The Development of Compartmentation of cAMP Signaling in Cardiomyocytes: The Role of T-Tubules and Caveolae Microdomains

{kind=link}
{kind=link}
Abstract
1. Introduction
2. β-Adrenergic Pathway
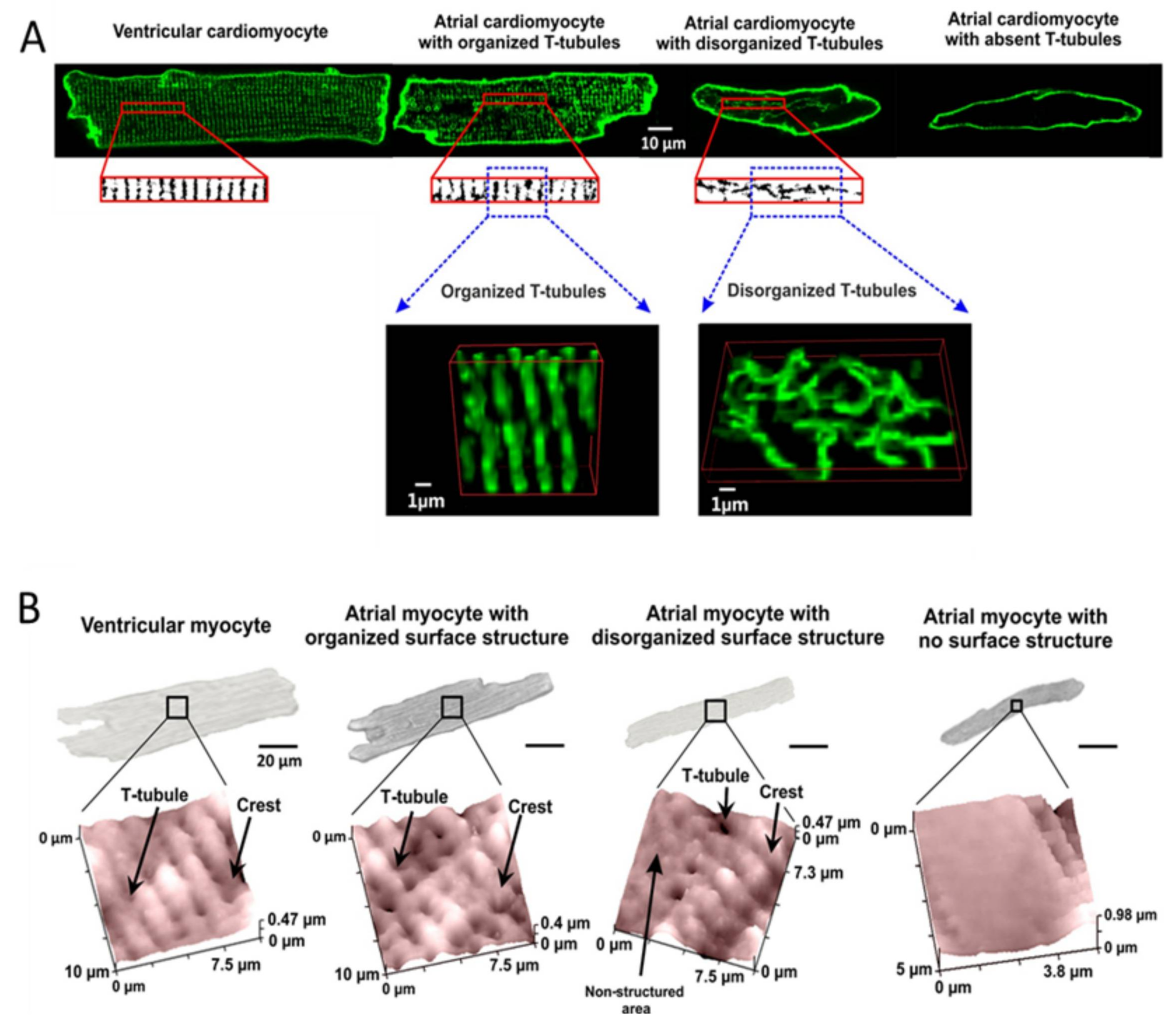
3. Transverse Axial Tubular System
4. Caveolae
5. Microdomains of cAMP Signalling in Healthy Ventricular Myocytes: T-Tubules and Caveolae
6. PDEs and cAMP Compartmentation in Ventricular Myocytes
7. cAMP Compartmentation and PDEs in Atrial Myocytes
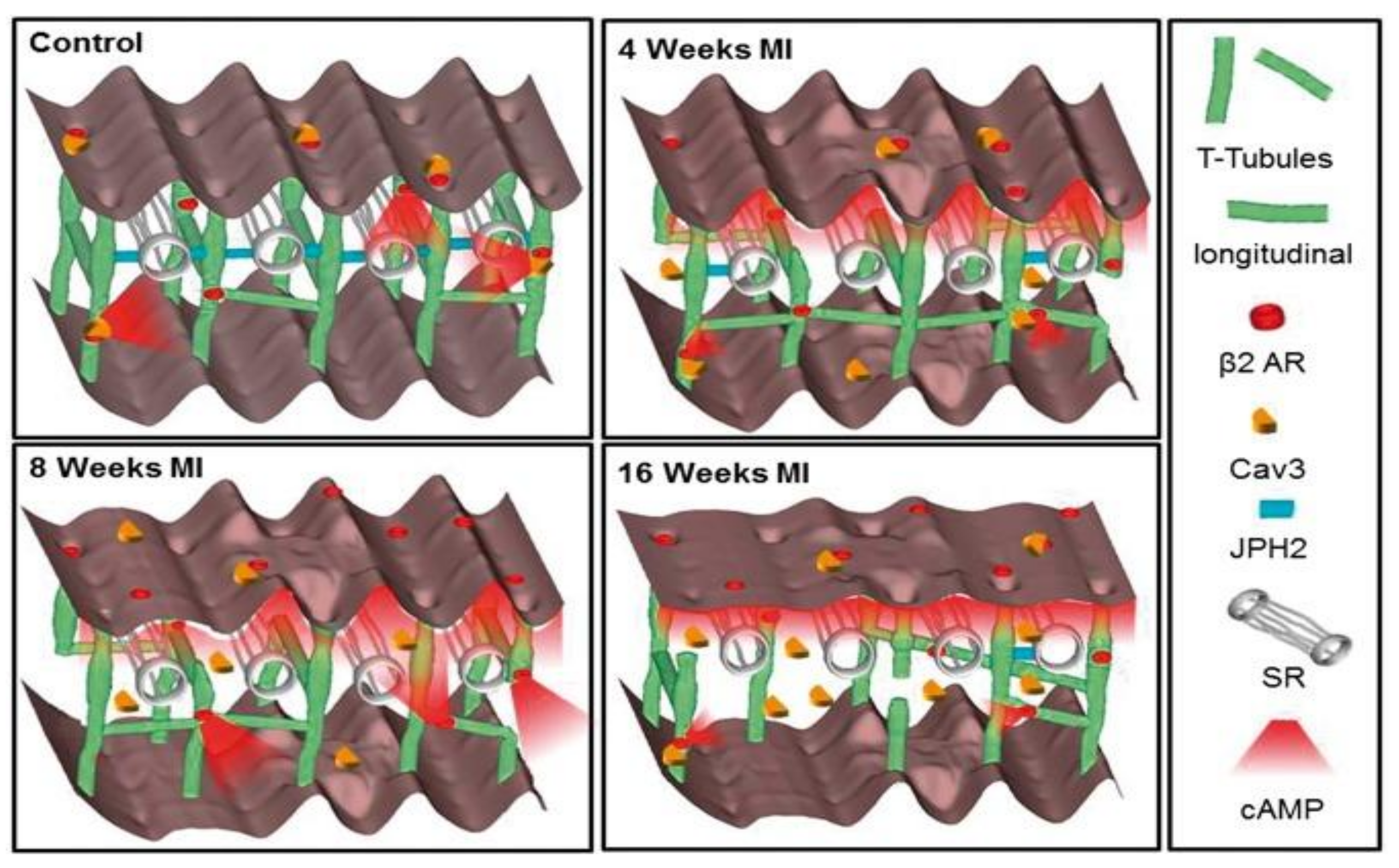
8. Failing Myocardium Represents Progressive Reduction in the Complexity of Structure and Receptor Function
9. Biogenesis and Maturation of Structural Microdomains during Differentiation of Myocytes from Stem Cells
10. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Brunton, L.L.; Hayes, J.S.; Mayer, S.E. Hormonally specific phosphorylation of cardiac troponin I and activation of glycogen phosphorylase. Nature 1979, 280, 78. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.S.; Brunton, L.L.; Brown, J.H.; Reese, J.B.; Mayer, S.E. Hormonally specific expression of cardiac protein kinase activity. Proc. Natl. Acad. Sci. USA 1979, 76, 1570–1574. [Google Scholar] [CrossRef] [PubMed]
- Buxton, I.L.; Brunton, L.L. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem. 1983, 258, 10233–10239. [Google Scholar] [PubMed]
- Steinberg, S.F.; Brunton, L.L. Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 751–773. [Google Scholar] [CrossRef] [PubMed]
- Saucerman, J.J.; Greenwald, E.C.; Polanowska-Grabowska, R. Mechanisms of cyclic AMP compartmentation revealed by computational models. J. Gen. Physiol. 2014, 143, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R. Beta-adrenergic signaling in the heart: Dual coupling of the beta2-adrenergic receptor to G (s) and G (i) proteins. Sci. Stke. 2001, 104, re15. [Google Scholar]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Zoccarato, A.; Lissandron, V.; Terrin, A.; Li, X.; Houslay, M.D.; Baillie, G.S.; Zaccolo, M. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ. Res. 2008, 103, 836–844. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D.; Tortora, P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 1991, 351, 145. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.; Breher, S.S.; Waldeyer, C.; Schindler, R.F.; Nikolaev, V.O.; Rinné, S.; Wischmeyer, E.; Schlueter, J.; Becher, J.; Simrick, S. Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J. Clin. Investig. 2012, 122, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Lands, A.M.; Luduena, F.P.; Buzzo, H.J. Differentiation of receptors responsive to isoproterenol. Life Sci. 1967, 6, 2241–2249. [Google Scholar] [CrossRef]
- Lands, A.; ARNOLD, A.; McAuliff, J.P.; Luduena, F.P.; Jun, T.B. Differentiation of receptor systems activated by sympathomimetic amines. Nature 1967, 214, 597. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, D.K.; Desai, K.H.; Jasper, J.R.; Stevens, M.E.; Regula, D.P.; Barsh, G.S.; Bernstein, D.; Kobilka, B.K. Targeted disruption of the mouse beta1-adrenergic receptor gene: developmental and cardiovascular effects. Proc. Natl. Acad. Sci. USA 1996, 93, 7375–7380. [Google Scholar] [CrossRef] [PubMed]
- Chruscinski, A.J.; Rohrer, D.K.; Schauble, E.; Desai, K.H.; Bernstein, D.; Kobilka, B.K. Targeted disruption of the β2 adrenergic receptor gene. J. Biol. Chem. 1999, 274, 16694–16700. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Ginsburg, R.; Umans, V.; Fowler, M.; Minobe, W.; Rasmussen, R.; Zera, P.; Menlove, R.; Shah, P.; Jamieson, S. Beta 1-and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: Coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ. Res. 1986, 59, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Timofeyev, V.; Myers, R.E.; Kim, H.J.; Woltz, R.; Sirish, P.; Heiserman, J.; Li, N.; Singapuri, A.; Tang, T.; Yarov-Yarovoy, V. Adenylyl cyclase subtype-specific compartmentalization: Differential regulation of L-type Ca2+ current in ventricular myocytes. Circ. Res. 2013, 112, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Takagi, G.; Kawabe, J.; Yang, G.; Lee, M.; Hong, C.; Liu, J.; Vatner, D.E.; Sadoshima, J.; Vatner, S.F. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc. Natl. Acad. Sci. USA 2003, 100, 9986–9990. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.M.; Gao, M.H.; Lai, N.C.; Drumm, J.; Dalton, N.; Zhou, J.Y.; Zhu, J.; Entrikin, D.; Hammond, H.K. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation 1999, 99, 3099–3102. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.M.; Bayat, H.; Drumm, J.D.; Gao, M.H.; Swaney, J.S.; Ander, A.; Hammond, H.K. Adenylyl cyclase increases survival in cardiomyopathy. Circulation 2002, 105, 1989–1994. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Gao, M.H.; Lai, N.C.; Firth, A.L.; Takahashi, T.; Guo, T.; Yuan, J.X.; Roth, D.M.; Hammond, H.K. Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation 2008, 117, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Pinali, C.; Bennett, H.; Davenport, J.B.; Trafford, A.W.; Kitmitto, A. Three-Dimensional Reconstruction of Cardiac Sarcoplasmic Reticulum Reveals a Continuous Network Linking Transverse-TubulesNovelty and Significance: This Organization Is Perturbed in Heart Failure. Circ. Res. 2013, 113, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Balijepalli, R.C.; Foell, J.D.; Hall, D.D.; Hell, J.W.; Kamp, T.J. Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 7500–7505. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Brandenburg, S.; Kohl, T.; Lehnart, S.E. Analysis of tubular membrane networks in cardiac myocytes from atria and ventricles. J. Vis. Exp. 2014, 92, 51823. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.H.; Rog-Zielinska, E.A.; Orchard, C.H.; Kohl, P.; Cannell, M.B. Sub-microscopic analysis of t-tubule geometry in living cardiac ventricular myocytes using a shape-based analysis method. J. Mol. Cell. Cardiol. 2017, 108, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, F.R.; Bito, V.; Volders, P.G.; Antoons, G.; Mubagwa, K.; Sipido, K.R. Spatial and temporal inhomogeneities during Ca2+ release from the sarcoplasmic reticulum in pig ventricular myocytes. Circ. Res. 2002, 91, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.; Chien, E.Y.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A specific cholesterol binding site is established by the 2.8 Å structure of the human β 2-adrenergic receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, A.; Lin, X.; Novak, P.; Mehta, K.; Korchev, Y.; Delmar, M.; Gorelik, J. Super-resolution Scanning Patch Clamp Reveals Clustering of Functional Ion Channels in Adult Ventricular MyocyteNovelty and Significance. Circ. Res. 2013, 112, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, S.; Kohl, T.; Williams, G.S.; Gusev, K.; Wagner, E.; Rog-Zielinska, E.A.; Hebisch, E.; Dura, M.; Didié, M.; Gotthardt, M. Axial tubule junctions control rapid calcium signaling in atria. J. Clin. Investig. 2016, 126, 3999–4015. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.A.; Clarke, J.D.; Saravanan, P.; Voigt, N.; Dobrev, D.; Eisner, D.A.; Trafford, A.W.; Dibb, K.M. Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H1996–H2005. [Google Scholar] [CrossRef] [PubMed]
- Gadeberg, H.C.; Bond, R.C.; Kong, C.H.; Chanoit, G.P.; Ascione, R.; Cannell, M.B.; James, A.F. Heterogeneity of T-tubules in pig hearts. PLoS ONE 2016, 11, e0156862. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Nakajima, T.; Hazama, H.; Hamada, E.; Murakawa, Y.; Sawada, K.; Omata, M. Regional differences in transient outward current density and inhomogeneities of repolarization in rabbit right atrium. Circulation 1995, 92, 3061–3069. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, A.V.; Balycheva, M.; Sanchez-Alonso, J.L.; Ilkan, Z.; Alvarez-Laviada, A.; Bhogal, N.; Diakonov, I.; Schobesberger, S.; Sikkel, M.B.; Bhargava, A. Direct evidence for microdomain-specific localization and remodeling of functional L-type calcium channels in rat and human atrial myocytes. Circulation 2015, 132, 2372–2384. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhang, R.; Kim, B.; Ma, A.; Philipson, K.D.; Goldhaber, J.I. Heterogeneity of transverse-axial tubule system in mouse atria: Remodeling in atrial-specific Na+–Ca2+ exchanger knockout mice. J. Mol. Cell. Cardiol. 2017, 108, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Frisk, M.; Koivumäki, J.T.; Norseng, P.A.; Maleckar, M.M.; Sejersted, O.M.; Louch, W.E. Variable t-tubule organization and Ca2+ homeostasis across the atria. Am. J. Physiol.-Heart Circ. Physiol. 2014, 307, H609–H620. [Google Scholar] [CrossRef] [PubMed]
- Hayer, A.; Stoeber, M.; Bissig, C.; Helenius, A. Biogenesis of caveolae: Stepwise assembly of large caveolin and cavin complexes. Traffic 2010, 11, 361–382. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Köster, D.; Ruez, R.; Gonnord, P.; Bastiani, M.; Abankwa, D.; Stan, R.V.; Butler-Browne, G.; Vedie, B.; Johannes, L. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 2011, 144, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Kohl, P.; Cooper, P.J.; Holloway, H. Effects of acute ventricular volume manipulation on in situ cardiomyocyte cell membrane configuration. Prog. Biophys. Mol. Biol. 2003, 82, 221–227. [Google Scholar] [CrossRef]
- Kozera, L.; White, E.; Calaghan, S. Caveolae act as membrane reserves which limit mechanosensitive ICl, swell channel activation during swelling in the rat ventricular myocyte. PLoS ONE 2009, 4, e8312. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Scarlata, S. Super-resolution Visualization of Caveola Deformation in Response to Osmotic Stress. J. Biol. Chem. 2017, 292, 3779–3788. [Google Scholar] [CrossRef] [PubMed]
- Mohan, J.; Morén, B.; Larsson, E.; Holst, M.R.; Lundmark, R. Cavin3 interacts with cavin1 and caveolin1 to increase surface dynamics of caveolae. J. Cell. Sci. 2015, 128, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Calaghan, S.; Kozera, L.; White, E. Compartmentalisation of cAMP-dependent signalling by caveolae in the adult cardiac myocyte. J. Mol. Cell. Cardiol. 2008, 45, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bünemann, M.; Schmitteckert, E.; Lohse, M.J.; Engelhardt, S. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching β1-adrenergic but locally confined β2-adrenergic receptor–mediated signaling. Circ. Res. 2006, 99, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Schobesberger, S.; Wright, P.; Tokar, S.; Bhargava, A.; Mansfield, C.; Glukhov, A.V.; Poulet, C.; Buzuk, A.; Monszpart, A.; Sikkel, M. T-tubule remodelling disturbs localized β2-adrenergic signalling in rat ventricular myocytes during the progression of heart failure. Cardiovasc. Res. 2017, 113, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.T.; Nikolaev, V.O.; O’Hara, T.; Diakonov, I.; Bhargava, A.; Tokar, S.; Schobesberger, S.; Shevchuk, A.I.; Sikkel, M.B.; Wilkinson, R. Caveolin-3 regulates compartmentation of cardiomyocyte β2-adrenergic receptor-mediated cAMP signaling. J. Mol. Cell. Cardiol. 2014, 67, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Levin, K.R.; Page, E. Quantitative studies on plasmalemmal folds and caveolae of rabbit ventricular myocardial cells. Circ. Res. 1980, 46, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Burton, R.A.; Rog-Zielinska, E.A.; Corbett, A.D.; Peyronnet, R.; Bodi, I.; Fink, M.; Sheldon, J.; Hoenger, A.; Calaghan, S.C.; Bub, G. Caveolae in rabbit ventricular myocytes: distribution and dynamic diminution after cell isolation. Biophys. J. 2017, 113, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.D.; Calaghan, S.C. Caveolae create local signalling domains through their distinct protein content, lipid profile and morphology. J. Mol. Cell. Cardiol. 2012, 52, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Rybin, V.O.; Pak, E.; Alcott, S.; Steinberg, S.F. Developmental changes in β2-adrenergic receptor signaling in ventricular myocytes: the role of Gi proteins and caveolae microdomains. Mol. Pharmacol. 2003, 63, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Head, B.P.; Patel, H.H.; Roth, D.M.; Lai, N.C.; Niesman, I.R.; Farquhar, M.G.; Insel, P.A. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J. Biol. Chem. 2005, 280, 31036–31044. [Google Scholar] [CrossRef] [PubMed]
- Calaghan, S.; White, E. Caveolae modulate excitation–contraction coupling and β2-adrenergic signalling in adult rat ventricular myocytes. Cardiovasc. Res. 2006, 69, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.R.; MacDougall, D.A.; Tyser, R.; Pugh, S.D.; Calaghan, S.C.; Harvey, R.D. Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 50, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Lefkimmiatis, K.; Zaccolo, M. cAMP signaling in subcellular compartments. Pharmacol. Ther. 2014, 143, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M. cAMP signal transduction in the heart: understanding spatial control for the development of novel therapeutic strategies. Br. J. Pharmacol. 2009, 158, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Abi-Gerges, A.; Nikolaev, V.O.; Richter, W.; Lechêne, P.; Mazet, J.; Conti, M.; Fischmeister, R.; Vandecasteele, G. Spatiotemporal Dynamics of β-Adrenergic cAMP Signals and L-Type Ca2+ Channel Regulation in Adult Rat Ventricular Myocytes. Circ. Res. 2008, 102, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Mika, D.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. PDEs create local domains of cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M. Phosphodiesterases and compartmentalized cAMP signalling in the heart. Eur. J. Cell Biol. 2006, 85, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; Zaccolo, M. A complex phosphodiesterase system controls beta-adrenoceptor signalling in cardiomyocytes. Biochem. Soc. Trans. 2006, 34, 510–511. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Mika, D.; Richter, W. Cyclic AMP compartments and signaling specificity: Role of cyclic nucleotide phosphodiesterases. J. Gen. Physiol. 2014, 143, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Lomas, O.; Zaccolo, M. Phosphodiesterases maintain signaling fidelity via compartmentalization of cyclic nucleotides. Physiology 2014, 29, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Rapundalo, S.T.; Solaro, R.J.; Kranias, E.G. Inotropic responses to isoproterenol and phosphodiesterase inhibitors in intact guinea pig hearts: Comparison of cyclic AMP levels and phosphorylation of sarcoplasmic reticulum and myofibrillar proteins. Circ. Res. 1989, 64, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Corbin, J.D. Structure and function of cyclic nucleotide-dependent protein kinases. Annu. Rev. Physiol. 1994, 56, 237–272. [Google Scholar] [CrossRef] [PubMed]
- Stangherlin, A.; Zaccolo, M. Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, H379–H390. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Kotera, J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Verde, I.; Vandecasteele, G.; Lezoualc’h, F.; Fischmeister, R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br. J. Pharmacol. 1999, 127, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C.; Muller, B.; Le Bec, A.; Beaudry, C.; Rousseau, E. Characterization of indolidan-and rolipram-sensitive cyclic nucleotide phosphodiesterases in canine and human cardiac microsomal fractions. J. Pharmacol. Exp. Ther. 1993, 265, 1142–1151. [Google Scholar] [PubMed]
- Sun, B.; Li, H.; Shakur, Y.; Hensley, J.; Hockman, S.; Kambayashi, J.; Manganiello, V.C.; Liu, Y. Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell. Signal. 2007, 19, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Beca, S.; Ahmad, F.; Shen, W.; Liu, J.; Makary, S.; Polidovitch, N.; Sun, J.; Hockman, S.; Chung, Y.W.; Movsesian, M. Phosphodiesterase Type 3A Regulates Basal Myocardial Contractility Through Interacting With Sarcoplasmic Reticulum Calcium ATPase Type 2a Signaling Complexes in Mouse HeartNovelty and Significance. Circ. Res. 2013, 112, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Movsesian, M. New pharmacologic interventions to increase cardiac contractility: Challenges and opportunities. Curr. Opin. Cardiol. 2015, 30, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Xie, M.; Scheitrum, C.; Krall, J.; Movsesian, M.A.; Conti, M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res. Cardiol. 2011, 106, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Molina, C.E.; Leroy, J.; Richter, W.; Xie, M.; Scheitrum, C.; Lee, I.; Maack, C.; Rucker-Martin, C.; Donzeau-Gouge, P.; Verde, I. Cyclic adenosine monophosphate phosphodiesterase type 4 protects against atrial arrhythmias. J. Am. Coll. Cardiol. 2012, 59, 2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Melsom, C.B.; Hussain, R.I.; Ørstavik, Ø.; Aronsen, J.M.; Sjaastad, I.; Skomedal, T.; Osnes, J.; Levy, F.O.; Krobert, K.A. Non-classical regulation of β1-and β2-adrenoceptor-mediated inotropic responses in rat heart ventricle by the G protein Gi. Naunyn Schmiedeberg’s Arch. Pharmacol. 2014, 387, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Conti, M. Subcellular Targeting of PDE4 in Cardiac Myocytes and Generation of Signaling Compartments. In Microdomains in the Cardiovascular System; Springer: Cham, Switzerland, 2017; pp. 143–160. [Google Scholar]
- Beca, S.; Helli, P.B.; Simpson, J.A.; Zhao, D.; Farman, G.P.; Jones, P.; Tian, X.; Wilson, L.S.; Ahmad, F.; Chen, S.W. Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ. Res. 2011, 109, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.C.; Conti, M.; Marks, A.R. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Vettel, C.; Lindner, M.; Dewenter, M.; Lorenz, K.; Schanbacher, C.; Riedel, M.; Lämmle, S.; Meinecke, S.; Mason, F.E.; Sossalla, S. Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ. Res. 2016. [Google Scholar] [CrossRef]
- Kajimoto, K.; Hagiwara, N.; Kasanuki, H.; Hosoda, S. Contribution of phosphodiesterase isozymes to the regulation of the L-type calcium current in human cardiac myocytes. Br. J. Pharmacol. 1997, 121, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Hua, R.; Adamczyk, A.; Robbins, C.; Ray, G.; Rose, R.A. Distinct patterns of constitutive phosphodiesterase activity in mouse sinoatrial node and atrial myocardium. PLoS ONE 2012, 7, e47652. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Guo, A.; Chen, B.; Kutschke, W.; Xie, Y.; Zimmerman, K.; Weiss, R.M.; Anderson, M.E.; Cheng, H.; Song, L. T-Tubule Remodeling During Transition From Hypertrophy to Heart Failure Novelty and Significance. Circ. Res. 2010, 107, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Seidel, T.; Navankasattusas, S.; Ahmad, A.; Diakos, N.A.; Xu, W.D.; Tristani-Firouzi, M.; Bonios, M.J.; Taleb, I.; Li, D.Y.; Selzman, C.H. Sheet-Like Remodeling of the Transverse Tubular System in Human Heart Failure Impairs Excitation-Contraction Coupling and Functional Recovery by Mechanical Unloading Clinical Perspective. Circulation 2017, 135, 1632–1645. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; MacLeod, K.T.; Zhang, Y.; Garcia, E.; Kanda, G.K.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. USA 2009, 106, 6854–6859. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Al Masri, A.; Navaratnarajah, M.; Siedlecka, U.; Soppa, G.K.; Moshkov, A.; Al-Saud, S.A.; Gorelik, J.; Yacoub, M.H.; Terracciano, C.M. Prolonged mechanical unloading affects cardiomyocyte excitation-contraction coupling, transverse-tubule structure, and the cell surface. FASEB J. 2010, 24, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Dibb, K.M.; Clarke, J.D.; Horn, M.A.; Richards, M.A.; Graham, H.K.; Eisner, D.A.; Trafford, A.W. Characterization of an Extensive Transverse Tubular Network in Sheep Atrial Myocytes and its Depletion in Heart FailureCLINICAL PERSPECTIVE. Circ. Heart Fail. 2009, 2, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Crossman, D.J.; Young, A.A.; Ruygrok, P.N.; Nason, G.P.; Baddelely, D.; Soeller, C.; Cannell, M.B. T-tubule disease: Relationship between t-tubule organization and regional contractile performance in human dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2015, 84, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, F.; Engelman, J.A.; Volonte, D.; Zhang, X.L.; Minetti, C.; Li, M.; Hou, H.; Kneitz, B.; Edelmann, W.; Lisanti, M.P. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J. Biol. Chem. 2001, 276, 21425–21433. [Google Scholar] [CrossRef] [PubMed]
- Makarewich, C.A.; Correll, R.N.; Gao, H.; Zhang, H.; Yang, B.; Berretta, R.M.; Rizzo, V.; Molkentin, J.D.; Houser, S.R. A Caveolae-Targeted L-Type Ca2+ Channel Antagonist Inhibits Hypertrophic Signaling Without Reducing Cardiac Contractility. Circ. Res. 2012, 110, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V.O. In vivo model with targeted cAMP biosensor reveals changes in receptor-microdomain communication in cardiac disease. Nat. Commun. 2015, 6, 6965. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, V.A.; Ji, G.J.; Wobus, A.M.; Fleischmann, B.K.; Hescheler, J. Establishment of β-adrenergic modulation of L-type Ca2+ current in the early stages of cardiomyocyte development. Circ. Res. 1999, 84, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Mehel, H.; Emons, J.; Vettel, C.; Wittköpper, K.; Seppelt, D.; Dewenter, M.; Lutz, S.; Sossalla, S.; Maier, L.S.; Lechêne, P. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts β-adrenergic responses in cardiomyocytes. J. Am. Coll. Cardiol. 2013, 62, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Seki, S.; Nagashima, M.; Yamada, Y.; Tsutsuura, M.; Kobayashi, T.; Namiki, A.; Tohse, N. Fetal and postnatal development of Ca2+ transients and Ca2+ sparks in rat cardiomyocytes. Cardiovasc. Res. 2003, 58, 535–548. [Google Scholar] [CrossRef]
- Haddock, P.S.; Coetzee, W.A.; Cho, E.; Porter, L.; Katoh, H.; Bers, D.M.; Jafri, M.S.; Artman, M. Subcellular [Ca2+]i gradients during excitation-contraction coupling in newborn rabbit ventricular myocytes. Circ. Res. 1999, 85, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hong, T. BIN1 regulates dynamic t-tubule membrane. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, F.; Ceriotti, P.; Miragoli, M.; Carullo, P.; Salvarani, N.; Rocchetti, M.; Di Pasquale, E.; Rossi, S.; Tessari, M.; Caprari, S. Peptidomimetic Targeting of Cavβ2 Overcomes Dysregulation of the L-Type Calcium Channel Density and Recovers Cardiac FunctionClinical Perspective. Circulation 2016, 134, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cui, Y.; Tang, M.; Hu, X.; Luo, H.; Hescheler, J.; Xi, J. Puerarin facilitates T-tubule development of murine embryonic stem cell-derived cardiomyocytes. Cell. Physiol. Biochem. 2014, 34, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Carozzi, A.J.; Ikonen, E.; Lindsay, M.R.; Parton, R.G. Role of cholesterol in developing T-Tubules: Analogous mechanisms for T-tubule and caveolae biogenesis. Traffic 2000, 1, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Fajardo, G.; Ribeiro, A.J.; Kooiker, K.B.; Coronado, M.; Zhao, M.; Hu, D.; Reddy, S.; Kodo, K.; Sriram, K. Time-dependent evolution of functional vs. remodeling signaling in induced pluripotent stem cell-derived cardiomyocytes and induced maturation with biomechanical stimulation. FASEB J. 2016, 30, 1464–1479. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.J.; Ang, Y.; Fu, J.; Rivas, R.N.; Mohamed, T.M.; Higgs, G.C.; Srivastava, D.; Pruitt, B.L. Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc. Natl. Acad. Sci. USA 2015, 112, 12705–12710. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I.; Kenyagin-Karsenti, D.; Snir, M.; Segev, H.; Amit, M.; Gepstein, A.; Livne, E.; Binah, O.; Itskovitz-Eldor, J.; Gepstein, L. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J. Clin. Investig. 2001, 108, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lee, J.; Vincent, L.G.; Wang, Q.; Gu, M.; Lan, F.; Churko, J.M.; Sallam, K.I.; Matsa, E.; Sharma, A. Epigenetic regulation of phosphodiesterases 2A and 3A underlies compromised β-adrenergic signaling in an iPSC model of dilated cardiomyopathy. Cell Stem Cell 2015, 17, 89–100. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhogal, N.K.; Hasan, A.; Gorelik, J. The Development of Compartmentation of cAMP Signaling in Cardiomyocytes: The Role of T-Tubules and Caveolae Microdomains. J. Cardiovasc. Dev. Dis. 2018, 5, 25. https://doi.org/10.3390/jcdd5020025
Bhogal NK, Hasan A, Gorelik J. The Development of Compartmentation of cAMP Signaling in Cardiomyocytes: The Role of T-Tubules and Caveolae Microdomains. Journal of Cardiovascular Development and Disease. 2018; 5(2):25. https://doi.org/10.3390/jcdd5020025
Chicago/Turabian StyleBhogal, Navneet K., Alveera Hasan, and Julia Gorelik. 2018. "The Development of Compartmentation of cAMP Signaling in Cardiomyocytes: The Role of T-Tubules and Caveolae Microdomains" Journal of Cardiovascular Development and Disease 5, no. 2: 25. https://doi.org/10.3390/jcdd5020025
APA StyleBhogal, N. K., Hasan, A., & Gorelik, J. (2018). The Development of Compartmentation of cAMP Signaling in Cardiomyocytes: The Role of T-Tubules and Caveolae Microdomains. Journal of Cardiovascular Development and Disease, 5(2), 25. https://doi.org/10.3390/jcdd5020025