PDE4-Mediated cAMP Signalling

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. cAMP Signalling and Compartmentalisation
3. PDEs and PDE4-Ology
4. Technological Approaches to Defining Roles for PDE4 Isoforms
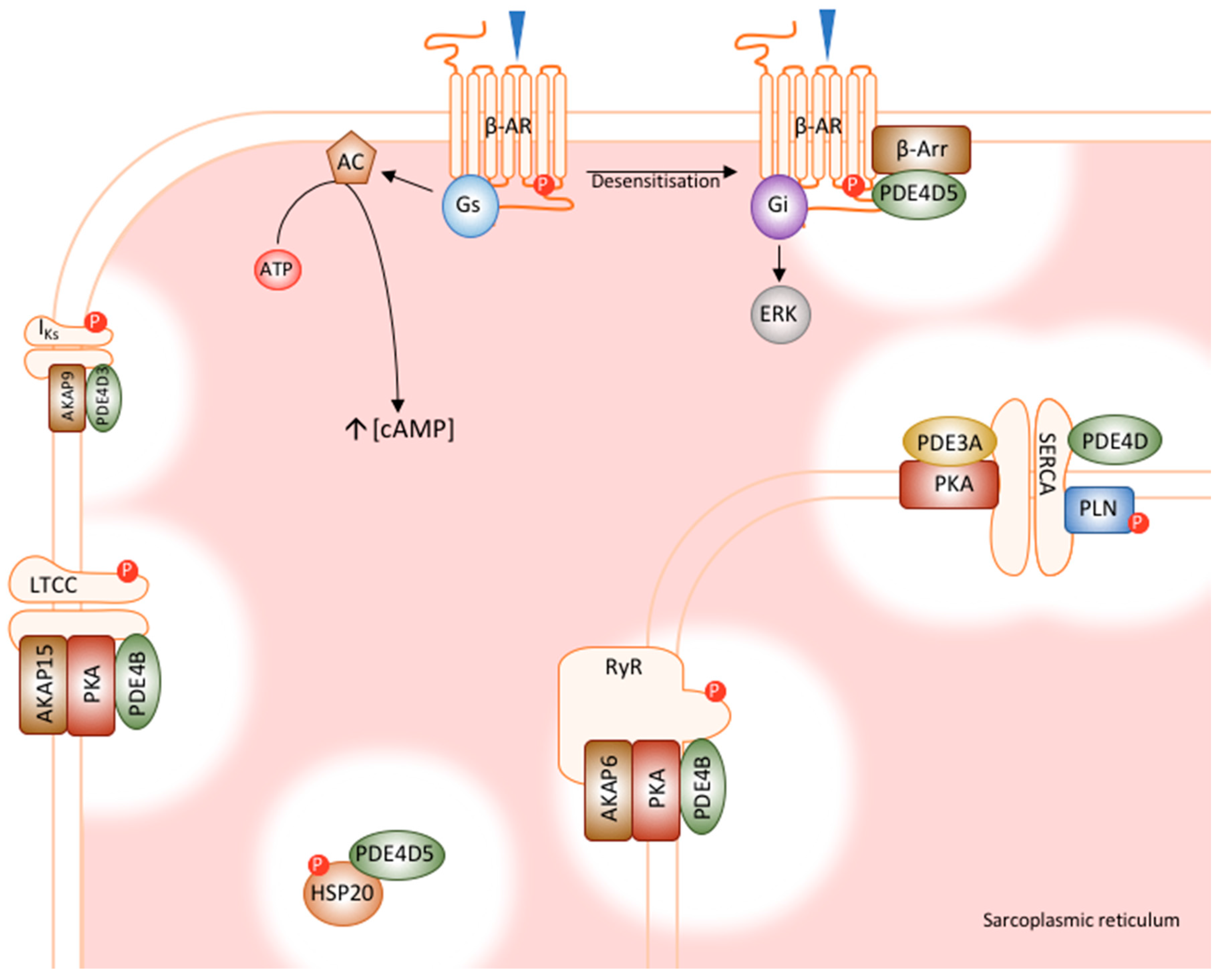
5. PDE4 in the Heart
5.1. PDE4D5’s Role in β-Adrenoceptor Desensitisation
5.2. PDE4D5 Regulates Cardioprotection by HSP20
5.3. PDE4D3’s Regulation of RyR Phosphorylation
5.4. PDE4D3 Regulates Basal IKs Activity
5.5. PDE4B Has a Dominant Role in LTCC Regulation
5.6. PDE4D and PDE3A Regulate PLN Phosphorylation and SERCA Function
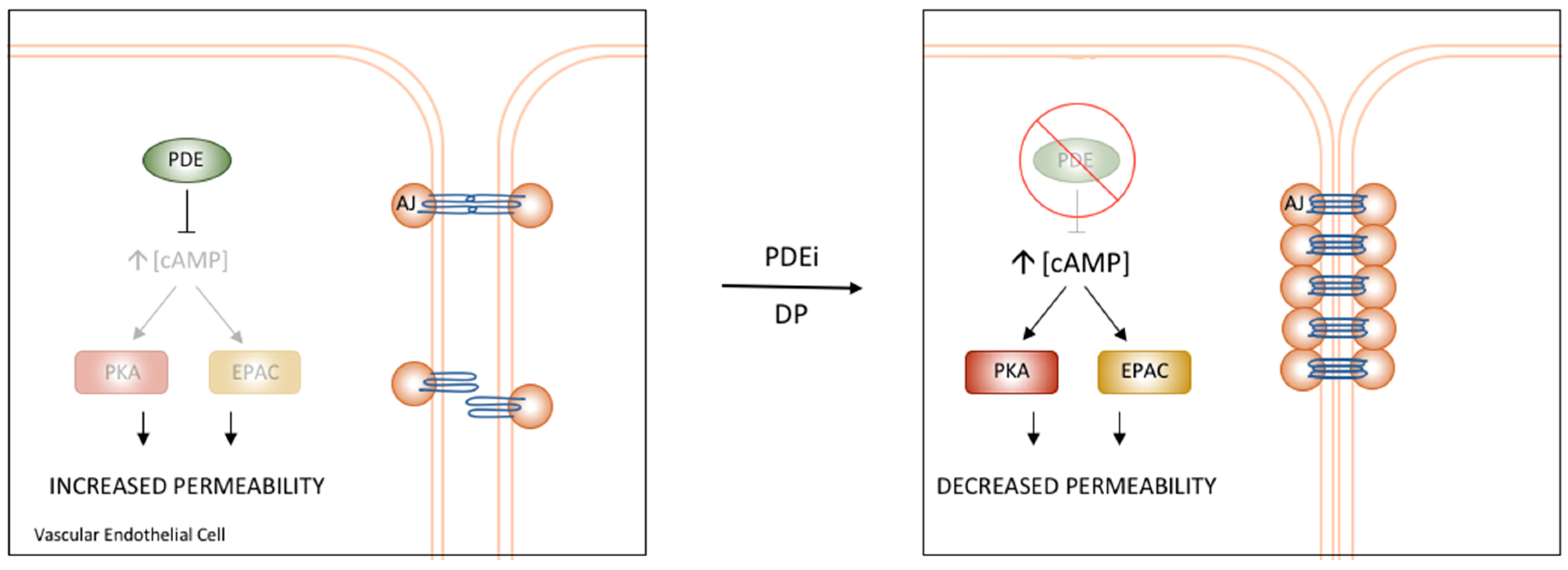
6. Vascular cAMP Signalling and PDEs
PDE4D’s Role in the Regulation of Vascular Permeability
7. Future Directions and Outlook
Conflicts of Interest
References
- Houslay, M.D.; Baillie, G.S.; Maurice, D.H. cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: A molecular toolbox for generating compartmentalized cAMP signaling. Circ. Res. 2007, 100, 950–966. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Nazmy, M.; Kajimoto, K.; Yehia, G.; Molina, C.A.; Sadoshima, J. Inducible cAMP early repressor (ICER) is a negative-feedback regulator of cardiac hypertrophy and an important mediator of cardiac myocyte apoptosis in response to beta-adrenergic receptor stimulation. Circ. Res. 2003, 93, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Rampersad, S.N.; Ovens, J.D.; Huston, E.; Umana, M.B.; Wilson, L.S.; Netherton, S.J.; Lynch, M.J.; Baillie, G.S.; Houslay, M.D.; Maurice, D.H. Cyclic AMP phosphodiesterase 4D (PDE4D) Tethers EPAC1 in a vascular endothelial cadherin (VE-Cad)-based signaling complex and controls cAMP-mediated vascular permeability. J. Biol. Chem. 2010, 285, 33614–33622. [Google Scholar] [CrossRef] [PubMed]
- Fetalvero, K.M.; Shyu, M.; Nomikos, A.P.; Chiu, Y.F.; Wagner, R.J.; Powell, R.J.; Hwa, J.; Martin, K.A. The prostacyclin receptor induces human vascular smooth muscle cell differentiation via the protein kinase A pathway. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1337–H1346. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Webb, J.G.; Newman, W.H.; Wang, Z. Regulation of human vascular smooth muscle cell migration by beta-adrenergic receptors. Am. Surg. 2006, 72, 51–54. [Google Scholar] [PubMed]
- Li, R.C.; Cindrova-Davies, T.; Skepper, J.N.; Sellers, L.A. Prostacyclin induces apoptosis of vascular smooth muscle cells by a cAMP-mediated inhibition of extracellular signal-regulated kinase activity and can counteract the mitogenic activity of endothelin-1 or basic fibroblast growth factor. Circ. Res. 2004, 94, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Bond, M.; Sala-Newby, G.B.; Newby, A.C. Altered S-phase kinase-associated protein-2 levels are a major mediator of cyclic nucleotide-induced inhibition of vascular smooth muscle cell proliferation. Circ. Res. 2006, 98, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. (Shanghai) 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Levin, L.R.; Buck, J. Role of soluble adenylyl cyclase in the heart. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H538–H543. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.S.; Brunton, L.L.; Mayer, S.E. Selective activation of particulate cAMP-dependent protein kinase by isoproterenol and prostaglandin E1. J. Biol. Chem. 1980, 255, 5113–5119. [Google Scholar] [PubMed]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research–still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.S.; Brunton, L.L. Functional compartments in cyclic nucleotide action. J. Cycl. Nucleotide Res. 1982, 8, 1–16. [Google Scholar]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S. Compartmentalized signalling: Spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. FEBS J. 2009, 276, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Wang, H. Crystal structures of phosphodiesterases and implications on substrate specificity and inhibitor selectivity. Curr. Top. Med. Chem. 2007, 7, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Bolger, G.B. Molecular biology of the cyclic AMP-specific cyclic nucleotide phosphodiesterases: A diverse family of regulatory enzymes. Cell. Signal. 1994, 6, 851–859. [Google Scholar] [CrossRef]
- Houslay, M.D. PDE4 cAMP-specific phosphodiesterases. Prog. Nucleic Acid Res. Mol. Biol. 2001, 69, 249–315. [Google Scholar] [PubMed]
- Hoffmann, R.; Baillie, G.S.; MacKenzie, S.J.; Yarwood, S.J.; Houslay, M.D. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 1999, 18, 893–903. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.J.; Baillie, G.S.; McPhee, I.; MacKenzie, C.; Seamons, R.; McSorley, T.; Millen, J.; Beard, M.B.; van Heeke, G.; Houslay, M.D. Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1). Br. J. Pharmacol. 2002, 136, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Sette, C.; Conti, M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J. Biol. Chem. 1996, 271, 16526–16534. [Google Scholar] [CrossRef] [PubMed]
- Burgin, A.B.; Magnusson, O.T.; Singh, J.; Witte, P.; Staker, B.L.; Bjornsson, J.M.; Thorsteinsdottir, M.; Hrafnsdottir, S.; Hagen, T.; Kiselyov, A.S.; et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat. Biotechnol. 2010, 28, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Murata, T.; Shimizu, K.; Degerman, E.; Maurice, D.; Manganiello, V. Cyclic nucleotide phosphodiesterases: Important signaling modulators and therapeutic targets. Oral Dis. 2015, 21, e25–e50. [Google Scholar] [CrossRef] [PubMed]
- McCahill, A.C.; Huston, E.; Li, X.; Houslay, M.D. PDE4 associates with different scaffolding proteins: Modulating interactions as treatment for certain diseases. Handb. Exp. Pharmacol. 2008, 186, 125–166. [Google Scholar]
- Houslay, M.D.; Adams, D.R. PDE4 cAMP phosphodiesterases: Modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem. J. 2003, 370, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Huston, E.; Gall, I.; Houslay, T.M.; Houslay, M.D. Helix-1 of the cAMP-specific phosphodiesterase PDE4A1 regulates its phospholipase-D-dependent redistribution in response to release of Ca2+. J. Cell Sci. 2006, 119, 3799–3810. [Google Scholar] [CrossRef] [PubMed]
- Houslay, K.F.; Christian, F.; MacLeod, R.; Adams, D.R.; Houslay, M.D.; Baillie, G.S. Identification of a multifunctional docking site on the catalytic unit of phosphodiesterase-4 (PDE4) that is utilised by multiple interaction partners. Biochem. J. 2017, 474, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Wills, L.; Ehsan, M.; Whiteley, E.L.; Baillie, G.S. Location, location, location: PDE4D5 function is directed by its unique N-terminal region. Cell. Signal. 2016, 28, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.C.; Maurice, D.H.; Baillie, G.S. Targeting protein-protein interactions within the cyclic AMP signaling system as a therapeutic strategy for cardiovascular disease. Future Med. Chem. 2013, 5, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.J.; Baillie, G.S.; Mohamed, A.; Li, X.; Maisonneuve, C.; Klussmann, E.; van Heeke, G.; Houslay, M.D. RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with beta arrestin to control the protein kinase A/AKAP79-mediated switching of the beta2-adrenergic receptor to activation of ERK in HEK293B2 cells. J. Biol. Chem. 2005, 280, 33178–33189. [Google Scholar] [CrossRef] [PubMed]
- Banan, M.; Puri, N. The ins and outs of RNAi in mammalian cells. Curr. Pharm. Biotechnol. 2004, 5, 441–450. [Google Scholar] [CrossRef] [PubMed]
- McCahill, A.; McSorley, T.; Huston, E.; Hill, E.V.; Lynch, M.J.; Gall, I.; Keryer, G.; Lygren, B.; Tasken, K.; van Heeke, G.; et al. In resting COS1 cells a dominant negative approach shows that specific, anchored PDE4 cAMP phosphodiesterase isoforms gate the activation, by basal cyclic AMP production, of AKAP-tethered protein kinase A type II located in the centrosomal region. Cell. Signal. 2005, 17, 1158–1173. [Google Scholar] [CrossRef] [PubMed]
- Frank, R. The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports–principles and applications. J. Immunol. Methods 2002, 267, 13–26. [Google Scholar] [CrossRef]
- Bolger, G.B.; Baillie, G.S.; Li, X.; Lynch, M.J.; Herzyk, P.; Mohamed, A.; Mitchell, L.H.; McCahill, A.; Hundsrucker, C.; Klussmann, E.; et al. Scanning peptide array analyses identify overlapping binding sites for the signalling scaffold proteins, beta-arrestin and RACK1, in cAMP-specific phosphodiesterase PDE4D5. Biochem. J. 2006, 398, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S. George Baillie on peptide array, a technique that transformed research on phosphodiesterases. Future Sci. OA 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Sin, Y.Y.; Edwards, H.V.; Li, X.; Day, J.P.; Christian, F.; Dunlop, A.J.; Adams, D.R.; Zaccolo, M.; Houslay, M.D.; Baillie, G.S. Disruption of the cyclic AMP phosphodiesterase-4 (PDE4)-HSP20 complex attenuates the beta-agonist induced hypertrophic response in cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 50, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Berrera, M.; Dodoni, G.; Monterisi, S.; Pertegato, V.; Zamparo, I.; Zaccolo, M. A toolkit for real-time detection of cAMP: Insights into compartmentalized signaling. Handb. Exp. Pharmacol. 2008, 186, 285–298. [Google Scholar]
- Fischmeister, R.; Castro, L.R.; Abi-Gerges, A.; Rochais, F.; Jurevicius, J.; Leroy, J.; Vandecasteele, G. Compartmentation of cyclic nucleotide signaling in the heart: The role of cyclic nucleotide phosphodiesterases. Circ. Res. 2006, 99, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Y.; Greenstein, J.L.; Winslow, R.L. Interaction between phosphodiesterases in the regulation of the cardiac beta-adrenergic pathway. J. Mol. Cell. Cardiol. 2015, 88, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; McSorley, T.; Evellin, S.; Sood, A.; Lissandron, V.; Terrin, A.; Huston, E.; Hannawacker, A.; Lohse, M.J.; Pozzan, T.; et al. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ. Res. 2004, 95, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Marks, A.R. Phosphodiesterase 4D and heart failure: A cautionary tale. Expert Opin. Ther. Targets 2006, 10, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Carver, J.R.; Rodeheffer, R.J.; Ivanhoe, R.J.; DiBianco, R.; Zeldis, S.M.; Hendrix, G.H.; Bommer, W.J.; Elkayam, U.; Kukin, M.L.; et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N. Engl. J. Med. 1991, 325, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.L.; Conti, M.; Marks, A.R. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Abi-Gerges, A.; Richter, W.; Lefebvre, F.; Mateo, P.; Varin, A.; Heymes, C.; Samuel, J.L.; Lugnier, C.; Conti, M.; Fischmeister, R.; et al. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ. Res. 2009, 105, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Bolger, G.B.; McCahill, A.; Huston, E.; Cheung, Y.F.; McSorley, T.; Baillie, G.S.; Houslay, M.D. The unique amino-terminal region of the PDE4D5 cAMP phosphodiesterase isoform confers preferential interaction with beta-arrestins. J. Biol. Chem. 2003, 278, 49230–49238. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Richter, W.; Mika, D.; Castro, L.R.; Abi-Gerges, A.; Xie, M.; Scheitrum, C.; Lefebvre, F.; Schittl, J.; Mateo, P.; et al. Phosphodiesterase 4B in the cardiac L-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias in mice. J. Clin. Invest. 2011, 121, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Beca, S.; Helli, P.B.; Simpson, J.A.; Zhao, D.; Farman, G.P.; Jones, P.; Tian, X.; Wilson, L.S.; Ahmad, F.; Chen, S.R.W.; et al. Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ. Res. 2011, 109, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Terrenoire, C.; Houslay, M.D.; Baillie, G.S.; Kass, R.S. The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J. Biol. Chem. 2009, 284, 9140–9146. [Google Scholar] [CrossRef] [PubMed]
- Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 1997, 390, 88–91. [Google Scholar] [PubMed]
- Krupnick, J.G.; Benovic, J.L. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 289–319. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.J.; Baillie, G.S.; Kohout, T.A.; McPhee, I.; Magiera, M.M.; Ang, K.L.; Miller, W.E.; McLean, A.J.; Conti, M.; Houslay, M.D.; et al. Targeting of cyclic AMP degradation to β2-adrenergic receptors by β-arrestins. Science 2002, 298, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S.; Sood, A.; McPhee, I.; Gall, I.; Perry, S.J.; Lefkowitz, R.J.; Houslay, M.D. β-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates β-adrenoceptor switching from Gs to Gi. Proc. Natl. Acad. Sci. USA 2003, 100, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; MacRae, T.H. The small heat shock proteins and their role in human disease. FEBS J. 2005, 272, 2613–2627. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C.; Chu, G.; Mitton, B.; Song, Q.; Yuan, Q.; Kranias, E.G. Small heat-shock protein Hsp20 phosphorylation inhibits β-agonist-induced cardiac apoptosis. Circ. Res. 2004, 94, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, P.; Knoll, R.; Haghighi, K.; Fan, G.C.; Dorn, G.W., 2nd; Hasenfub, G.; Kranias, E.G. Human mutation in the anti-apoptotic heat shock protein 20 abrogates its cardioprotective effects. J. Biol. Chem. 2008, 283, 33465–33471. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Ren, X.; Wang, X.; Zhang, P.; Jones, W.K.; Molkentin, J.D.; Fan, G.C.; Kranias, E.G. Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ. Res. 2009, 105, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zingarelli, B.; O’Connor, M.; Zhang, P.; Adeyemo, A.; Kranias, E.G.; Wang, Y.; Fan, G.C. Overexpression of Hsp20 prevents endotoxin-induced myocardial dysfunction and apoptosis via inhibition of NF-κB activation. J. Mol. Cell. Cardiol. 2009, 47, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C.; Zhou, X.; Wang, X.; Song, G.; Qian, J.; Nicolaou, P.; Chen, G.; Ren, X.; Kranias, E.G. Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ. Res. 2008, 103, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Tessier, D.J.; Komalavilas, P.; Panitch, A.; Joshi, L.; Brophy, C.M. The small heat shock protein (HSP) 20 is dynamically associated with the actin cross-linking protein actinin. J. Surg. Res. 2003, 111, 152–157. [Google Scholar] [CrossRef]
- Zhu, Y.H.; Ma, T.M.; Wang, X. Gene transfer of heat-shock protein 20 protects against ischemia/reperfusion injury in rat hearts. Acta Pharmacol. Sin. 2005, 26, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Dohke, T.; Wada, A.; Isono, T.; Fujii, M.; Yamamoto, T.; Tsutamoto, T.; Horie, M. Proteomic analysis reveals significant alternations of cardiac small heat shock protein expression in congestive heart failure. J. Card. Fail. 2006, 12, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C.; Yuan, Q.; Song, G.; Wang, Y.; Chen, G.; Qian, J.; Zhou, X.; Lee, Y.J.; Ashraf, M.; Kranias, E.G. Small heat-shock protein Hsp20 attenuates β-agonist-mediated cardiac remodeling through apoptosis signal-regulating kinase 1. Circ. Res. 2006, 99, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Edwards, H.V.; Scott, J.D.; Baillie, G.S. PKA phosphorylation of the small heat-shock protein Hsp20 enhances its cardioprotective effects. Biochem. Soc. Trans. 2012, 40, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.P.; Hortigon-Vinagre, M.P.; Findlay, J.E.; Elliott, C.; Currie, S.; Baillie, G.S. Targeted disruption of the heat shock protein 20-phosphodiesterase 4D (PDE4D) interaction protects against pathological cardiac remodelling in a mouse model of hypertrophy. FEBS Open Bio 2014, 4, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef]
- Antos, C.L.; Frey, N.; Marx, S.O.; Reiken, S.; Gaburjakova, M.; Richardson, J.A.; Marks, A.R.; Olson, E.N. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase A. Circ. Res. 2001, 89, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Reiken, S.; Gaburjakova, M.; Guatimosim, S.; Gomez, A.M.; D’Armiento, J.; Burkhoff, D.; Wang, J.; Vassort, G.; Lederer, W.J.; Marks, A.R. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J. Biol. Chem. 2003, 278, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Reiken, S.; Wehrens, X.H.; Vest, J.A.; Barbone, A.; Klotz, S.; Mancini, D.; Burkhoff, D.; Marks, A.R. β-Blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation 2003, 107, 2459–2466. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, X.H.; Lehnart, S.E.; Huang, F.; Vest, J.A.; Reiken, S.R.; Mohler, P.J.; Sun, J.; Guatimosim, S.; Song, L.S.; Rosemblit, N.; et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003, 113, 829–840. [Google Scholar] [CrossRef]
- Wehrens, X.H.; Lehnart, S.E.; Reiken, S.R.; Deng, S.X.; Vest, J.A.; Cervantes, D.; Coromilas, J.; Landry, D.W.; Marks, A.R. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 2004, 304, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Jin, S.L.C.; Conti, M. Splice variants of the cyclic nucleotide phosphodiesterase PDE4D are differentially expressed and regulated in rat tissue. Biochem. J. 2005, 388, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Regitz-Zagrosek, V.; Hertrampf, R.; Steffen, C.; Hildebrandt, A.; Fleck, E. Myocardial cyclic AMP and norepinephrine content in human heart failure. Eur. Heart J. 1994, 15 (Suppl. D), 7–13. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Curran, M.E.; Zou, A.; Shen, J.; Spector, P.S.; Atkinson, D.L.; Keating, M.T. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKS potassium channel. Nature 1996, 384, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. KVLQT1 and lsK (minK) proteins associate to form the IKS cardiac potassium current. Nature 1996, 384, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Kass, R.S.; Dessauer, C.W. The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKS potassium channel in heart. J. Biol. Chem. 2012, 287, 29815–29824. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Kurokawa, J.; Reiken, S.; Motoike, H.; D’Armiento, J.; Marks, A.R.; Kass, R.S. Requirement of a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 2002, 295, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.D.; Emrick, M.A.; Sadilek, M.; Scheuer, T.; Catterall, W.A. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci. Signal. 2010, 3, ra70. [Google Scholar] [CrossRef] [PubMed]
- Bunemann, M.; Gerhardstein, B.L.; Gao, T.; Hosey, M.M. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the beta(2) subunit. J. Biol. Chem. 1999, 274, 33851–33854. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Beca, S.; Ahmad, F.; Shen, W.; Liu, J.; Makary, S.; Polidovitch, N.; Sun, J.; Hockman, S.; Chung, Y.W.; Movsesian, M.; et al. Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ. Res. 2013, 112, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H. Subcellular signaling in the endothelium: Cyclic nucleotides take their place. Curr. Opin. Pharmacol. 2011, 11, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Sayner, S.L.; Alexeyev, M.; Dessauer, C.W.; Stevens, T. Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier. Circ. Res. 2006, 98, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Tournier-Lasserve, E.; Weinstein, B.M. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Dev. Cell 2009, 16, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Dejana, E. Adhesion molecule signalling: Not always a sticky business. Nat. Rev. Mol. Cell Biol. 2011, 12, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Surdo, N.C.; Berrera, M.; Koschinski, A.; Brescia, M.; Machado, M.R.; Carr, C.; Wright, P.; Gorelik, J.; Morotti, S.; Grandi, E.; et al. FRET biosensor uncovers cAMP nano-domains at beta-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun. 2017, 8, 15031. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fertig, B.A.; Baillie, G.S. PDE4-Mediated cAMP Signalling. J. Cardiovasc. Dev. Dis. 2018, 5, 8. https://doi.org/10.3390/jcdd5010008
Fertig BA, Baillie GS. PDE4-Mediated cAMP Signalling. Journal of Cardiovascular Development and Disease. 2018; 5(1):8. https://doi.org/10.3390/jcdd5010008
Chicago/Turabian StyleFertig, Bracy A., and George S. Baillie. 2018. "PDE4-Mediated cAMP Signalling" Journal of Cardiovascular Development and Disease 5, no. 1: 8. https://doi.org/10.3390/jcdd5010008
APA StyleFertig, B. A., & Baillie, G. S. (2018). PDE4-Mediated cAMP Signalling. Journal of Cardiovascular Development and Disease, 5(1), 8. https://doi.org/10.3390/jcdd5010008