ridges [11]. The trabeculae then undergo a process of remodeling/compaction where the bases of the trabeculation thicken and collapse into the myocardial wall. Proliferation of the compact myocardium is concomitant with remodeling. By E14.5, mature trabeculation is formed, along with a thick, compact myocardium [12,13]. Numerous signaling pathways have been implicated in development of the ventricular chambers including Notch, BMP, FGF, retinoic acid and planar cell polarity (PCP) signaling [10,14]. In addition, various mouse models of ventricular noncompaction defect have been generated but the ventricular defects between the mouse models are not consistent, reflecting the complex process of ventricular myocardium development. To date, the complete signaling mechanisms underlying ventricular myocardium development are still not completely understood [15,16].
The small GTPase, Rac1, acts as a crucial regulator of numerous developmental events including proliferation, cell cycle progression, cell survival, differentiation and regulation of cell shape, morphology and polarity [17]. Specifically, the importance of Rac1 signaling in embryonic heart development has emerged in recent studies [18,19]. Our previous work has demonstrated a critical role of Rac1 signaling in the anterior SHF and anterior-SHF derived structures including the RV, interventricular septum and the OFT. Rac1 regulates cardiomyocyte polarization in the RV and formation of the cardiac apex [18]. In order to study the cardiomyocyte specific role of Rac1 in heart development, the Rac1 gene is deleted in the myocardium using the Nkx2.5-Cre transgenic mouse, which drives Cre recombinase activity in both the RV and LV [20]. We show that Rac1 signaling is crucial for ventricular myocardium development and cardiomyocyte specific deficiency of Rac1 leads to a spectrum of CHDs including a thin compact myocardium and hypertrabeculation, similar to clinical features of LVNC. Rac1 deficient hearts also had a bifid cardiac apex and OFT alignment defects. Furthermore, we showed that a Rac1 deficiency in the myocardium disrupts proliferation along with the organization and polarization of cardiomyocytes.
2. Methods
2.1. Mice
The Rac1f/f mouse line (Stock #5550) and mT/mG mouse line (Stock #7676) were purchased from Jackson Laboratory, Bar Harbor, ME, USA [21,22]. The Nkx2.5-Cre transgenic mouse was provided by Dr. Chi-Chung Hui at The Hospital for Sick Children, University of Toronto, originally generated by McFadden and colleagues [20]. The mT/mG mouse is a global double-fluorescent Cre reporter mouse. This reporter mouse expresses membrane-targeted Tomato (mT) before Cre-excision and membrane-targeted GFP (mG) after excision of mT [22]. A breeding program to generate Nkx2.5-Cre;Rac1f/f (Rac1Nkx2.5) and Nkx2.5-Cre;mT/mG mice was carried out and genotyping was performed as described previously [18]. Genotyping primer sequences are listed in Table 1. Mouse experiments and procedures were approved by the Animal Care Committee at Western University in accordance with the guidelines of the Canadian Council of Animal Care.

Table 1.
PCR primer sequences for genotyping genetically modified mice.
2.2. Histological Analysis
Embryonic samples (thoracic cavity) were fixed overnight in 4% paraformaldehyde at 4 °C, dehydrated and paraffin embedded. Samples were serially sectioned at 5 μm from the top of the aortic arch to the apex of the heart with a Leica RM2255 microtome. Sections were mounted onto positively charged albumin/glycerin coated microslides. Slides were stained with hematoxylin and eosin (H/E) for histological analysis and images were captured using a light microscope (Observer D1, Zeiss, Oberkochen, Germany).
2.3. Immunohistochemistry
Immunohistochemical staining was performed on paraffin heart sections. Antigen retrieval was carried out in sodium citrate buffer (pH 6.0) at 92 °C using a BP-111 laboratory microwave (Microwave Research & Applications, Carol Stream, IL, USA). Immunostaining was performed with primary antibodies for GFP (Abcam, Cambridge, MA, USA) and Scrib (Santa Cruz, Santa Cruz, CA, USA) followed by incubation with biotinylated secondary antibody and avidin and biotinylated HRP (Santa Cruz). 3-3′ diaminobenzidine tetrahydrochloride (DAB, Dallas, TX, USA) substrate solution was used to visualize the substrate and slides were counterstained with hematoxylin. Images were captured with Zeiss Observer D1 microscope using AxioVision Rel 4.7 software. For proliferation and apoptosis analysis, E9.5 heart samples were fixed in 4% paraformaldehyde for one hour, cryoprotected in 30% sucrose and embedded in FSC22 frozen section media (Leica, Wetzlar, Germany). Samples were sectioned in a sagittal orientation with a Leica cryostat at 10 μm thick onto glass slides. Slides were incubated with anti-phosphohistone-H3 (phospho S10) (Abcam), anti-cleaved caspase-3 (Cell Signaling) primary antibody, Alexa Fluor 647 wheat germ agglutinin (Invitrogen, Waltham, MA, USA), Alexa Fluor 488 phalloidin (Life Technologies, Carlsbad, CA, USA) and counterstained with Hoechst 33342 (Invitrogen). Confocal images were obtained at the Biotron Research Centre, Western University with a Zeiss LSM 510 Duo microscope. Cardiomyocyte cell size was assessed by measuring the cross-sectional diameter at the nuclear level in ~50 cardiomyocytes per section and 5 sections per heart in the LV free wall using AxioVision software (Zeiss, Oberkochen, Germany) [23].
2.4. Quantitative Real Time RT-PCR
Total RNA was isolated from E12.5 ventricular myocardium using the RNeasy Mini Kit (QIAGEN). Reverse transcription reaction was performed, as described previously [18]. Briefly, M-MLV Reverse Transcriptase (Invitrogen) and EvaGreen qPCR Mastermix (Abm, Vancouver, BC, Canada) were used for real time thermal cycling. 28S rRNA was used as an internal control. Samples were amplified for 35 cycles using the Eppendorf Mastercycler Realplex Real-Time PCR machine (Hamburg, Germany). Real time RT-PCR primer sequences are listed in Table 2. The mRNA level of Rac1 in relation to 28S rRNA was determined using a comparative CT method [24,25].
Table 2.
Primer sequences for real time RT-PCR analysis.
2.5. Western Blot Analysis
Rac1 protein expression from E12.5 ventricular myocardium was measured by western blot analysis. Briefly, 25 μg of protein from isolated ventricular tissue was separated by 12% SDS-Page gel and transferred to nitrocellulose membranes. Blots were probed with antibodies against Rac1 (1:500, Santa Cruz) and α-actinin (1:5000, Sigma, St. Louis, MO, USA). Blots were then washed and probed with horseradish peroxidase conjugated secondary antibodies (1:2500, Bio-Rad, Hercules, CA, USA) and detected using an ECL detection method. Densitometry was then performed to quantify the signal.
2.6. Statistical Analysis
Data are presented as means ± SEM. An unpaired Student’s t test was employed to determine statistical significance between the two groups. Differences were considered significant at p < 0.05.
3. Results
3.1. Generation of a Cardiomyocyte Specific Rac1 Knockout Mouse
The Cre recombinase in the Nkx2.5-Cre mouse line is activated after E8.5 and initial specification of cardiac progenitors [20]. Nkx2.5-Cre transgenic mice and Rac1f/f mice were crossed to generate Nkx2.5-Cre;Rac1f/f (Rac1Nkx2.5) offspring (Figure 1A,B). To confirm a knockdown in Rac1 mRNA expression, a real-time RT-PCR analysis was performed in RNAs isolated from E12.5 ventricles. The Rac1 mRNA expression was significantly decreased by approximately 35% in Rac1Nkx2.5 ventricular myocardium compared to littermate Rac1f/f controls (Figure 1C). Rac1 protein levels in the ventricular myocardium were analyzed using western blotting. The ratio of Rac1 to α-actinin protein levels was reduced by 49% in E12.5 Rac1Nkx2.5 ventricular myocardium compared to littermate Rac1f/f controls (Figure 1D). These results confirm that Nkx2.5-Cre-mediated recombination sufficiently downregulates Rac1 mRNA and protein expression in the ventricular myocardium of the developing heart.
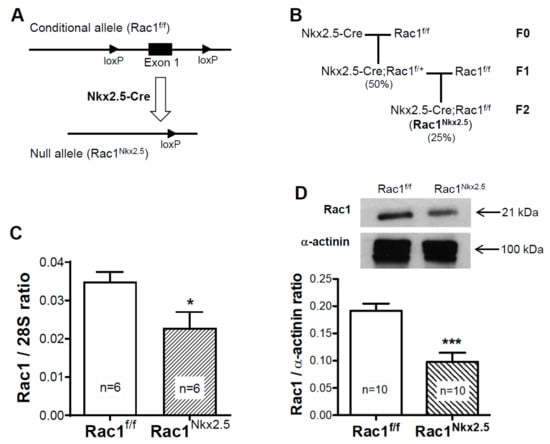
Figure 1.
Generation of mouse line with a myocardium specific deletion of Rac1 (Rac1Nkx2.5). A schema of the floxed Rac1 allele and Nkx2.5-Cre mediated generation of the Rac1 null allele (A). Rac1Nkx2.5 mice were generated by crossing Nkx2.5-Cre transgenic mice with Rac1f/f mice for 2 generations with the expected genotype frequencies indicated in brackets (B). Rac1 mRNA expression was significantly reduced in E12.5 Rac1Nkx2.5 ventricular myocardium compared to Rac1f/f littermates (C). Western blot analysis of E12.5 ventricular myocardium showed a significant decrease in Rac1 protein levels in Rac1Nkx2.5 hearts compared to controls (D). * p < 0.05, *** p < 0.001 by unpaired Student’s t-test.
3.2. Lineage Tracing of Nkx2.5-Cre Transgenic Mouse
To trace where the Cre recombinase is active in Nkx2.5-Cre transgenic hearts, Nkx2.5-Cre mice were crossed to mT/mG reporter mice, which marks all tissues possessing Cre recombinase activity with GFP. McFadden et al., the group who first created the Nkx2.5-Cre transgenic mouse, used a lacZ reporter to show that the Cre recombinase is active throughout the ventricular myocardium with minimal recombination in the OFT and atria [20]. Using the Nkx2.5-Cre;mT/mG mouse, we showed that the Cre recombinase was active throughout the ventricular myocardium and a large portion of the atria at E12.5 (Figure 2A). In addition, a majority of the pulmonary artery myocardium and part of the aorta were GFP+ in Nkx2.5-Cre;mT/mG hearts (Figure 2B). Closer analysis of the aortic valves showed that some of the cells in the early aortic valves were also GFP+ in Nkx2.5-Cre;mT/mG hearts (Figure 2C), indicating the contribution of Nkx2.5 expressing cells to the development of the OFT and atria. Furthermore, closer examination of the epicardium and endocardial cells at E18.5 showed that these cells remained RFP+, indicating no Cre recombinase activity in these cell types (Figure 2D–F). Thus, the Nkx2.5-Cre mouse drives recombination in the ventricular myocardium, atria and part of the OFT.
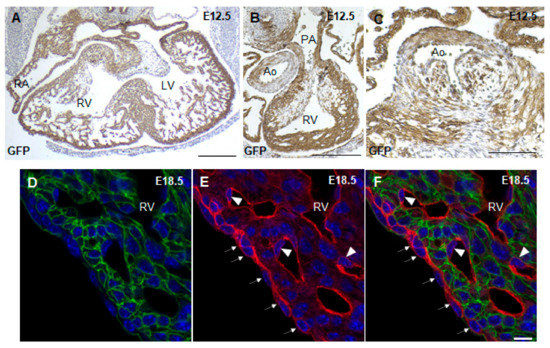
Figure 2.
Lineage tracing with Nkx2.5-Cre;mT/mG mice. GFP immunostaining of E12.5 Nkx2.5-Cre;mT/mG paraffin heart sections showed Cre recombinase activity in the atrial and ventricular myocardium (A), OFT (B) and aortic valve leaflets (C). Fluorescence imaging of cryosections of E18.5 Nkx2.5-Cre;mT/mG hearts showed that the epicardium (arrows) and endothelial cells (arrowhead) remain RFP+ in Nkx2.5-Cre;mT/mG hearts (D–F). RV, right ventricle; LV, left ventricle; PA, pulmonary artery; Ao, aorta; epi, epicardium. Scale bars: 250 µm (A–C), 10 µm (D–F).
3.3. Congenital Heart Defects in Rac1Nkx2.5 Mice
Rac1Nkx2.5 embryos were alive from E11.5–18.5, but all neonates were found dead at P0 (n = 5). Gross morphological analysis of these P0 Rac1Nkx2.5 hearts revealed a bifid cardiac apex, similar to what was observed and reported in our previous study with a Mef2c-Cre anterior second heart field-specific deletion of Rac1 [18]. Examination of all Rac1Nkx2.5 hearts at earlier embryonic time points showed evidence of a bifid cardiac apex as well (Figure 3A). In addition, Rac1Nkx2.5 hearts had incomplete development of the interventricular septum, resulting in a ventricular septal defect (VSD) (Figure 3B,C, Table 3). Alignment of the outflow tract (OFT) to the ventricles was also defective in Rac1Nkx2.5 hearts compared to littermate Rac1f/f controls. A double outlet right ventricle (DORV) was observed in 11 of the 17 Rac1Nkx2.5 hearts (Figure 3D–G, Table 3) and 6 of the 17 Rac1Nkx2.5 hearts exhibited an overriding aorta (Table 3). In addition, both the left ventricle (LV) and right ventricle (RV) of Rac1Nkx2.5 hearts show a thin compact myocardium and hypertrabeculation (Figure 4A–D). The compact myocardium of both Rac1Nkx2.5 ventricles at E15.5 was poorly formed and significantly thinner while the trabecular to compact myocardium ratio increased by more than 2.5-fold compared to littermate Rac1f/f controls (Figure 4E,F). These findings suggest a critical role for Rac1 in interventricular septum formation, OFT alignment and development of the trabecular and compact ventricular myocardium.
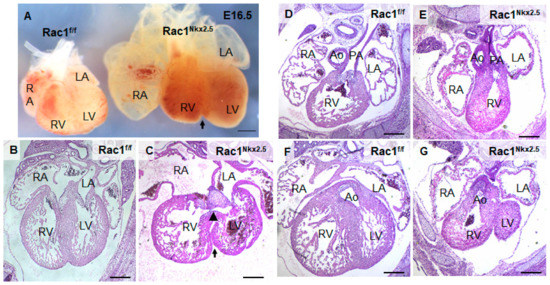
Figure 3.
Congenital heart defects in Rac1Nkx2.5. (A) Bifid cardiac apex in an E16.5 Rac1Nkx2.5 heart. Arrow indicates bifurcation between the RV and LV. (B,C) Ventricular septal defect (arrowhead) was found in E15.5 Rac1Nkx2.5 hearts with an arrow indicating the bifid cardiac apex. (D–G) Double outlet right ventricle (DORV) was found in E15.5 Rac1Nkx2.5 hearts. The pulmonary artery and aorta were connected to the RV and LV, respectively in Rac1f/f control hearts (D,F). However, both pulmonary artery and aorta were connected to the RV in Rac1Nkx2.5 hearts (E,G). RA, right atrium; LA, left atrium. Scale bars: 500 µm.
Table 3.
Congenital heart defects in Rac1Nkx2.5 mice (E14.5—P0).
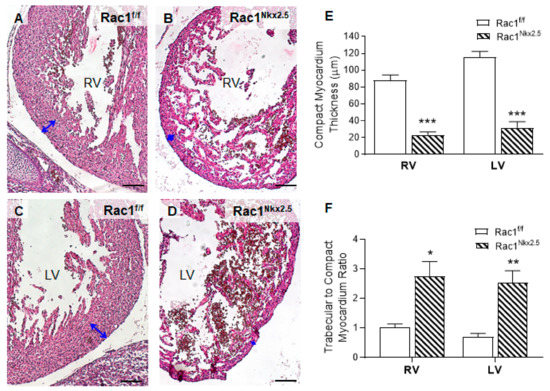
Figure 4.
Ventricular myocardium abnormalities in Rac1Nkx2.5 hearts at E15.5. In comparison to Rac1f/f controls (A,C), the Rac1Nkx2.5 mice (B,D) show thin compact myocardium and hypertrabeculation in the RV and LV wall. The thickness of the compact myocardium in Rac1Nkx2.5 hearts (n = 5) was decreased while trabecular to compact myocardium ratio was increased as compared to littermate controls (n = 6) (E,F). Double-headed arrows in (A–D) indicate measurements of compact myocardium thickness. * p < 0.05, ** p < 0.01, *** p< 0.001 vs. Rac1f/f by Student’s t-test. Scale bars: 100 µm.
3.4. Loss of F-Actin Filament Organization and Cardiomyocyte Polarity in Rac1Nkx2.5 Hearts
To analyze F-actin filament organization and cardiomyocyte polarity in the ventricular myocardium, E18.5 Rac1Nkx2.5 heart sections were double stained with phalloidin and wheat germ agglutinin (WGA) to mark F-actin filaments and cell borders, respectively. Our data show that Rac1Nkx2.5 hearts had severely disrupted F-actin filament organization compared to controls, which had long, parallel running F-actin filaments throughout the myocardium (Figure 5A,B). Additionally, WGA staining revealed rounded, spherically shaped cardiomyocytes in Rac1Nkx2.5 hearts, in both the RV and LV (Figure 5D). In comparison, littermate ventricular myocardium had cardiomyocytes that underwent polarization with an elongated shape and were well organized/aligned in both the RV and LV (Figure 5C). Furthermore, quantitative analysis revealed that the cardiomyocyte cell size was significantly larger in Rac1Nkx2.5 compared to the littermate controls (Figure 5E). This data suggests a crucial role for Rac1 in F-actin filament organization, polarization and elongation of cardiomyocytes during embryonic heart development.
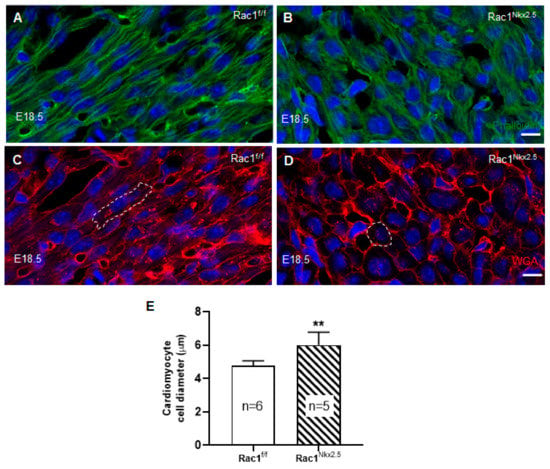
Figure 5.
Disruption of F-actin organization and cardiomyocyte polarity in Rac1Nkx2.5 hearts. E18.5 cryosections of the heart were double stained for phalloidin and wheat germ agglutinin (WGA) to assess F-actin organization and cardiomyocyte cell polarity, respectively. The images were taken from the corresponding areas of LV myocardium free wall in Rac1f/f and Rac1Nkx2.5 mice. F-actin filament organization was disrupted in Rac1Nkx2.5 myocardium (B) compared to littermate controls (A). WGA staining shows rounded cardiomyocytes in Rac1Nkx2.5 ventricular myocardium (D) compared to the elongated cardiomyocytes in littermate controls (C). The cell borders of a cardiomyocyte in (C,D) are outlined. The short axis of cardiomyocyte diameter was significantly larger in Rac1Nkx2.5 compared to littermates (E). ** p < 0.01 by unpaired Student’s t-test. Scale bars: 10 µm.
3.5. Decreased Scrib Protein Expression in Rac1Nkx2.5 Hearts
Scrib plays an important role in cell polarity through interacting with Rac1 in the developing myocardium. Loss of either Scrib or Rac1 leads to a reduction in membrane association of the other [19]. To analyze the expression of Scrib, immunostaining was performed on E15.5 Rac1Nkx2.5 hearts. Scrib was highly expressed in the myocardium surrounding the opening of the aorta in control E15.5 hearts (Figure 6A). In comparison, the expression of Scrib in this area was reduced in E15.5 Rac1Nkx2.5 heart sections (Figure 6B). Similar to what was described in our previous study [18], Scrib protein expression was abundant in the interventricular junction in control hearts at E15.5. However, expression of Scrib was reduced in E15.5 Rac1Nkx2.5 the myocardium of interventricular junction compared to littermate controls (Figure 6C–F). Overall, Scrib protein expression was significantly decreased in E15.5 Rac1Nkx2.5 hearts compared to littermate controls (Figure 6G). The loss of Scrib expression in Rac1Nkx2.5 hearts suggests a disruption in cell polarity and the PCP pathway, further supporting a failure of cardiomyocytes to undergo polarization.
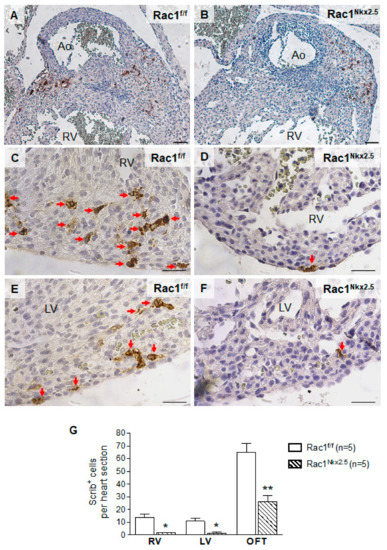
Figure 6.
Loss of Scrib expression in Rac1Nkx2.5 hearts. Scrib immunostaining was performed on E15.5 Rac1Nkx2.5 and Rac1f/f heart sections. The areas analyzed included the myocardium surrounding the aorta (A,B), RV (C,D) and the LV (E,F). Five sections per heart were used. The number of Scrib-expressing positive cells was significantly decreased in the myocardium surrounding the aorta (OFT), RV and LV of Rac1Nkx2.5 hearts (G). * p < 0.05, ** p < 0.01 by Student’s t-test. Scale bars: 50 µm.
3.6. Decreased Cell Proliferation in Rac1Nkx2.5 Hearts
The observed defects in Rac1Nkx2.5 ventricular myocardium development could also be attributed to a decrease in cell proliferation and/or aberrant apoptosis. Cell proliferation has been shown to be highest at E9.5 in the developing mouse heart [26]. Thus, proliferation of E9.5 Rac1Nkx2.5 hearts was analyzed by immunostaining for phospho-histone H3 (pHH3) and cyclin D1, which are a marker of the mitotic phase of cell division and a cell cycle regulator, respectively. The cell proliferation rate assessed by pHH3+ and cyclin D1+ cells in the ventricular myocardium was significantly reduced in E9.5 Rac1Nkx2.5 hearts compared to littermate Rac1f/f controls (Figure 7A–F). Immunostaining for cleaved caspase-3 (CC3), a marker of activated apoptosis, showed little to no apoptosis in both control E9.5 and Rac1Nkx2.5 ventricular myocardium (Figure 7G,H). However, apoptosis was detected in tissues outside the heart (Figure 7I,J). The decreased proliferation rate in Rac1Nkx2.5 hearts suggests a critical role for Rac1 in regulating cardiomyocyte proliferation in the ventricular myocardium, after initial specification of cardiac progenitors.
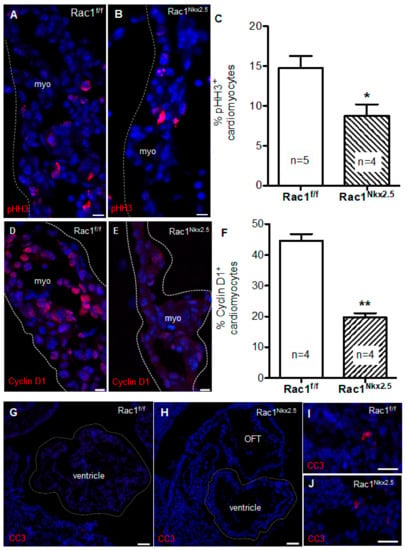
Figure 7.
Decreased proliferation rate in Rac1Nkx2.5 hearts. Phospho-histone H3 (pHH3) immunostaining to mark proliferating cells undergoing mitosis in ventricular myocardium (myo) of E9.5 Rac1Nkx2.5 and Rac1f/f hearts (A,B). Proliferation rate was significantly decreased in E9.5 Rac1Nkx2.5 ventricular myocardium compared to littermate controls (C). Cyclin D1 immunostaining in E9.5 Rac1f/f and Rac1Nkx2.5 ventricular myocardium marked cell progression through G1 (D,E). Cyclin D1 expression was significantly decreased in E9.5 Rac1Nkx2.5 ventricular myocardium compared to littermate controls (F). Cleaved caspase-3 (CC3) immunostaining to mark apoptotic cells in ventricular myocardium of E9.5 Rac1Nkx2.5 and Rac1f/f hearts. No apoptosis was detected in E9.5 Rac1Nkx2.5 and Rac1f/f ventricular myocardium (G,H). Apoptotic cells were detected in tissues outside of the heart in Rac1f/f and Rac1Nkx2.5 embryos (I,J). * p < 0.05, ** p < 0.01 by unpaired Student’s t-test. Scale bars: 10 µm (A,B), 50 µm (D–G).
3.7. Decreased Scrib and Cardiac Transcription Factor Expression in Rac1Nkx2.5 Hearts
To assess the genetic pathways regulated by Rac1 signaling, we analyzed the mRNA expression of factors critical to embryonic heart development in E12.5 hearts. Since Scrib-Rac1 interaction is crucial for normal heart development, Scrib mRNA levels were assessed. Consistent with its protein expression (Figure 6), Scrib mRNA levels were significantly reduced in E12.5 Rac1Nkx2.5 compared to Rac1f/f hearts (Figure 8A). Additionally, cardiac transcription and growth factors including Nkx2.5, Gata4, Tbx5, Tbx20, Hand1, Hand2 and Bmp10, except Mef2c, were all significantly decreased in E12.5 Rac1Nkx2.5 compared to Rac1f/f hearts (Figure 8B–I). These results indicate that transcriptional regulation of heart development was severely disrupted in the Rac1Nkx2.5 hearts.
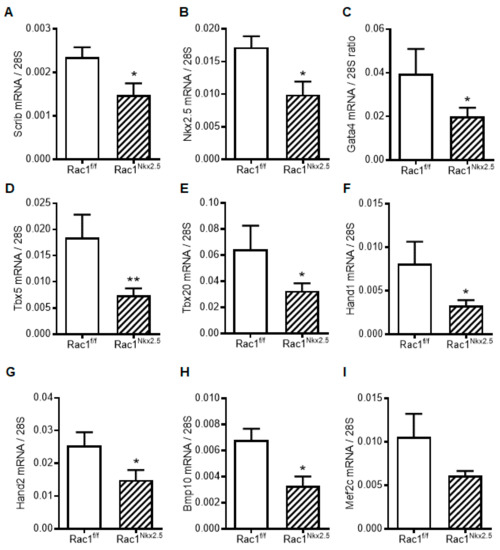
Figure 8.
Myocardial mRNA expression of transcription and growth factors in E12.5 Rac1f/f and Rac1Nkx2.5 hearts. (A) Scrib. (B) Nkx2.5. (C) Gata4. (D) Tbx5. (E) Tbx20. (F) Hand1. (G) Hand2. (H) Bmp10. (I) Mef2c. Data are expressed as mRNA to 28S ratios. n = 5–6 per group. * p < 0.05, ** p < 0.01 by unpaired Student’s t test.
4. Discussion
Recent studies have implicated PCP signaling and cell polarity as critical regulators of compact myocardium development. Vangl2, Scrib and Dishevelled mouse mutants have cardiomyocytes that are not polarized and the ventricular myocardium is thinned, resembling LVNC [27,28,29]. Rac1 is a known downstream effector of PCP signaling; however, the cardiomyocyte specific role of Rac1 in heart development is unclear [30]. In the present study, we demonstrated that downregulation of Rac1 signaling in the ventricular myocardium disrupted formation of a trabecular network and development of the compact myocardium. In addition, Rac1Nkx2.5 mice had a bifid cardiac apex, defects in ventricular septum formation and OFT alignment. The F-actin filament organization and polarization of cardiomyocytes in the Rac1 deficient ventricular myocardium was also abnormal. Overall, cell proliferation was decreased in Rac1Nkx2.5 hearts, along with expression of the PCP protein, Scrib. Our study demonstrates a critical role for Rac1 signaling in outflow tract and compact myocardium development (Figure 9).
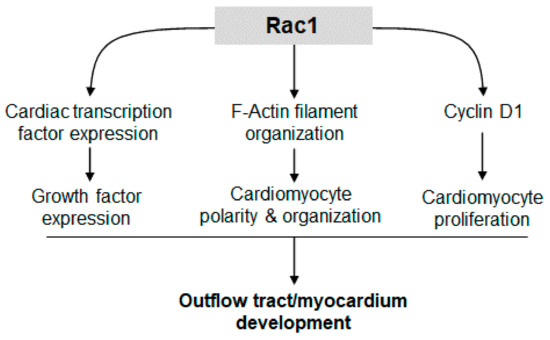
Figure 9.
Rac1 signaling promotes cardiac transcription/growth factor expression, cardiomyocyte polarity and proliferation, leading to normal development of the outflow tract and compact myocardium.
Cell proliferation is a regulated spatially and temporally during heart development. The rate of cell proliferation in the ventricular myocardium peaks at E9.5 and gradually decreases during development [26]. Studies have shown that cardiomyocyte proliferation is the major determinant of overall cardiac size during heart development. The mass of the heart must increase to match the increasing circulatory demands of the growing embryo [31]. Rac1 has been shown to regulate cell proliferation through various pathways. Our data showed that a Rac1 deficiency in the ventricular myocardium decreased the expression of factors critical to cardiogenesis, cardiomyocyte differentiation and proliferation including Nkx2.5, Gata4, Tbx5, Tbx20, Hand 1, Hand2 and Bmp10. Additionally, Cyclin D1 protein levels and cell proliferation were reduced in E9.5 Rac1Nkx2.5 hearts. These results suggest that Rac1 promotes cardiac transcription and growth factor expression, leading to cell cycle progression and cardiomyocyte proliferation in the developing heart (Figure 9).
An earlier report by Boczonadi et al. also used a Nkx2.5-Cre;Rac1f/f mouse line in their studies [19]. However, the Nkx2.5-Cre mouse used by Boczonadi et al. was an Nkx2.5 heterozygous mouse since the Cre recombinase gene was knocked into the Nkx2.5 genetic locus [32]. In contrast, the Nkx2.5-Cre mouse used in the present study is a transgenic mouse and retains two intact alleles of Nkx2.5 [20]. The Nkx2.5-Cre;Rac1f/f mice used in Boczonadi et al. were embryonically lethal due to their interaction with E13.5, which precluded analysis of ventricular septation and OFT alignment with the developing ventricles [19]. However, at E12.5 these mice did show underdeveloped ventricles with a thin ventricular wall. The earlier lethality of the Nkx2.5-Cre;Rac1f/f mouse line compared to the Rac1Nkx2.5 used in the current study is likely due to the heterozygous expression of Nkx2.5 compounded with decreased Rac1 signaling in the ventricular myocardium. Furthermore, whether the underdeveloped heart reported in the study by Boczonadi et al. [19] is exclusively due to deficient Rac1 signaling or also due to downregulation of Nkx2.5 is unclear. Our data implicates Rac1 signaling specifically and we were able to demonstrate that Rac1Nkx2.5 hearts exhibit VSDs and OFT alignment defects.
Molecular players in the planar cell polarity (PCP) pathway are not only critical for normal myocardial development but also govern OFT development. For example, mutation in Vangl2, a member of the highly conserved non-canonical Wnt signaling cascade, results in cardiac malalignment and DORV [28]. In addition, Dishevelled 2 knockout mice show transposition of the great arteries, persistent truncus arteriosus and DORV, with the latter being the most common cardiac defect [33]. Notably, anterior SHF specific deletion of Rac1 results in a spectrum of OFT alignment defects including DORV via a disruption of migration of neural crest cells and cardiomyocytes into the OFT [34]. In the present study, Nkx2.5-Cre-driven GFP expression is seen in pulmonary and aortic walls, and in aortic valve leaflets, suggesting cardiomyocyte migration to the OFT. The OFT myocardium produces axon guidance molecules such as Sema3c, a chemoattractant that navigates neural crest cells to the OFT [34]. Reductions in cardiomyocyte proliferation and migration to the OFT may diminish cardiomyocyte contribution to the OFT and impede neural crest cell migration, leading to OFT alignment defects.
Our previous work reported a bifid cardiac apex when Rac1 was specifically deleted in the anterior SHF and all anterior SHF-derived cells [18]. We had postulated that loss of Rac1 signaling in the RV led to an inability of the nonpolarized and disorganized cardiomyocytes to bridge the interventricular junction to unify the two developing ventricles and form a distinct cardiac apex. However, in the present study using the Rac1Nkx2.5 hearts, we show that development of normal cardiac apex is also dependent on polarity and organization of the cardiomyocytes in the LV myocardium. Interestingly, other reports where PCP signaling was disrupted did not report observations of a bifid cardiac apex, despite similar reports of cardiomyocyte disorganization [27,28]. Rac1 is a known downstream effector of PCP signaling, regulating actin cytoskeleton dynamics and cell polarity [35,36]. Our findings suggest that development of a bifid cardiac apex involves a pathway specific to a disruption of Rac1 signaling. In addition, since Rac1 is a pleiotropic effector of numerous cellular events, the concomitant disruption of several cellular mechanisms is likely responsible for bifid cardiac apex, along with the other observed CHDs in Rac1Nkx2.5 hearts.
Our lineage tracing analysis with the mT/mG global double florescence mouse showed similar GFP+ expression in domains of the heart that were reported by McFadden et al, who used a lacZ reporter. However, we also showed Cre recombinase activity in the OFT and atria, which were reported to be minimal by McFadden et al [20]. This discrepancy in Cre recombinase activity may be due to the additional β-galactosidase enzymatic reaction step that is required to visualize the blue color of lacZ staining, making it a less efficient reporter compared to a GFP reporter. These results suggest that future lineage tracing analysis should use a florescence reporter as a more reliable readout of expression compared to a lacZ reporter.
Rac1 is involved in reactive oxygen species (ROS) generation through activation of NADPH oxidase [37]. ROS mediates numerous cellular functions including proliferation, cell survival, differentiation and migration [37,38]. The levels of ROS are finely tuned in a cell to regulate these diverse functions. For example, excess ROS induces apoptosis while basal levels of ROS regulate gene expression and proliferation [37,39]. Work in our lab has shown that ROS levels must be tightly regulated to facilitate normal cardiac development. Excess ROS induced by pregestational maternal diabetes and decreased levels of ROS in the NADPH oxidase Nox2 knockout mouse both have a detrimental effect on heart development, leading to a spectrum of CHDs [40,41,42]. Whether the CHDs observed in the Rac1Nkx2.5 hearts can also be attributed to decreased ROS levels should be determined in future studies.
5. Conclusions
A Rac1 deficiency in the myocardium disrupts cardiomyocyte organization and proliferation, leading to bifid cardiac apex, VSDs and OFT alignment and ventricular myocardial compaction defects. Our study suggests a critical role for Rac1 regulation of cardiomyocyte proliferation, organization and polarization in development of the outflow tract and ventricular myocardium. Whether perturbed Rac1 signaling in the ventricular myocardium underlies human cases of LVNC warrants further investigation.
Author Contributions
C.L. and Q.F. conceived the experiments. C.L., A.E., X.L. and Q.F. designed the experiments. C.L., A.E., M.Y.K. and X.L. performed the experiments and data analyses. C.L. drafted the manuscript. C.L., A.E., X.L. and Q.F. revised the manuscript. All authors contributed to the interpretation of results and proofreading of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by operating grants to Q.F. from Natural Sciences and Engineering Research Council of Canada (NSERC) and Canadian Institutes of Health Research (CIHR). C.L. was supported by an NSERC Scholarship. Q.F. is a Richard and Jean Ivy Chair in Molecular Toxicology at Western University.
Institutional Review Board Statement
Animal use was approved by the Animal Care Committee at Western University (protocol #2007-011, date of approval: 12 March 2014) in accordance with the guidelines of the Canadian Council of Animal Care.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Pierpont, M.E.; Basson, C.T.; Benson, D.W.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L. Genetic Basis for Congenital Heart Defects: Current Knowledge: A Scientific Statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef]
- Hoffman, J.I.; Kaplan, S.; Liberthson, R.R. Prevalence of Congenital Heart Disease. Am. Heart J. 2004, 147, 425–439. [Google Scholar] [CrossRef]
- Sun, R.; Liu, M.; Lu, L.; Zheng, Y.; Zhang, P. Congenital Heart Disease: Causes, Diagnosis, Symptoms, and Treatments. Cell Biochem. Biophys. 2015, 72, 857–860. [Google Scholar] [CrossRef]
- Warnes, C.A. The Adult with Congenital Heart Disease: Born to Be Bad? J. Am. Coll. Cardiol. 2005, 46, 1–8. [Google Scholar] [CrossRef]
- Guha, K.; Treibel, T.A.; Roussin, I.; Prasad, S.K.; Duncan, A.M.; Brookes, C.; McDonagh, T.A.; Sharma, R. Treatment of Left Ventricular Non-Compaction with Cardiac Resynchronization Therapy. QJM 2013, 106, 575–579. [Google Scholar] [CrossRef]
- Chin, T.K.; Perloff, J.K.; Williams, R.G.; Jue, K.; Mohrmann, R. Isolated Noncompaction of Left Ventricular Myocardium. A Study of Eight Cases. Circulation 1990, 82, 507–513. [Google Scholar] [CrossRef]
- Finsterer, J. Left Ventricular Non-Compaction and Its Cardiac and Neurologic Implications. Heart Fail. Rev. 2010, 15, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, E.; Marshalko, S.J.; Jaffe, C.C.; Hui, P. Isolated Noncompaction of the Ventricular Myocardium: Clinical and Molecular Aspects of a Rare Cardiomyopathy. Lab. Investig. 2002, 82, 117–122. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ritter, M.; Oechslin, E.; Sutsch, G.; Attenhofer, C.; Schneider, J.; Jenni, R. Isolated Noncompaction of the Myocardium in Adults. Mayo Clin. Proc. 1997, 72, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Samsa, L.A.; Yang, B.; Liu, J. Embryonic Cardiac Chamber Maturation: Trabeculation, Conduction, and Cardiomyocyte Proliferation. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Sedmera, D.; Pexieder, T.; Vuillemin, M.; Thompson, R.P.; Anderson, R.H. Developmental Patterning of the Myocardium. Anat. Rec. 2000, 258, 319–337. [Google Scholar] [CrossRef]
- Risebro, C.A.; Riley, P.R. Formation of the Ventricles. Sci. World J. 2006, 6, 1862–1880. [Google Scholar] [CrossRef]
- Pasumarthi, K.B.; Field, L.J. Cardiomyocyte Cell Cycle Regulation. Circ. Res. 2002, 90, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, H.; Qu, X.; Chang, C.P.; Shou, W. Molecular Mechanism of Ventricular Trabeculation/Compaction and the Pathogenesis of the Left Ventricular Noncompaction Cardiomyopathy (LVNC). Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, E.; Jenni, R. Left Ventricular Non-Compaction Revisited: A Distinct Phenotype with Genetic Heterogeneity? Eur. Heart J. 2011, 32, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Luedde, M.; Ehlermann, P.; Weichenhan, D.; Will, R.; Zeller, R.; Rupp, S.; Muller, A.; Steen, H.; Ivandic, B.T.; Ulmer, H.E.; et al. Severe Familial Left Ventricular Non-Compaction Cardiomyopathy Due to a Novel Troponin T (TNNT2) Mutation. Cardiovasc. Res. 2010, 86, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Duquette, P.M.; Lamarche-Vane, N. Rho GTPases in Embryonic Development. Small GTPases 2014, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Lu, X.; Liu, M.; Feng, Q. Rac1 Signaling Is Critical to Cardiomyocyte Polarity and Embryonic Heart Development. J. Am. Heart Assoc. 2014, 3, e001271. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; Gillespie, R.; Keenan, I.; Ramsbottom, S.A.; Donald-Wilson, C.; Al Nazer, M.; Humbert, P.; Schwarz, R.J.; Chaudhry, B.; Henderson, D.J. Scrib: Rac1 Interactions Are Required for the Morphogenesis of the Ventricular Myocardium. Cardiovasc. Res. 2014, 104, 103–115. [Google Scholar] [CrossRef][Green Version]
- McFadden, D.G.; Barbosa, A.C.; Richardson, J.A.; Schneider, M.D.; Srivastava, D.; Olson, E.N. The Hand1 and Hand2 Transcription Factors Regulate Expansion of the Embryonic Cardiac Ventricles in a Gene Dosage-Dependent Manner. Development 2005, 132, 189–201. [Google Scholar] [CrossRef]
- Glogauer, M.; Marchal, C.C.; Zhu, F.; Worku, A.; Clausen, B.E.; Foerster, I.; Marks, P.; Downey, G.P.; Dinauer, M.; Kwiatkowski, D.J. Rac1 Deletion in Mouse Neutrophils Has Selective Effects on Neutrophil Functions. J. Immunol. 2003, 170, 5652–5657. [Google Scholar] [CrossRef]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A Global Double-Fluorescent Cre Reporter Mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Xiang, F.L.; Lu, X.; Hammoud, L.; Zhu, P.; Chidiac, P.; Robbins, J.; Feng, Q. Cardiomyocyte-Specific Overexpression of Human Stem Cell Factor Improves Cardiac Function and Survival Post Myocardial Infarction in Mice. Circulation 2009, 120, 1065–1074. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, X.; Xiang, F.L.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; Robbins, J.; Feng, Q. Nitric Oxide Synthase-3 Deficiency Results in Hypoplastic Coronary Arteries and Postnatal Myocardial Infarction. Eur. Heart J. 2014, 35, 920–931. [Google Scholar] [CrossRef]
- Engineer, A.; Saiyin, T.; Lu, X.; Kucey, A.S.; Urquhart, B.L.; Drysdale, T.A.; Norozi, K.; Feng, Q. Sapropterin Treatment Prevents Congenital Heart Defects Induced by Pregestational Diabetes Mellitus in Mice. J. Am. Heart Assoc. 2018, 7, e009624. [Google Scholar] [CrossRef] [PubMed]
- De Boer, B.A.; van den Berg, G.; de Boer, P.A.; Moorman, A.F.; Ruijter, J.M. Growth of the Developing Mouse Heart: An Interactive Qualitative and Quantitative 3D Atlas. Dev. Biol. 2012, 368, 203–213. [Google Scholar] [CrossRef]
- Phillips, H.M.; Hildreth, V.; Peat, J.D.; Murdoch, J.N.; Kobayashi, K.; Chaudhry, B.; Henderson, D.J. Non-Cell-Autonomous Roles for the Planar Cell Polarity Gene Vangl2 in Development of the Coronary Circulation. Circ. Res. 2008, 102, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.M.; Rhee, H.J.; Murdoch, J.N.; Hildreth, V.; Peat, J.D.; Anderson, R.H.; Copp, A.J.; Chaudhry, B.; Henderson, D.J. Disruption of Planar Cell Polarity Signaling Results in Congenital Heart Defects and Cardiomyopathy Attributable to Early Cardiomyocyte Disorganization. Circ. Res. 2007, 101, 137–145. [Google Scholar] [CrossRef]
- Sinha, T.; Wang, B.; Evans, S.; Wynshaw-Boris, A.; Wang, J. Disheveled Mediated Planar Cell Polarity Signaling Is Required in the Second Heart Field Lineage for Outflow Tract Morphogenesis. Dev. Biol. 2012, 370, 135–144. [Google Scholar] [CrossRef]
- Simons, M.; Mlodzik, M. Planar Cell Polarity Signaling: From Fly Development to Human Disease. Annu. Rev. Genet. 2008, 42, 517–540. [Google Scholar] [CrossRef]
- Sedmera, D.; Hu, N.; Weiss, K.M.; Keller, B.B.; Denslow, S.; Thompson, R.P. Cellular Changes in Experimental Left Heart Hypoplasia. Anat. Rec. 2002, 267, 137–145. [Google Scholar] [CrossRef]
- Moses, K.A.; DeMayo, F.; Braun, R.M.; Reecy, J.L.; Schwartz, R.J. Embryonic Expression of an Nkx2-5/Cre Gene Using ROSA26 Reporter Mice. Genesis 2001, 31, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Hamblet, N.S.; Lijam, N.; Ruiz-Lozano, P.; Wang, J.; Yang, Y.; Luo, Z.; Mei, L.; Chien, K.R.; Sussman, D.J.; Wynshaw-Boris, A. Dishevelled 2 Is Essential for Cardiac Outflow Tract Development, Somite Segmentation and Neural Tube Closure. Development 2002, 129, 5827–5838. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Liu, Y.; Lu, X.; Kim, M.; Drysdale, T.A.; Feng, Q. Rac1 Signaling Is Required for Anterior Second Heart Field Cellular Organization and Cardiac Outflow Tract Development. J. Am. Heart Assoc. 2016, 5, e002508. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt Signaling Pathways Meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The Complex World of WNT Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Engineer, A.; Saiyin, T.; Greco, E.R.; Feng, Q. Say NO to ROS: Their Roles in Embryonic Heart Development and Pathogenesis of Congenital Heart Defects in Maternal Diabetes. Antioxidants 2019, 8, 436. [Google Scholar] [CrossRef]
- Covarrubias, L.; Hernandez-Garcia, D.; Schnabel, D.; Salas-Vidal, E.; Castro-Obregon, S. Function of Reactive Oxygen Species during Animal Development: Passive or Active? Dev. Biol. 2008, 320, 1–11. [Google Scholar] [CrossRef]
- Chowdhury, A.R.; Ghosh, I.; Datta, K. Excessive Reactive Oxygen Species Induces Apoptosis in Fibroblasts: Role of Mitochondrially Accumulated Hyaluronic Acid Binding Protein 1 (HABP1/P32/GC1qR). Exp. Cell Res. 2008, 314, 651–667. [Google Scholar] [CrossRef]
- Moazzen, H.; Lu, X.; Liu, M.; Feng, Q. Pregestational Diabetes Induces Fetal Coronary Artery Malformation via Reactive Oxygen Species Signaling. Diabetes 2015, 64, 1431–1443. [Google Scholar] [CrossRef]
- Moazzen, H.; Lu, X.; Ma, N.L.; Velenosi, T.J.; Urquhart, B.L.; Wisse, L.J.; Groot, A.C.; Feng, Q. N-Acetylcysteine Prevents Congenital Heart Defects Induced by Pregestational Diabetes. Cardiovasc. Diabetol. 2014, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Moazzen, H.; Wu, Y.; Engineer, A.; Lu, X.; Aulakh, S.; Feng, Q. NOX2 Is Critical to Endocardial to Mesenchymal Transition and Heart Development. Oxid. Med. Cell Longev. 2020, 2020, 1679045. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).