Vulnerable Plaque, Characteristics, Detection, and Potential Therapies


Abstract
1. Introduction
2. Biology of Atheromatous Plaque Formation and Progression
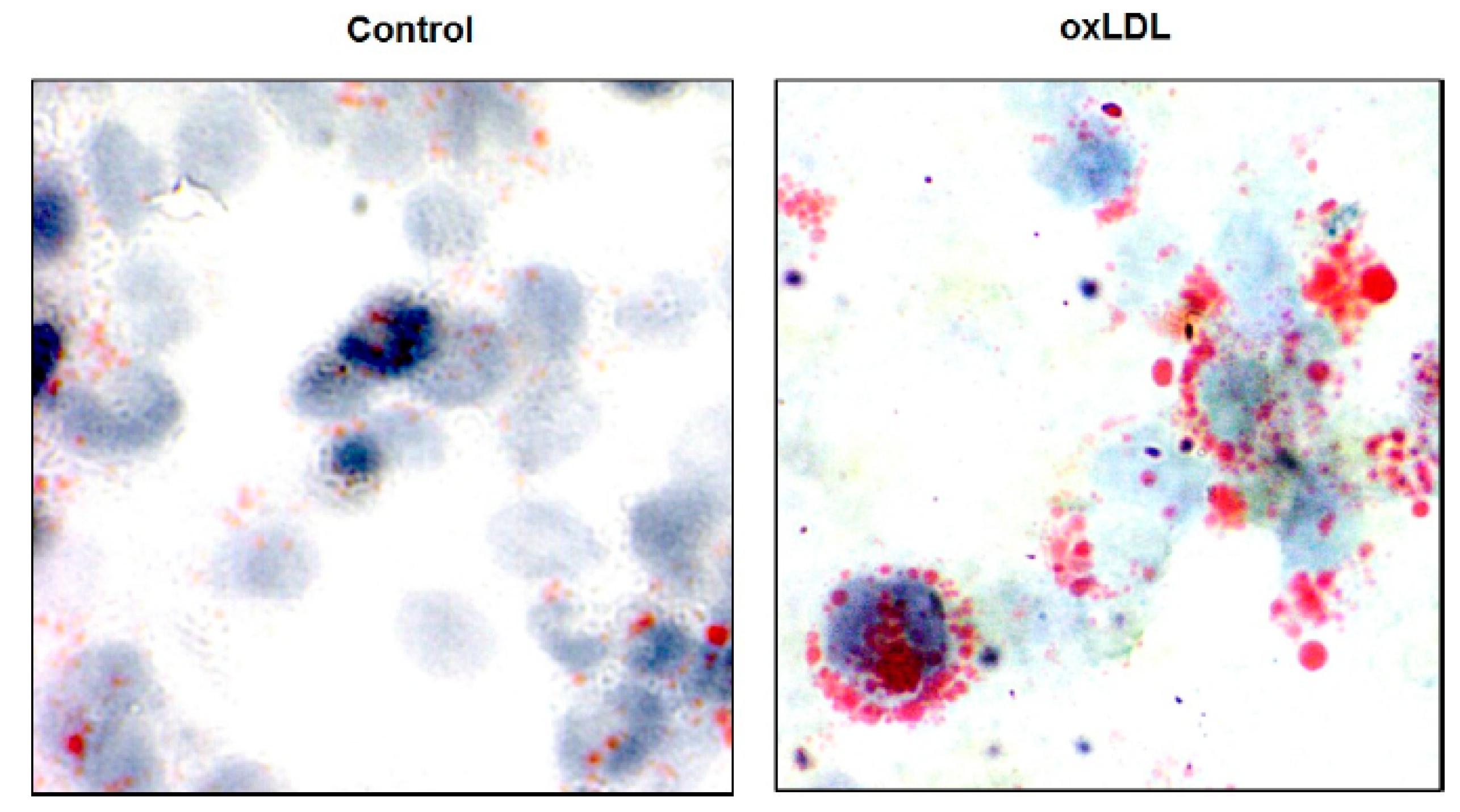
2.1. Lipid Retention
2.2. Plaque Progression
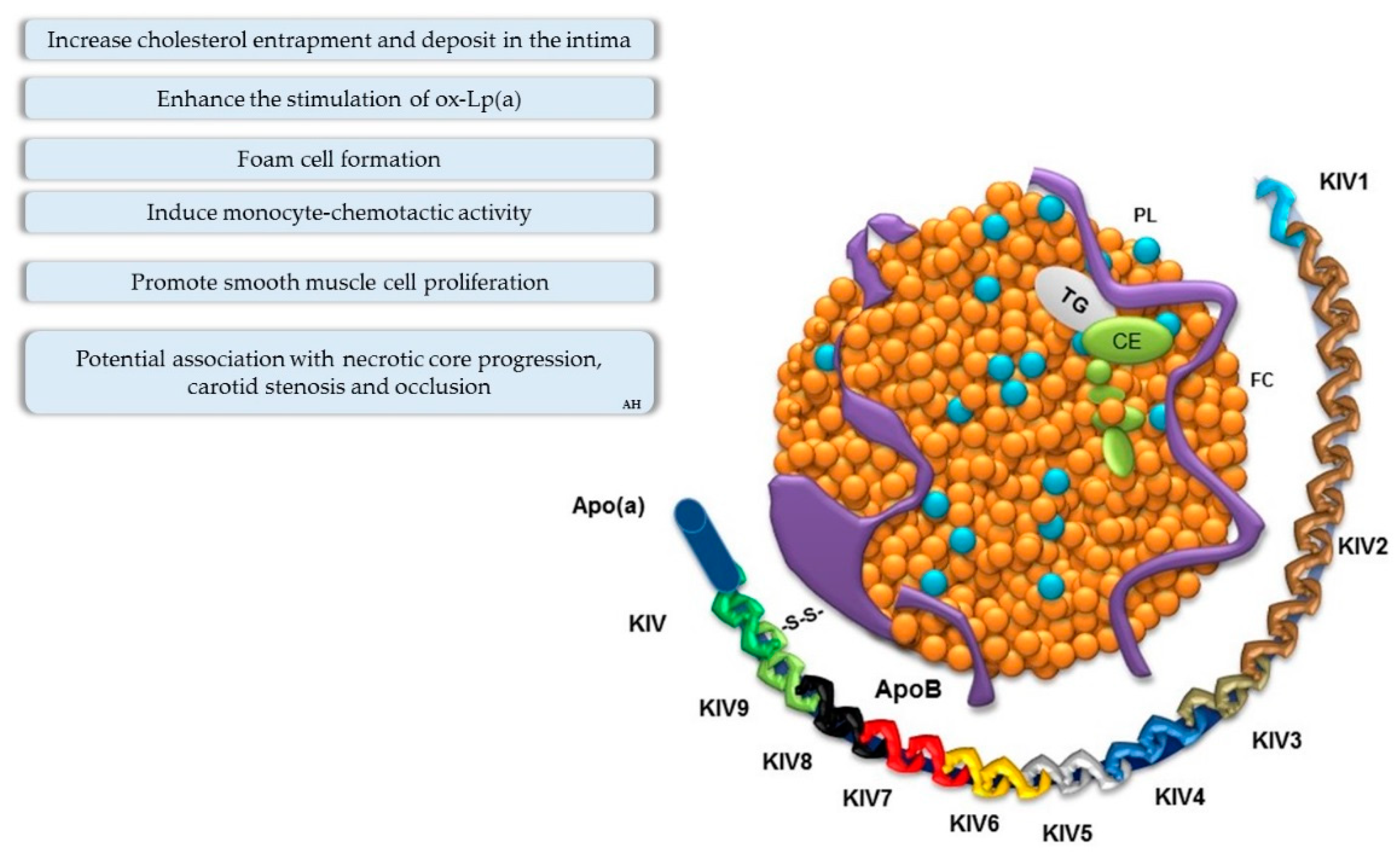
Role of Lipoprotein(a)
3. Vulnerable Plaque Controversy
4. Biochemical and Genetic Markers of Vulnerable Atherosclerotic Plaque
4.1. Biochemical Markers
4.2. Genetic Markers
5. Imaging Biomarkers for Vulnerable Plaque
Limitations and Advantages
6. Plaque Rupture Prediction and Challenges
7. Potential Therapies for Promoting Plaque Regression
7.1. Evidence of Plaque Regression
7.2. Lipid-Lowering Therapy
7.2.1. Anacetrapib
7.2.2. Probucol
7.2.3. Therapies against PCSK9
7.2.4. Therapies against Lp(a)
7.3. Potential Molecular Targets for Plaque Regression
7.4. Angiogenesis Inhibitors
7.5. HDL Biogenesis and Plaque
7.6. Current Perepectives for Reducing Atherosclerotic Plaque
Locally Applied Therapy for Vulnerable Plaque
8. Conclusion
Funding
Conflicts of Interest
References
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Theelen, T.L.; Lappalainen, J.P.; Sluimer, J.C.; Gurzeler, E.; Cleutjens, J.P.; Gijbels, M.J.; Biessen, E.A.; Daemen, M.J.; Alitalo, K.; Yla-Herttuala, S. Angiopoietin-2 blocking antibodies reduce early atherosclerotic plaque development in mice. Atherosclerosis 2015, 241, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Garcia-Cardena, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Stefanadis, C.; Antoniou, C.K.; Tsiachris, D.; Pietri, P. Coronary Atherosclerotic Vulnerable Plaque: Current Perspectives. J. Am. Heart Assoc. 2017, 6, e005543. [Google Scholar] [CrossRef] [PubMed]
- Tay, C.; Liu, Y.H.; Hosseini, H.; Kanellakis, P.; Cao, A.; Peter, K.; Tipping, P.; Bobik, A.; Toh, B.H.; Kyaw, T. B-cell-specific depletion of tumour necrosis factor alpha inhibits atherosclerosis development and plaque vulnerability to rupture by reducing cell death and inflammation. Cardiovasc. Res. 2016, 111, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Wu, M.Y.; Li, C.J.; Hou, M.F.; Chu, P.Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef]
- Tang, X.; Yang, Y.; Luo, S.; Zhao, Y.; Lu, C.; Luo, Y.; Zhang, F.; Xiao, H. The effect of statin therapy on plaque regression following acute coronary syndrome: A meta-analysis of prospective trials. Coron. Artery Dis. 2016, 27, 636–649. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Tonti, L.; Roma, P.; Catapano, A.L. Apoptosis and proliferation of endothelial cells in early atherosclerotic lesions: Possible role of oxidised LDL. Nutr. Metab. Cardiovasc. Dis. NMCD 2002, 12, 297–305. [Google Scholar] [PubMed]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 2006, 47, C13–C18. [Google Scholar] [CrossRef] [PubMed]
- Langley, S.R.; Willeit, K.; Didangelos, A.; Matic, L.P.; Skroblin, P.; Barallobre-Barreiro, J.; Lengquist, M.; Rungger, G.; Kapustin, A.; Kedenko, L.; et al. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J. Clin. Investig. 2017, 127, 1546–1560. [Google Scholar] [CrossRef] [PubMed]
- Felton, C.V.; Crook, D.; Davies, M.J.; Oliver, M.F. Relation of Plaque Lipid Composition and Morphology to the Stability of Human Aortic Plaques. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Humphries, S.E. Guidelines for the identification and management of patients with familial hypercholesterolaemia (FH): Are we coming to a consensus? Atheroscler. Suppl. 2011, 12, 217–220. [Google Scholar] [CrossRef]
- Blanco-Colio, L.M.; Martin-Ventura, J.L.; Vivanco, F.; Michel, J.B.; Meilhac, O.; Egido, J. Biology of atherosclerotic plaques: What we are learning from proteomic analysis. Cardiovasc. Res. 2006, 72, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.H.; Rennick, R.E.; Kalevitch, S.G.; Campbell, G.R. Heparan sulfate-degrading enzymes induce modulation of smooth muscle phenotype. Exp. Cell Res. 1992, 200, 156–167. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jorgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Wissler, R.W. The arterial medial cell, smooth muscle, or multifunctional mesenchyme? Circulation 1967, 36, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, O.A.; Gomez, D.; Shankman, L.S.; Swiatlowska, P.; Williams, J.; Sarmento, O.F.; Alencar, G.F.; Hess, D.L.; Bevard, M.H.; Greene, E.S.; et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat. Med. 2016, 22, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Owens, G.K. Reconciling Smooth Muscle Cell Oligoclonality and Proliferative Capacity in Experimental Atherosclerosis. Circ. Res. 2016, 119, 1262–1264. [Google Scholar] [CrossRef] [PubMed]
- Spacek, M.; Zemanek, D.; Hutyra, M.; Sluka, M.; Taborsky, M. Vulnerable atherosclerotic plaque—A review of current concepts and advanced imaging. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2018, 162, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Vergallo, R.; Porto, I.; D’Amario, D.; Annibali, G.; Galli, M.; Benenati, S.; Bendandi, F.; Migliaro, S.; Fracassi, F.; Aurigemma, C.; et al. Coronary Atherosclerotic Phenotype and Plaque Healing in Patients with Recurrent Acute Coronary Syndromes Compared with Patients with Long-term Clinical Stability: An In Vivo Optical Coherence Tomography StudyAtherosclerotic Phenotype, Plaque Healing, and Recurrent Acute Coronary SyndromesAtherosclerotic Phenotype, Plaque Healing, and Recurrent Acute Coronary Syndromes. JAMA Cardiol. 2019, 4, 321–329. [Google Scholar]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons from sudden coronary death: A comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1262–1275. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Boren, J.; Andreotti, F.; Watts, G.F.; Ginsberg, H.; Amarenco, P.; Catapano, A.; Descamps, O.S.; et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur. Heart J. 2010, 31, 2844–2853. [Google Scholar] [CrossRef]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef]
- Kronenberg, F.; Kronenberg, M.F.; Kiechl, S.; Trenkwalder, E.; Santer, P.; Oberhollenzer, F.; Egger, G.; Utermann, G.; Willeit, J. Role of Lipoprotein(a) and Apolipoprotein(a) Phenotype in Atherogenesis. Circulation 1999, 100, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.B. Atherogenecity of lipoprotein(a) and oxidized low density lipoprotein: Insight from in vivo studies of arterial wall influx, degradation and efflux. Atherosclerosis 1999, 143, 229–243. [Google Scholar] [CrossRef]
- Zheng, K.H.; Tsimikas, S.; Pawade, T.; Kroon, J.; Jenkins, W.S.A.; Doris, M.K.; White, A.C.; Timmers, N.K.L.M.; Hjortnaes, J.; Rogers, M.A.; et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients With Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 2150–2162. [Google Scholar] [CrossRef] [PubMed]
- Van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; Van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Vacante, M.; Russo, C.; Malaguarnera, G.; Antic, T.; Malaguarnera, L.; Bella, R.; Pennisi, G.; Galvano, F.; Frigiola, A. Lipoprotein(a) in cardiovascular diseases. BioMed Res. Int. 2013, 2013, 650989. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.J.; Irimpen, A.M.; Siebenlist, U.; Chandrasekar, B. OxLDL induces endothelial dysfunction and death via TRAF3IP2: Inhibition by HDL3 and AMPK activators. Free Radic. Biol. Med. 2014, 70, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, M.I.; Gimbrone, M.A., Jr. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science 1991, 251, 788–791. [Google Scholar] [CrossRef]
- Legler, D.F.; Matti, C.; Laufer, J.M.; Jakobs, B.D.; Purvanov, V.; Uetz-von Allmen, E.; Thelen, M. Modulation of Chemokine Receptor Function by Cholesterol: New Prospects for Pharmacological Intervention. Mol. Pharmacol. 2017, 91, 331–338. [Google Scholar] [CrossRef]
- Brown, M.S.; Ho, Y.K.; Goldstein, J.L. The cholesteryl ester cycle in macrophage foam cells. Continual hydrolysis and re-esterification of cytoplasmic cholesteryl esters. J. Biol. Chem. 1980, 255, 9344–9352. [Google Scholar]
- Woollard, K.J.; Geissmann, F. Monocytes in atherosclerosis: subsets and functions. Nat. Rev. Cardiol. 2010, 7, 77–86. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013, 339, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Sharif, F.; Murphy, R.T. Current status of vulnerable plaque detection. Catheter. Cardiovasc. Interv. 2010, 75, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Maenhaut, N.; Van de Voorde, J. Regulation of vascular tone by adipocytes. BMC Med. 2011, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.R. Vulnerable plaque: Definition, diagnosis, and treatment. Cardiol. Clin. 2010, 28, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Nissen, S.E.; Shao, M.; Elshazly, M.B.; Kataoka, Y.; Kapadia, S.R.; Tuzcu, E.M.; Nicholls, S.J. Non-HDL Cholesterol and Triglycerides: Implications for Coronary Atheroma Progression and Clinical Events. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- Yonetsu, T.; Jang, I.-K. Advances in Intravascular Imaging: New Insights into the Vulnerable Plaque from Imaging Studies. Korean Circ. J. 2018, 48, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.; Ishibashi, F.; Muller, J.E. Detection and Treatment of Vulnerable Plaques and Vulnerable Patients. Circulation 2006, 114, 2390–2411. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, S.S.; Stone, G.W.; Singh, M.; Virmani, R.; Olin, J.; Akasaka, T.; Narula, J. Acute coronary syndromes without coronary plaque rupture. Nat. Rev. Cardiol. 2016, 13, 257–265. [Google Scholar] [CrossRef]
- Otsuka, F.; Yasuda, S.; Noguchi, T.; Ishibashi-Ueda, H. Pathology of coronary atherosclerosis and thrombosis. Cardiovasc. Diagn. Ther. 2016, 6, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.R. Pathophysiology of plaque disruption and thrombosis in acute ischemic syndromes. J. Stroke Cerebrovasc. Dis. 2001, 10, 2–9. [Google Scholar] [CrossRef]
- Kramer, M.C.; Rittersma, S.Z.; de Winter, R.J.; Ladich, E.R.; Fowler, D.R.; Liang, Y.H.; Kutys, R.; Carter-Monroe, N.; Kolodgie, F.D.; van der Wal, A.C.; et al. Relationship of thrombus healing to underlying plaque morphology in sudden coronary death. J. Am. Coll. Cardiol. 2010, 55, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Yamamoto, E.; Sugiyama, T.; Jia, H.; Ma, L.; Hu, S.; Wang, C.; Zhu, Y.; Li, L.; Xu, M.; et al. EROSION Study (Effective Anti-Thrombotic Therapy Without Stenting: Intravascular Optical Coherence Tomography-Based Management in Plaque Erosion): A 1-Year Follow-Up Report. Circ. Cardiovasc. Interv. 2017, 10, e005860. [Google Scholar] [CrossRef] [PubMed]
- Costopoulos, C.; Huang, Y.; Brown, A.J.; Calvert, P.A.; Hoole, S.P.; West, N.E.J.; Gillard, J.H.; Teng, Z.; Bennett, M.R. Plaque Rupture in Coronary Atherosclerosis Is Associated With Increased Plaque Structural Stress. JACC Cardiovasc. Imaging 2017, 10, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Koide, M.; Matsuo, A.; Shimoo, S.; Takamatsu, K.; Kyodo, A.; Tsuji, Y.; Mera, K.; Tsubakimoto, Y.; Isodono, K.; Sakatani, T.; et al. Cholesterol crystal depth in coronary atherosclerotic plaques: A novel index of plaque vulnerability using optical frequency domain imaging. PLoS ONE 2017, 12, e0180303. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E. The vulnerable plaque "hypothesis": Promise, but little progress. JACC Cardiovasc. Imaging 2009, 2, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Abela, G.S.; Aziz, K.; Vedre, A.; Pathak, D.R.; Talbott, J.D.; Dejong, J. Effect of cholesterol crystals on plaques and intima in arteries of patients with acute coronary and cerebrovascular syndromes. Am. J. Cardiol. 2009, 103, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Fish, P.M.; Strawn, T.L.; Lohman, A.W.; Wu, J.; Szalai, A.J.; Fay, W.P. C-reactive protein induces expression of tissue factor and plasminogen activator inhibitor-1 and promotes fibrin accumulation in vein grafts. J. Thromb. Haemost. JTH 2014, 12, 1667–1677. [Google Scholar] [CrossRef]
- Van der Meer Irene, M.; de Maat Moniek, P.M.; Hak, A.E.; Kiliaan Amanda, J.; del Sol Antonio, I.; van der Kuip Deirdre, A.M.; Nijhuis Rogier, L.G.; Hofman, A.; Witteman Jacqueline, C.M. C-Reactive Protein Predicts Progression of Atherosclerosis Measured at Various Sites in the Arterial Tree. Stroke 2002, 33, 2750–2755. [Google Scholar] [CrossRef][Green Version]
- Khera, A.; de Lemos James, A.; Peshock Ronald, M.; Lo Hao, S.; Stanek Harold, G.; Murphy Sabina, A.; Wians Frank, H.; Grundy Scott, M.; McGuire Darren, K. Relationship Between C-Reactive Protein and Subclinical Atherosclerosis. Circulation 2006, 113, 38–43. [Google Scholar] [CrossRef]
- Elhage, R.; Jawien, J.; Rudling, M.; Ljunggren, H.G.; Takeda, K.; Akira, S.; Bayard, F.; Hansson, G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 2003, 59, 234–240. [Google Scholar] [CrossRef]
- Blankenberg, S.; Tiret, L.; Bickel, C.; Peetz, D.; Cambien, F.; Meyer, J.; Rupprecht Hans, J. Interleukin-18 Is a Strong Predictor of Cardiovascular Death in Stable and Unstable Angina. Circulation 2002, 106, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kofoed, S.C.; Wittrup, H.H.; Sillesen, H.; Nordestgaard, B.G. Fibrinogen predicts ischaemic stroke and advanced atherosclerosis but not echolucent, rupture-prone carotid plaques: The Copenhagen City Heart Study. Eur. Heart J. 2003, 24, 567–576. [Google Scholar] [CrossRef]
- Miller, A.M.; Xu, D.; Asquith, D.L.; Denby, L.; Li, Y.; Sattar, N.; Baker, A.H.; McInnes, I.B.; Liew, F.Y. IL-33 reduces the development of atherosclerosis. J. Exp. Med. 2008, 205, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Dieplinger, B.; Egger, M.; Haltmayer, M.; Kleber, M.E.; Scharnagl, H.; Silbernagel, G.; de Boer, R.A.; Maerz, W.; Mueller, T. Increased soluble ST2 predicts long-term mortality in patients with stable coronary artery disease: Results from the Ludwigshafen risk and cardiovascular health study. Clin. Chem. 2014, 60, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Ojrzanowski, M.; Figiel, L.; Peruga, J.Z.; Sahni, S.; Kasprzak, J.D. Relative value of serum pregnancy-associated plasma protein A (PAPP-A) and GRACE score for a 1-year prognostication: A complement to calculation in patients with suspected acute coronary syndrome. Adv. Clin. Exp. Med. 2018, 27, 1573–1580. [Google Scholar] [CrossRef]
- Wu, X.F.; Yang, M.; Qu, A.J.; Mintz, G.S.; Yang, Y.; Shang, Y.P.; Gao, H.; Zhang, Y.C.; Ge, C.J.; Wang, L.Y.; et al. Level of Pregnancy-associated Plasma Protein-A Correlates With Coronary Thin-cap Fibroatheroma Burden in Patients With Coronary Artery Disease: Novel Findings From 3-Vessel Virtual Histology Intravascular Ultrasound Assessment. Medicine 2016, 95, e2563. [Google Scholar] [CrossRef]
- Abdo, A.I.; Rayner, B.S.; van Reyk, D.M.; Hawkins, C.L. Low-density lipoprotein modified by myeloperoxidase oxidants induces endothelial dysfunction. Redox Biol. 2017, 13, 623–632. [Google Scholar] [CrossRef]
- Wang, J.; Tan, G.J.; Han, L.N.; Bai, Y.Y.; He, M.; Liu, H.B. Novel biomarkers for cardiovascular risk prediction. J. Geriatr. Cardiol. JGC 2017, 14, 135–150. [Google Scholar]
- Lahdentausta, L.; Leskela, J.; Winkelmann, A.; Tervahartiala, T.; Sorsa, T.; Pesonen, E.; Pussinen, P.J. Serum MMP-9 Diagnostics, Prognostics, and Activation in Acute Coronary Syndrome and Its Recurrence. J. Cardiovasc. Transl. Res. 2018, 11, 210–220. [Google Scholar] [CrossRef]
- Eldrup, N.; Grønholdt Marie-Louise, M.; Sillesen, H.; Nordestgaard Børge, G. Elevated Matrix Metalloproteinase-9 Associated With Stroke or Cardiovascular Death in Patients With Carotid Stenosis. Circulation 2006, 114, 1847–1854. [Google Scholar] [CrossRef]
- Heider, P.; Pelisek, J.; Poppert, H.; Eckstein, H.H. Evaluation of serum matrix metalloproteinases as biomarkers for detection of neurological symptoms in carotid artery disease. Vasc. Endovasc. Surg. 2009, 43, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Gough, P.J.; Gomez, I.G.; Wille, P.T.; Raines, E.W. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J. Clin. Investig. 2006, 116, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Schaub, N.; Reichlin, T.; Meune, C.; Twerenbold, R.; Haaf, P.; Hochholzer, W.; Niederhauser, N.; Bosshard, P.; Stelzig, C.; Freese, M.; et al. Markers of Plaque Instability in the Early Diagnosis and Risk Stratification of Acute Myocardial Infarction. Clin. Chem. 2012, 58, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.; Stewart, A.F. Genes and coronary artery disease: Where are we? J. Am. Coll. Cardiol. 2012, 60, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, E.; Hultman, K.; Duner, P.; Asciutto, G.; Almgren, P.; Orho-Melander, M.; Melander, O.; Nilsson, J.; Hultgardh-Nilsson, A.; Goncalves, I. ADAMTS-7 is associated with a high-risk plaque phenotype in human atherosclerosis. Sci. Rep. 2017, 7, 3753. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, J.; Chen, J.; Su, S.; Chen, R.; Gu, D. Haplotype analysis of the matrix metalloproteinase 3 gene and myocardial infarction in a Chinese Han population. The Beijing atherosclerosis study. Thromb. Haemost. 2004, 92, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.; Xiao, Q.; Kiechl, S.; Chan, K.; Ng, F.L.; Gor, S.; Poston, R.N.; Fang, C.; Patel, A.; Senver, E.C.; et al. ADAMTS7 cleavage and vascular smooth muscle cell migration is affected by a coronary-artery-disease-associated variant. Am. J. Hum. Genet. 2013, 92, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Colige, A.; Monseur, C.; Crawley, J.T.B.; Santamaria, S.; de Groot, R. Proteomic discovery of substrates of the cardiovascular protease ADAMTS7. J. Biol. Chem. 2019, 294, 8037–8045. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Labreuche, J.; Touboul, P.J.; Schmidt-Petersen, K.; Poirier, O.; Perret, C.; Schonfelder, J.; Combadiere, C.; Lathrop, M.; Cambien, F.; et al. Cytokine polymorphisms associated with carotid intima-media thickness in stroke patients. Stroke 2006, 37, 1691–1696. [Google Scholar] [CrossRef]
- Celeng, C.; Takx, R.A.; Ferencik, M.; Maurovich-Horvat, P. Non-invasive and invasive imaging of vulnerable coronary plaque. Trends Cardiovasc. Med. 2016, 26, 538–547. [Google Scholar] [CrossRef]
- Gonzalo, N.; Garcia-Garcia, H.M.; Regar, E.; Barlis, P.; Wentzel, J.; Onuma, Y.; Ligthart, J.; Serruys, P.W. In vivo assessment of high-risk coronary plaques at bifurcations with combined intravascular ultrasound and optical coherence tomography. JACC Cardiovasc. Imaging 2009, 2, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, C.; Zheng, J.; Bach, R.; Muccigrosso, D.; Woodard, P.K.; Tang, D. 3D MRI-based multicomponent thin layer structure only plaque models for atherosclerotic plaques. J. Biomech. 2016, 49, 2726–2733. [Google Scholar] [CrossRef]
- Stefanadis, C.; Diamantopoulos, L.; Vlachopoulos, C.; Tsiamis, E.; Dernellis, J.; Toutouzas, K.; Stefanadi, E.; Toutouzas, P. Thermal Heterogeneity Within Human Atherosclerotic Coronary Arteries Detected In Vivo: A New Method of Detection by Application of a Special Thermography Catheter. Circulation 1999, 99, 1965–1971. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Geng, Y.J.; Guo, B.; Klima, T.; Lal, B.N.; Willerson, J.T.; Casscells, W. Near-infrared spectroscopic characterization of human advanced atherosclerotic plaques. J. Am. Coll. Cardiol. 2002, 39, 1305–1313. [Google Scholar] [CrossRef]
- Brezinski, M.E.; Tearney, G.J.; Weissman, N.J.; Boppart, S.A.; Bouma, B.E.; Hee, M.R.; Weyman, A.E.; Swanson, E.A.; Southern, J.F.; Fujimoto, J.G. Assessing atherosclerotic plaque morphology: Comparison of optical coherence tomography and high frequency intravascular ultrasound. Heart 1997, 77, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Schaar, J.A.; Regar, E.; Mastik, F.; McFadden, E.P.; Saia, F.; Disco, C.; de Korte, C.L.; de Feyter, P.J.; van der Steen, A.F.W.; Serruys, P.W. Incidence of High-Strain Patterns in Human Coronary Arteries: Assessment With Three-Dimensional Intravascular Palpography and Correlation With Clinical Presentation. Circulation 2004, 109, 2716–2719. [Google Scholar] [CrossRef]
- Nair, A.; Kuban, B.D.; Tuzcu, E.M.; Schoenhagen, P.; Nissen, S.E.; Vince, D.G. Coronary plaque classification with intravascular ultrasound radiofrequency data analysis. Circulation 2002, 106, 2200–2206. [Google Scholar] [CrossRef]
- Doonan, R.J.; Hafiane, A.; Lai, C.; Veinot, J.P.; Genest, J.; Daskalopoulou, S.S. Cholesterol efflux capacity, carotid atherosclerosis, and cerebrovascular symptomatology. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 921–926. [Google Scholar] [CrossRef]
- Patel, K.; Tarkin, J.; Serruys, P.W.; Tenekecioglu, E.; Foin, N.; Zhang, Y.-J.; Crake, T.; Moon, J.; Mathur, A.; Bourantas, C.V. Invasive or non-invasive imaging for detecting high-risk coronary lesions? Expert Rev. Cardiovasc. Ther. 2017, 15, 165–179. [Google Scholar] [CrossRef]
- Ge, J.; Chirillo, F.; Schwedtmann, J.; Görge, G.; Haude, M.; Baumgart, D.; Shah, V.; von Birgelen, C.; Sack, S.; Boudoulas, H.; et al. Screening of ruptured plaques in patients with coronary artery disease by intravascular ultrasound. Heart 1999, 81, 621–627. [Google Scholar] [CrossRef][Green Version]
- Rogers, W.J.; Prichard, J.W.; Hu, Y.-L.; Olson, P.R.; Benckart, D.H.; Kramer, C.M.; Vido, D.A.; Reichek, N. Characterization of Signal Properties in Atherosclerotic Plaque Components by Intravascular MRI. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Tearney, G.J.; Jang, I.K.; Bouma, B.E. Optical coherence tomography for imaging the vulnerable plaque. J. Biomed. Opt. 2006, 11, 021002. [Google Scholar] [CrossRef] [PubMed]
- Scarfe, L.; Brillant, N.; Kumar, J.D.; Ali, N.; Alrumayh, A.; Amali, M.; Barbellion, S.; Jones, V.; Niemeijer, M.; Potdevin, S.; et al. Preclinical imaging methods for assessing the safety and efficacy of regenerative medicine therapies. NPJ Regen. Med. 2017, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Kok, A.M.; Speelman, L.; Virmani, R.; van der Steen, A.F.W.; Gijsen, F.J.H.; Wentzel, J.J. Peak cap stress calculations in coronary atherosclerotic plaques with an incomplete necrotic core geometry. BioMed. Eng. OnLine 2016, 15, 48. [Google Scholar] [CrossRef] [PubMed]
- Ohayon, J.; Finet, G.; Gharib, A.M.; Herzka, D.A.; Tracqui, P.; Heroux, J.; Rioufol, G.; Kotys, M.S.; Elagha, A.; Pettigrew, R.I. Necrotic core thickness and positive arterial remodeling index: Emergent biomechanical factors for evaluating the risk of plaque rupture. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H717–H727. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Ren, X.; Vergallo, R.; Xing, L.; Yu, H.; Jia, H.; Soeda, T.; McNulty, I.; Hu, S.; Lee, H.; et al. Distinct morphological features of ruptured culprit plaque for acute coronary events compared to those with silent rupture and thin-cap fibroatheroma: A combined optical coherence tomography and intravascular ultrasound study. J. Am. Coll. Cardiol. 2014, 63, 2209–2216. [Google Scholar] [CrossRef]
- Uemura, S.; Ishigami, K.; Soeda, T.; Okayama, S.; Sung, J.H.; Nakagawa, H.; Somekawa, S.; Takeda, Y.; Kawata, H.; Horii, M.; et al. Thin-cap fibroatheroma and microchannel findings in optical coherence tomography correlate with subsequent progression of coronary atheromatous plaques. Eur. Heart J. 2012, 33, 78–85. [Google Scholar] [CrossRef]
- Zaghloul, A.; Iorgoveanu, C.; Balakumaran, K.; Balanescu, D.V.; Donisan, T. Limitations of Coronary Computed Tomography Angiography in Predicting Acute Coronary Syndrome in a Low to Intermediate-risk Patient with Chest Pain. Cureus 2018, 10, e2649. [Google Scholar] [CrossRef]
- Pohle, K.; Achenbach, S.; Macneill, B.; Ropers, D.; Ferencik, M.; Moselewski, F.; Hoffmann, U.; Brady, T.J.; Jang, I.K.; Daniel, W.G. Characterization of non-calcified coronary atherosclerotic plaque by multi-detector row CT: Comparison to IVUS. Atherosclerosis 2007, 190, 174–180. [Google Scholar] [CrossRef]
- Cai, J.M.; Hatsukami, T.S.; Ferguson, M.S.; Small, R.; Polissar, N.L.; Yuan, C. Classification of human carotid atherosclerotic lesions with in vivo multicontrast magnetic resonance imaging. Circulation 2002, 106, 1368–1373. [Google Scholar] [CrossRef]
- Teng, Z.; Canton, G.; Yuan, C.; Ferguson, M.; Yang, C.; Huang, X.; Zheng, J.; Woodard, P.K.; Tang, D. 3D critical plaque wall stress is a better predictor of carotid plaque rupture sites than flow shear stress: An in vivo MRI-based 3D FSI study. J. Biomech. Eng. 2010, 132, 031007. [Google Scholar] [PubMed]
- Andrews, J.P.M.; Fayad, Z.A.; Dweck, M.R. New methods to image unstable atherosclerotic plaques. Atherosclerosis 2018, 272, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, C.; Zheng, J.; Bach, R.; Muccigrosso, D.; Woodard, P.K.; Tang, D. Higher critical plaque wall stress in patients who died of coronary artery disease compared with those who died of other causes: A 3D FSI study based on ex vivo MRI of coronary plaques. J. Biomech. 2014, 47, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Waters, S.L.; Alastruey, J.; Beard, D.A.; Bovendeerd, P.H.; Davies, P.F.; Jayaraman, G.; Jensen, O.E.; Lee, J.; Parker, K.H.; Popel, A.S.; et al. Theoretical models for coronary vascular biomechanics: Progress & challenges. Prog. Biophys. Mol. Biol. 2011, 104, 49–76. [Google Scholar] [PubMed]
- Coolen, B.F.; Calcagno, C.; van Ooij, P.; Fayad, Z.A.; Strijkers, G.J.; Nederveen, A.J. Vessel wall characterization using quantitative MRI: What’s in a number? Magma 2018, 31, 201–222. [Google Scholar] [CrossRef]
- Osborn, E.A.; Jaffer, F.A. Imaging atherosclerosis and risk of plaque rupture. Curr. Atheroscler. Rep. 2013, 15, 359. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Warlow, C.P. Prediction of benefit from carotid endarterectomy in individual patients: A risk-modelling study. European Carotid Surgery Trialists’ Collaborative Group. Lancet 1999, 353, 2105–2110. [Google Scholar] [CrossRef]
- Jang, I.K.; Tearney, G.J.; MacNeill, B.; Takano, M.; Moselewski, F.; Iftima, N.; Shishkov, M.; Houser, S.; Aretz, H.T.; Halpern, E.F.; et al. In vivo characterization of coronary atherosclerotic plaque by use of optical coherence tomography. Circulation 2005, 111, 1551–1555. [Google Scholar] [CrossRef]
- Chu, B.; Kampschulte, A.; Ferguson, M.S.; Kerwin, W.S.; Yarnykh, V.L.; O’Brien, K.D.; Polissar, N.L.; Hatsukami, T.S.; Yuan, C. Hemorrhage in the Atherosclerotic Carotid Plaque: A High-Resolution MRI Study. Stroke 2004, 35, 1079–1084. [Google Scholar] [CrossRef]
- Cappendijk, V.C.; Cleutjens, K.B.; Heeneman, S.; Schurink, G.W.; Welten, R.J.; Kessels, A.G.; van Suylen, R.J.; Daemen, M.J.; van Engelshoven, J.M.; Kooi, M.E. In vivo detection of hemorrhage in human atherosclerotic plaques with magnetic resonance imaging. J. Magn. Reson. Imaging JMRI 2004, 20, 105–110. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Burke, A.P.; Farb, A.; Gold, H.K.; Yuan, J.; Narula, J.; Finn, A.V.; Virmani, R. The thin-cap fibroatheroma: A type of vulnerable plaque: The major precursor lesion to acute coronary syndromes. Curr. Opin. Cardiol. 2001, 16, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Grasso, M.; Diegoli, M.; Pucci, A.; Bramerio, M.; Ardissino, D.; Angoli, L.; de Servi, S.; Bramucci, E.; Mussini, A.; et al. Coronary atherosclerotic plaques with and without thrombus in ischemic heart syndromes: A morphologic, immunohistochemical, and biochemical study. Am. J. Cardiol. 1991, 68, 36b–50b. [Google Scholar] [CrossRef]
- Schaar, J.A.; De Korte, C.L.; Mastik, F.; Strijder, C.; Pasterkamp, G.; Boersma, E.; Serruys, P.W.; Van Der Steen, A.F. Characterizing vulnerable plaque features with intravascular elastography. Circulation 2003, 108, 2636–2641. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.M.; Davies, M.J. Vulnerable Plaque: Relation of Characteristics to Degree of Stenosis in Human Coronary Arteries. Circulation 1996, 94, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Hackett, D.; Davies, G.; Maseri, A. Pre-existing coronary stenoses in patients with first myocardial infarction are not necessarily severe. Eur. Heart J. 1988, 9, 1317–1323. [Google Scholar] [CrossRef]
- Stone, G.W.; Maehara, A.; Lansky, A.J.; de Bruyne, B.; Cristea, E.; Mintz, G.S.; Mehran, R.; McPherson, J.; Farhat, N.; Marso, S.P.; et al. A Prospective Natural-History Study of Coronary Atherosclerosis. N. Engl. J. Med. 2011, 364, 226–235. [Google Scholar] [CrossRef]
- Nissen, S.E.; Nicholls, S.J.; Sipahi, I.; Libby, P.; Raichlen, J.S.; Ballantyne, C.M.; Davignon, J.; Erbel, R.; Fruchart, J.C.; Tardif, J.-C.; et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: The asteroid trial. JAMA 2006, 295, 1556–1565. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Tuzcu, E.M.; Sipahi, I.; Grasso, A.W.; Schoenhagen, P.; Hu, T.; Wolski, K.; Crowe, T.; Desai, M.Y.; Hazen, S.L.; et al. Statins, high-density lipoprotein cholesterol, and regression of coronary atherosclerosis. JAMA 2007, 297, 499–508. [Google Scholar] [CrossRef]
- Karthikeyan, V.J.; Lip, G.Y. Statins and intra-plaque angiogenesis in carotid artery disease. Atherosclerosis 2007, 192, 455–456. [Google Scholar] [CrossRef]
- Nie, P.; Li, D.; Hu, L.; Jin, S.; Yu, Y.; Cai, Z.; Shao, Q.; Shen, J.; Yi, J.; Xiao, H.; et al. Atorvastatin improves plaque stability in ApoE-knockout mice by regulating chemokines and chemokine receptors. PLoS ONE 2014, 9, e97009. [Google Scholar] [CrossRef]
- Crisby, M.; Nordin-Fredriksson, G.; Shah, P.K.; Yano, J.; Zhu, J.; Nilsson, J. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: Implications for plaque stabilization. Circulation 2001, 103, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Bielicki, J.K.; Zhang, H.; Cortez, Y.; Zheng, Y.; Narayanaswami, V.; Patel, A.; Johansson, J.; Azhar, S. A new HDL mimetic peptide that stimulates cellular cholesterol efflux with high efficiency greatly reduces atherosclerosis in mice. J. Lipid Res. 2010, 51, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Ruchala, P.; Waring, A.J.; Lehrer, R.I.; Hama, S.; Hough, G.; Palgunachari, M.N.; Anantharamaiah, G.M.; Fogelman, A.M. A novel method for oral delivery of apolipoprotein mimetic peptides synthesized from all L-amino acids. J. Lipid Res. 2009, 50, 1538–1547. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.; Jahagirdar, R.; Gordon, A.; Hafiane, A.; Campbell, S.; Chatur, S.; Wagner, G.S.; Hansen, H.C.; Chiacchia, F.S.; Johansson, J.; et al. RVX-208: A small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J. Am. Coll. Cardiol. 2010, 55, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- Kuhnast, S.; van der Tuin, S.J.; van der Hoorn, J.W.; van Klinken, J.B.; Simic, B.; Pieterman, E.; Havekes, L.M.; Landmesser, U.; Luscher, T.F.; Willems van Dijk, K.; et al. Anacetrapib reduces progression of atherosclerosis, mainly by reducing non-HDL-cholesterol, improves lesion stability and adds to the beneficial effects of atorvastatin. Eur. Heart J. 2015, 36, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Takase, S.; Matoba, T.; Nakashiro, S.; Mukai, Y.; Inoue, S.; Oi, K.; Higo, T.; Katsuki, S.; Takemoto, M.; Suematsu, N.; et al. Ezetimibe in Combination with Statins Ameliorates Endothelial Dysfunction in Coronary Arteries After Stenting: The CuVIC Trial (Effect of Cholesterol Absorption Inhibitor Usage on Target Vessel Dysfunction After Coronary Stenting), a Multicenter Randomized Controlled Trial. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 350–358. [Google Scholar] [PubMed]
- Yamashita, S.; Masuda, D.; Ohama, T.; Arai, H.; Bujo, H.; Kagimura, T.; Kita, T.; Matsuzaki, M.; Saito, Y.; Fukushima, M.; et al. Rationale and Design of the PROSPECTIVE Trial: Probucol Trial for Secondary Prevention of Atherosclerotic Events in Patients with Prior Coronary Heart Disease. J. Atheroscler. Thromb. 2016, 23, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, G.; Zhao, X.; Yang, H. Effects of probucol on atherosclerotic plaque and soluble thrombomodulin in patients with coronary heart disease. Exp. Ther. Med. 2018, 16, 886–890. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.; Hogue, M.; Milne, R.W.; Tall, A.R.; Marcel, Y.L. Increase in plasma cholesteryl ester transfer protein during probucol treatment. Relation to changes in high density lipoprotein composition. Arterioscler. Thromb. J. Vasc. Biol. 1991, 11, 476–481. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Leiter, L.A.; Wiviott, S.D.; Giugliano, R.P.; Deedwania, P.; De Ferrari, G.M.; Murphy, S.A.; Kuder, J.F.; Gouni-Berthold, I.; Lewis, B.S.; et al. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: A prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 941–950. [Google Scholar] [CrossRef]
- Cannon, C.P.; Cariou, B.; Blom, D.; McKenney, J.M.; Lorenzato, C.; Pordy, R.; Chaudhari, U.; Colhoun, H.M. Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: The ODYSSEY COMBO II randomized controlled trial. Eur. Heart J. 2015, 36, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Giugliano, R.P.; Sabatine, M.S.; Koren, M.J.; Langslet, G.; Bays, H.; Blom, D.; Eriksson, M.; Dent, R.; Wasserman, S.M.; et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): A pooled analysis of more than 1300 patients in 4 phase II trials. J. Am. Coll. Cardiol. 2014, 63, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Kereiakes, D.J.; McKenney, J.M.; Roth, E.M.; Hanotin, C.; Gipe, D.; Du, Y.; Ferrand, A.C.; Ginsberg, H.N.; Stein, E.A. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am. J. Cardiol. 2014, 114, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Pharmaceutical, I. AKCEA-APO(a)-LRx Advances as Leading Pharmaceutical Company Exercises Option to License. IONIS 2019, 3. [Google Scholar]
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Gomes, F.L.T.; Maranhao, R.C.; Tavares, E.R.; Carvalho, P.O.; Higuchi, M.L.; Mattos, F.R.; Pitta, F.G.; Hatab, S.A.; Kalil-Filho, R.; Serrano, C.V., Jr. Regression of Atherosclerotic Plaques of Cholesterol-Fed Rabbits by Combined Chemotherapy with Paclitaxel and Methotrexate Carried in Lipid Core Nanoparticles. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 561–569. [Google Scholar] [CrossRef]
- Choudhury, R.P.; Birks, J.S.; Mani, V.; Biasiolli, L.; Robson, M.D.; L’Allier, P.L.; Gingras, M.A.; Alie, N.; McLaughlin, M.A.; Basson, C.T.; et al. Arterial Effects of Canakinumab in Patients With Atherosclerosis and Type 2 Diabetes or Glucose Intolerance. J. Am. Coll. Cardiol. 2016, 68, 1769–1780. [Google Scholar] [CrossRef]
- Lou, N. FDA Rejects Canakinumab in CVD Prevention. MED PAGE Today, 19 October 2018. [Google Scholar]
- Leow, C.C.; Coffman, K.; Inigo, I.; Breen, S.; Czapiga, M.; Soukharev, S.; Gingles, N.; Peterson, N.; Fazenbaker, C.; Woods, R.; et al. MEDI3617, a human anti-angiopoietin 2 monoclonal antibody, inhibits angiogenesis and tumor growth in human tumor xenograft models. Int. J. Oncol. 2012, 40, 1321–1330. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Q.; Ke, D.; Li, G. Ghrelin inhibits atherosclerotic plaque angiogenesis and promotes plaque stability in a rabbit atherosclerotic model. Peptides 2017, 90, 17–26. [Google Scholar] [CrossRef]
- Camare, C.; Pucelle, M.; Negre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, P.; Emini Veseli, B.; Van der Veken, B.; Roth, L.; Martinet, W.; De Meyer, G.R.Y. Pharmacological strategies to inhibit intra-plaque angiogenesis in atherosclerosis. Vasc. Pharmacol. 2019, 112, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, L.; Haller, V.; Ritsch, A. A Novel Candidate for Prevention and Treatment of Atherosclerosis: Urolithin B Decreases Lipid Plaque Deposition in apoE(-/-) Mice and Increases Early Stages of Reverse Cholesterol Transport in ox-LDL Treated Macrophages Cells. Mol. Nutr. Food Res. 2019, 63, e1800887. [Google Scholar] [CrossRef] [PubMed]
- Kasim, S.; Moran, D.; McFadden, E. Vulnerable plaque: From bench to bedside; local pacification versus systemic therapy. Heart Views 2012, 13, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Zellweger, M.; Wagnières, G.; Bergh, H.; Cook, S.; Giraud, M.-N. Photodynamic therapy for the treatment of atherosclerotic plaque: Lost in translation? Cardiovasc. Ther. 2017, 35, e12238. [Google Scholar] [CrossRef]
- Waksman, R.; Leitch, I.M.; Roessler, J.; Yazdi, H.; Seabron, R.; Tio, F.; Scott, R.W.; Grove, R.I.; Rychnovsky, S.; Robinson, B.; et al. Intracoronary photodynamic therapy reduces neointimal growth without suppressing re-endothelialisation in a porcine model. Heart 2006, 92, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Kharlamov, A.N. Plasmonic photothermal therapy for atheroregression below Glagov threshold. Future Cardiol. 2013, 9, 405–425. [Google Scholar] [CrossRef]
- Kharlamov, A.N.; Tyurnina, A.E.; Veselova, V.S.; Kovtun, O.P.; Shur, V.Y.; Gabinsky, J.L. Silica-gold nanoparticles for atheroprotective management of plaques: Results of the NANOM-FIM trial. Nanoscale 2015, 7, 8003–8015. [Google Scholar] [CrossRef]
- Feig, J.E. Regression of atherosclerosis: Insights from animal and clinical studies. Ann. Glob. Health 2014, 80, 13–23. [Google Scholar] [CrossRef]
- Keraliya, A.; Blankstein, R. Regression of Coronary Atherosclerosis with Medical Therapy. N. Engl. J. Med. 2017, 376, 1370. [Google Scholar] [CrossRef]
- Matsushita, K.; Hibi, K.; Komura, N.; Akiyama, E.; Maejima, N.; Iwahashi, N.; Tsukahara, K.; Kosuge, M.; Ebina, T.; Sumita, S.; et al. Effects of 4 Statins on Regression of Coronary Plaque in Acute Coronary Syndrome. Circ. J. 2016, 80, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Albers, J.J.; Fisher, L.D.; Schaefer, S.M.; Lin, J.T.; Kaplan, C.; Zhao, X.Q.; Bisson, B.D.; Fitzpatrick, V.F.; Dodge, H.T. Regression of coronary artery disease as a result of intensive lipid-lowering therapy in men with high levels of apolipoprotein B. N. Engl. J. Med. 1990, 323, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Raichlen, J.S.; Nicholls, S.J.; Erbel, R.; Tardif, J.C.; Brener, S.J.; Cain, V.A.; Nissen, S.E. Effect of rosuvastatin therapy on coronary artery stenoses assessed by quantitative coronary angiography: A study to evaluate the effect of rosuvastatin on intravascular ultrasound-derived coronary atheroma burden. Circulation 2008, 117, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Breslow, J.L. Mouse Models of Atherosclerosis. Science 1996, 272, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Trogan, E.; Feig, J.E.; Dogan, S.; Rothblat, G.H.; Angeli, V.; Tacke, F.; Randolph, G.J.; Fisher, E.A. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3781–3786. [Google Scholar] [CrossRef] [PubMed]
- Trogan, E.; Fayad, Z.A.; Itskovich, V.V.; Aguinaldo, J.G.; Mani, V.; Fallon, J.T.; Chereshnev, I.; Fisher, E.A. Serial studies of mouse atherosclerosis by in vivo magnetic resonance imaging detect lesion regression after correction of dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Libby, P.; Nissen, S.E.; Wolski, K.; Ballantyne, C.M.; Barter, P.J.; Chapman, M.J.; Erbel, R.; Raichlen, J.S.; Uno, K.; et al. Long-term effects of maximally intensive statin therapy on changes in coronary atheroma composition: Insights from SATURN. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 380–388. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ballantyne, C.M.; Barter, P.J.; Chapman, M.J.; Erbel, R.M.; Libby, P.; Raichlen, J.S.; Uno, K.; Borgman, M.; Wolski, K.; et al. Effect of two intensive statin regimens on progression of coronary disease. N. Engl. J. Med. 2011, 365, 2078–2087. [Google Scholar] [CrossRef]
- Bayturan, O.; Kapadia, S.; Nicholls, S.J.; Tuzcu, E.M.; Shao, M.; Uno, K.; Shreevatsa, A.; Lavoie, A.J.; Wolski, K.; Schoenhagen, P.; et al. Clinical predictors of plaque progression despite very low levels of low-density lipoprotein cholesterol. J. Am. Coll. Cardiol. 2010, 55, 2736–2742. [Google Scholar] [CrossRef]
- Gragnano, F.; Calabro, P. Role of dual lipid-lowering therapy in coronary atherosclerosis regression: Evidence from recent studies. Atherosclerosis 2018, 269, 219–228. [Google Scholar] [CrossRef]
- Li, T.; Chen, W.; An, F.; Tian, H.; Zhang, J.; Peng, J.; Zhang, Y.; Guo, Y. Probucol attenuates inflammation and increases stability of vulnerable atherosclerotic plaques in rabbits. Tohoku J. Exp. Med. 2011, 225, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Ikegami, C.; Tsujii, K.; Zhang, Z.; Matsuura, F.; Nakagawa-Toyama, Y.; Koseki, M.; Masuda, D.; Maruyama, T.; Shimomura, I.; et al. Probucol enhances the expression of human hepatic scavenger receptor class B. type, I.; possibly through a species-specific mechanism. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2422–2427. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Ben, D.O.A.; Spahis, S.; Sane, A.T.; Garofalo, C.; Grenier, E.; Emonnot, L.; Yara, S.; Couture, P.; Beaulieu, J.F.; et al. PCSK9 plays a significant role in cholesterol homeostasis and lipid transport in intestinal epithelial cells. Atherosclerosis 2013, 227, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Hiro, T.; Hirayama, A.; Komatsu, S.; Matsuoka, H.; Takayama, T.; Ishihara, M.; Hayashi, T.; Saito, S.; Kodama, K. Effect of Ezetimibe on Stabilization and Regression of Intracoronary Plaque—The ZIPANGU Study. Circ. J. 2017, 81, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Puri, R.; Anderson, T.; Ballantyne, C.M.; Cho, L.; Kastelein, J.J.; Koenig, W.; Somaratne, R.; Kassahun, H.; Yang, J.; et al. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA 2016, 316, 2373–2384. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.C.; Kaouache, M.; Grover, S.A. Evaluation of the cost-effectiveness of evolocumab in the fourier study: A canadian analysis. CMAJ Open 2018, 6, E162–E167. [Google Scholar] [CrossRef][Green Version]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef]
- Jaeger, B.R.; Richter, Y.; Nagel, D.; Heigl, F.; Vogt, A.; Roeseler, E.; Parhofer, K.; Ramlow, W.; Koch, M.; Utermann, G.; et al. Investigators ftGoC. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 229. [Google Scholar]
- Safarova, M.S.; Ezhov, M.V.; Afanasieva, O.I.; Matchin, Y.G.; Atanesyan, R.V.; Adamova, I.Y.; Utkina, E.A.; Konovalov, G.A.; Pokrovsky, S.N. Effect of specific lipoprotein(a) apheresis on coronary atherosclerosis regression assessed by quantitative coronary angiography. Atheroscler. Suppl. 2013, 14, 93–99. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Tall, A.R.; Westerterp, M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J. Lipid Res. 2019, 60, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Post, S.; Peeters, W.; Busser, E.; Lamers, D.; Sluijter, J.P.; Goumans, M.J.; de Weger, R.A.; Moll, F.L.; Doevendans, P.A.; Pasterkamp, G.; et al. Balance between angiopoietin-1 and angiopoietin-2 is in favor of angiopoietin-2 in atherosclerotic plaques with high microvessel density. J. Vasc. Res. 2008, 45, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.S.; Cronstein, B.N. Mechanisms of action of methotrexate. Bull. Hosp. Jt. Dis. 2013, 71, S5–S8. [Google Scholar]
- Tang, W.H.; Hazen, S.L. Atherosclerosis in 2016: Advances in new therapeutic targets for atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, M.; Fukushi, J.; Okamoto, M.; Nishie, A.; Goto, H.; Ishibashi, T.; Ono, M. Angiogenesis factors. Int. Med. 2001, 40, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 1999, 99, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Yuan, R.; Shi, W.L.; Xin, Q.Q.; Chen, K.J.; Cong, W.H. Holistic Regulation of Angiogenesis with Chinese Herbal Medicines as a New Option for Coronary Artery Disease. Evid.-Based Complement. Altern. Med. 2018, 2018, 3725962. [Google Scholar] [CrossRef]
- Libby, P. Mechanisms of Acute Coronary Syndromes and Their Implications for Therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Hafiane, A.; Kellett, S.; Genest, J. Treatment options for low high-density lipoproteins. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Hafiane, A.; Genest, J. HDL, Atherosclerosis, and Emerging Therapies. Cholesterol 2013, 2013, 891403. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Calabresi, L.; Franceschini, G.; Baldassarre, D.; Amato, M.; Johansson, J.; Salvetti, M.; Monteduro, C.; Zulli, R.; Muiesan, M.L.; et al. Cardiovascular status of carriers of the apolipoprotein A-I(Milano) mutant: The Limone sul Garda study. Circulation 2001, 103, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Bielicki, J.K. ABCA1 agonist peptides for the treatment of disease. Curr. Opin. Lipidol. 2016, 27, 40–46. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Ranalletta, M.; Wang, N.; Han, S.; Terasaka, N.; Li, R.; Welch, C.; Tall, A.R. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Investig. 2007, 117, 3900–3908. [Google Scholar] [CrossRef]
- Hafiane, A.; Jabor, B.; Ruel, I.; Ling, J.; Genest, J. High-density lipoprotein mediated cellular cholesterol efflux in acute coronary syndromes. Am. J. Cardiol. 2014, 113, 249–255. [Google Scholar] [CrossRef]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol Efflux Capacity, High-Density Lipoprotein Function, and Atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef]
- Zimetti, F.; Freitas, W.M.; Campos, A.M.; Daher, M.; Adorni, M.P.; Bernini, F.; Sposito, A.C.; Zanotti, I. Cholesterol efflux capacity does not associate with coronary calcium, plaque vulnerability, and telomere length in healthy octogenarians. J. Lipid Res. 2018, 59, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Mutharasan, R.K.; Thaxton, C.S.; Berry, J.; Daviglus, M.L.; Yuan, C.; Sun, J.; Ayers, C.; Lloyd-Jones, D.M.; Wilkins, J.T. HDL efflux capacity, HDL particle size, and high-risk carotid atherosclerosis in a cohort of asymptomatic older adults: The Chicago Healthy Aging Study. J. Lipid Res. 2017, 58, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Heidari, M.; Ebrahimian, T.; Simon, D.; Vali, H.; Mandato, C.A.; Lehoux, S. SEMAPHORIN3A REDUCES ATHEROSCLEROTIC PLAQUE FORMATION IN APOE-/-MICE. Can. J. Cardiol. 2015, 31, S281. [Google Scholar] [CrossRef]
- Ta, V.; Ebrahimian, T.; Simon, D.; Lehoux, S. Semaphorin-3A Promotes Foam Cell Migration. Atheroscler. Suppl. 2018, 32, 103–104. [Google Scholar] [CrossRef]
- Wanschel, A.; Seibert, T.; Hewing, B.; Ramkhelawon, B.; Ray, T.D.; van Gils, J.M.; Rayner, K.J.; Feig, J.E.; O’Brien, E.R.; Fisher, E.A.; et al. Neuroimmune guidance cue Semaphorin 3E is expressed in atherosclerotic plaques and regulates macrophage retention. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 886–893. [Google Scholar] [CrossRef]
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased lesion formation in CCR2-/-mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar] [CrossRef]
- Feig, J.E.; Shang, Y.; Rotllan, N.; Vengrenyuk, Y.; Wu, C.; Shamir, R.; Torra, I.P.; Fernandez-Hernando, C.; Fisher, E.A.; Garabedian, M.J. Statins promote the regression of atherosclerosis via activation of the CCR7-dependent emigration pathway in macrophages. PLoS ONE 2011, 6, e28534. [Google Scholar] [CrossRef]
- Gasbarrino, K.; Zheng, H.; Hafiane, A.; Veinot, J.P.; Lai, C.; Daskalopoulou, S.S. Decreased Adiponectin-Mediated Signaling Through the AdipoR2 Pathway Is Associated with Carotid Plaque Instability. Stroke 2017, 48, 915–924. [Google Scholar] [CrossRef]
- Kereiakes, D.J.; Szyniszewski, A.M.; Wahr, D.; Herrmann, H.C.; Simon, D.I.; Rogers, C.; Kramer, P.; Shear, W.; Yeung, A.C.; Shunk, K.A.; et al. Phase I Drug and Light Dose-Escalation Trial of Motexafin Lutetium and Far Red Light Activation (Phototherapy) in Subjects With Coronary Artery Disease Undergoing Percutaneous Coronary Intervention and Stent Deployment. Circulation 2003, 108, 1310–1315. [Google Scholar] [CrossRef]
- DiStasio, N.; Lehoux, S.; Khademhosseini, A.; Tabrizian, M. The Multifaceted Uses and Therapeutic Advantages of Nanoparticles for Atherosclerosis Research. Materials 2018, 11, 754. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Steps | Characteristics and Consequences | Ref. |
|---|---|---|
| (1) LDL binds to the subendothelial arterial matrix | Retention/accumulation of ApoB-containing lipoprotein in the arterial intima; Lp(a) binding to extracellular matrix; fatty streaks forming in the vessel wall. | [32] [36] [32] |
| (2) Chemical modification of LDL: oxidation of LDL, acetylated LDL | Activation of endothelial cells lining the vessel wall, damage to endothelial cells and macrophages and maintenance of leucocyte recruitment. | [37] |
| (3) Recruitment of monocytes–macrophages to the arterial wall | Activated VSMCs secrete proinflammatory chemokines and contribute to the recruitment of monocytes, which differentiate to macrophages. Expression of leukocyte adhesion molecules on the endothelial wall. | [38] [5] |
| (3.1) Adhesion of inflammatory cells to endothelium surface | VCAM-1 upregulation; migration of monocytes into the intima; monocyte differentiation. | [37] |
| (4) Uptake of oxidized LDL by family scavenger receptors (TLR, SR-A, CD 63, and others) Accumulation of Lp(a) in the vessel wall | Scavenger receptors bind and then internalize modified LDL into the media. Macrophages phagocytes, consumes LDL, and forms foam cells. Initiate macrophage buildup in the atherosclerotic plaque. Promote cholesterol accumulation in macrophages, forming foam cells and subsequent fatty streaks. | [39] [40] [33] |
| (4.1) Macrophage accumulation in arterial wall; cholesterol deposition | Crystallization of cholesterol in atherosclerotic plaques; increased ET-1 production and decreased nitric oxide production. | [41] [42] [43] |
| (5) Fibrous cap formation | Matrix deposition, migration, and proliferation of VSMCs; loss of proteoglycans or collagen; progressive narrowing and hardening of the arteries. | [44] |
| (5.1) Advanced plaque: Plaque-associated thrombosis | Plaque erosion/rupture. | [45] |
| Pharmacotherapeutic Strategy | Drug | Aim, Effects, and Clinical Phase | Ref. |
|---|---|---|---|
| Inhibit HMG–CoA reductase | Statin | Inhibit cholesterol synthesis; Increase the clearance of LDL-C; Promote plaque regression and reduce IP angiogenesis; Decrease monocyte recruitment to the plaque; Reduce macrophage accumulation in the plaque; Inhibit the production of MMP; FDA approval | [117] [118] [119] [120] [121] |
| HDL biogenesis | Fibrate RVX-208 HDL mimetic peptides* | Increase apoA-I production and promote the secretion of LDL. Approved; Plaque regression in mice: RVX phase III; Regression of coronary atherosclerosis; No studies | [122] [123] [124] |
| CETP inhibitors Combination therapy with statins | Anacetrapib | Regress atherosclerotic plaques; Improvement of lesion stability; Phase III | [125] |
| Cholesterol absorption reduction: inhibition of NPC1L1 | Ezetimibe Atorvastatin | Regress atherosclerosis; Reduction of plaque volume; Approved by FDA | [126] |
| Lipid-lowering by increasing the rate of LDL catabolism | Probucol | May inhibit early stages of cholesterol biosynthesis; Reduce atherosclerotic plaque; Inhibits ABCA1-mediated cellular lipid efflux; Reduce serum HDL-C; In use in Japan, and left by western countries | [127] [128] [129] |
| (PCSK9) inhibitors with human antibodies | Repatha® Praluent® | Regress atherosclerosis; Hypercholesterolemia therapy; Approved by EMA and FDA | [130] [131] |
| Lp(a)-lowering therapies | Evolocumab Alirocumab AKCEA-APO(a)-LRx Antisense RNA (ISIS-APO(a)Rx) | Phase II trials Phase III trials Phase II trials | [132] [133] [134] [135] |
| Targeting anti-inflammatory pathway IL-1β inhibition by monoclonal antibodies | Methotrexate Canakinumab | Residual major CV event risk reduced by 15%; May affect vascular disease progression Phase IV ongoing; Data from the CANTOS trial III were not approved by FDA | [136] [137] [138] [139] |
| Angiopoietin-2 blockage with monoclonal antibodies | Antibodies: MEDI3617 | Promote plaque neovascularization in vivo. No toxicity in vivo. Phase I trials. | [2] [140] |
| Angiogenesis inhibitors | Ghrelin Anti-VEGF/VEGFR: Bevacizumab | Promote plaque stability in vivo. Experimental phase. Controversy in preventing plaque instability. Inhibit IP angiogenesis. Reduce IP hemorrhage: approved. | [141] [142] [143] |
| Activate RCT | Urolithin B | Increase RCT in foam cells of apoE−/− mice; Experimental phase | [144] |
| Mechanical regression therapies: Plaque sealing Photodynamic Thermotherapy | Metallic Bioabsorbale Photosensitizer Plasmonic nanoparticles | Mechanically rupture plaque; Lack of clinical data; limited applications despite safety trials; Absence of clinical data; Promote atheroregression below 40%; Phase I safety trial | [145] [146] [147] [148] [149] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafiane, A. Vulnerable Plaque, Characteristics, Detection, and Potential Therapies. J. Cardiovasc. Dev. Dis. 2019, 6, 26. https://doi.org/10.3390/jcdd6030026
Hafiane A. Vulnerable Plaque, Characteristics, Detection, and Potential Therapies. Journal of Cardiovascular Development and Disease. 2019; 6(3):26. https://doi.org/10.3390/jcdd6030026
Chicago/Turabian StyleHafiane, Anouar. 2019. "Vulnerable Plaque, Characteristics, Detection, and Potential Therapies" Journal of Cardiovascular Development and Disease 6, no. 3: 26. https://doi.org/10.3390/jcdd6030026
APA StyleHafiane, A. (2019). Vulnerable Plaque, Characteristics, Detection, and Potential Therapies. Journal of Cardiovascular Development and Disease, 6(3), 26. https://doi.org/10.3390/jcdd6030026