Imaging of PDE2- and PDE3-Mediated cGMP-to-cAMP Cross-Talk in Cardiomyocytes


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. The cAMP and β-Adrenergic Pathway
- (a)
- cAMP-dependent protein kinase (PKA), which is responsible for phosphorylation of several calcium handling proteins involved in cardiac excitation-contraction coupling (ECC) including L-type Ca2+ channel (LTCC) at the plasmalemma, phospholamban, and ryanodine receptors at the sarcoplasmic reticulum (SR), myosin-binding protein C, and troponin I at the myofilaments [1,2]. PKA is the main effector protein in the cAMP cascade, while Ca2+-inhibited AC5 and AC6 are the predominant cAMP generating adenylyl cyclases in adult (AC5 and AC6) and fetal (AC6) ventricular cardiac tissue [3];
- (b)
- (c)
- (d)
1.2. NO/sGC/cGMP Pathway
1.3. NP/pGC/cGMP Pathway
2. Compartmentation of cAMP and cGMP Signaling
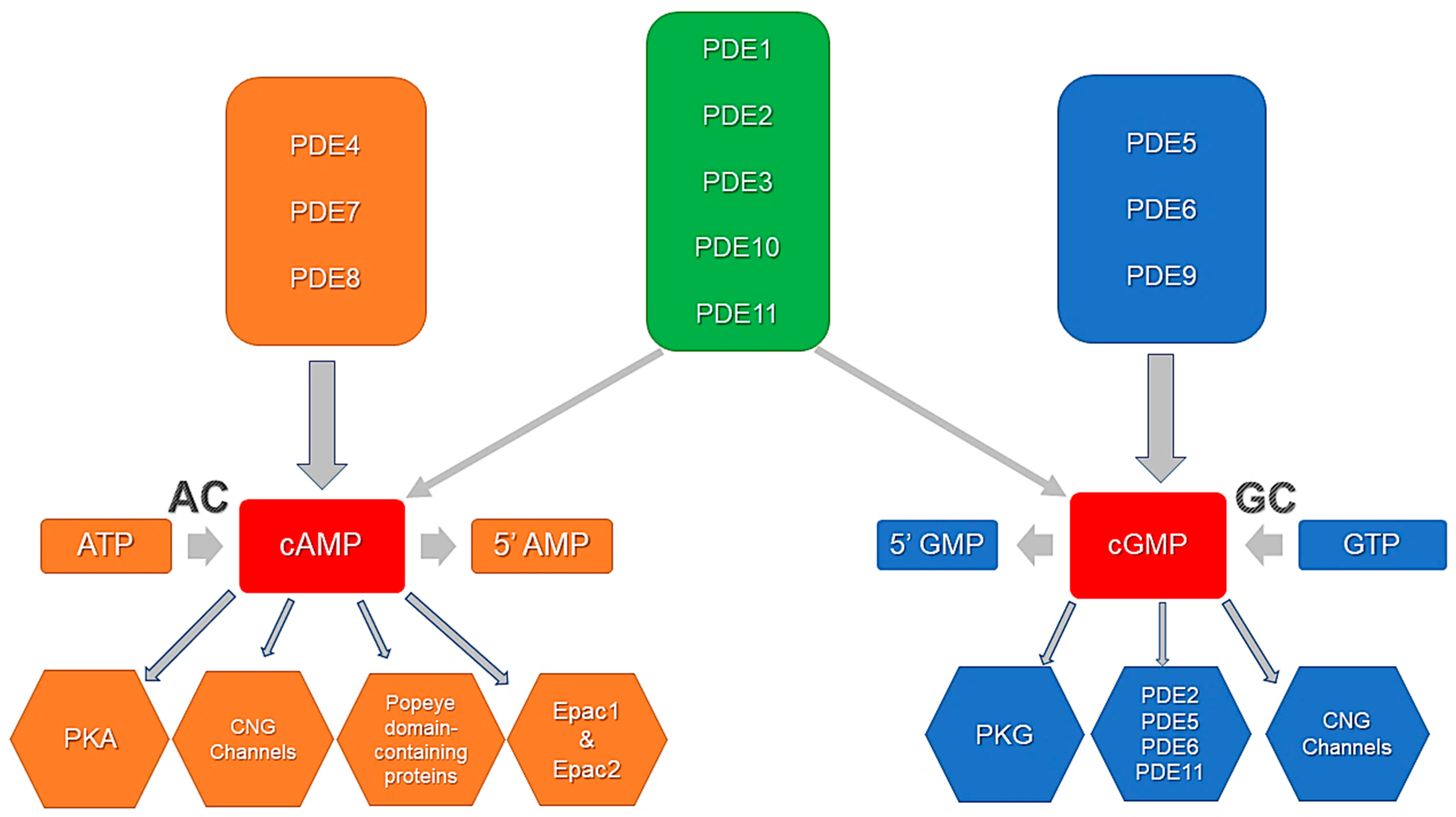
3. Phosphodiesterases (PDEs)
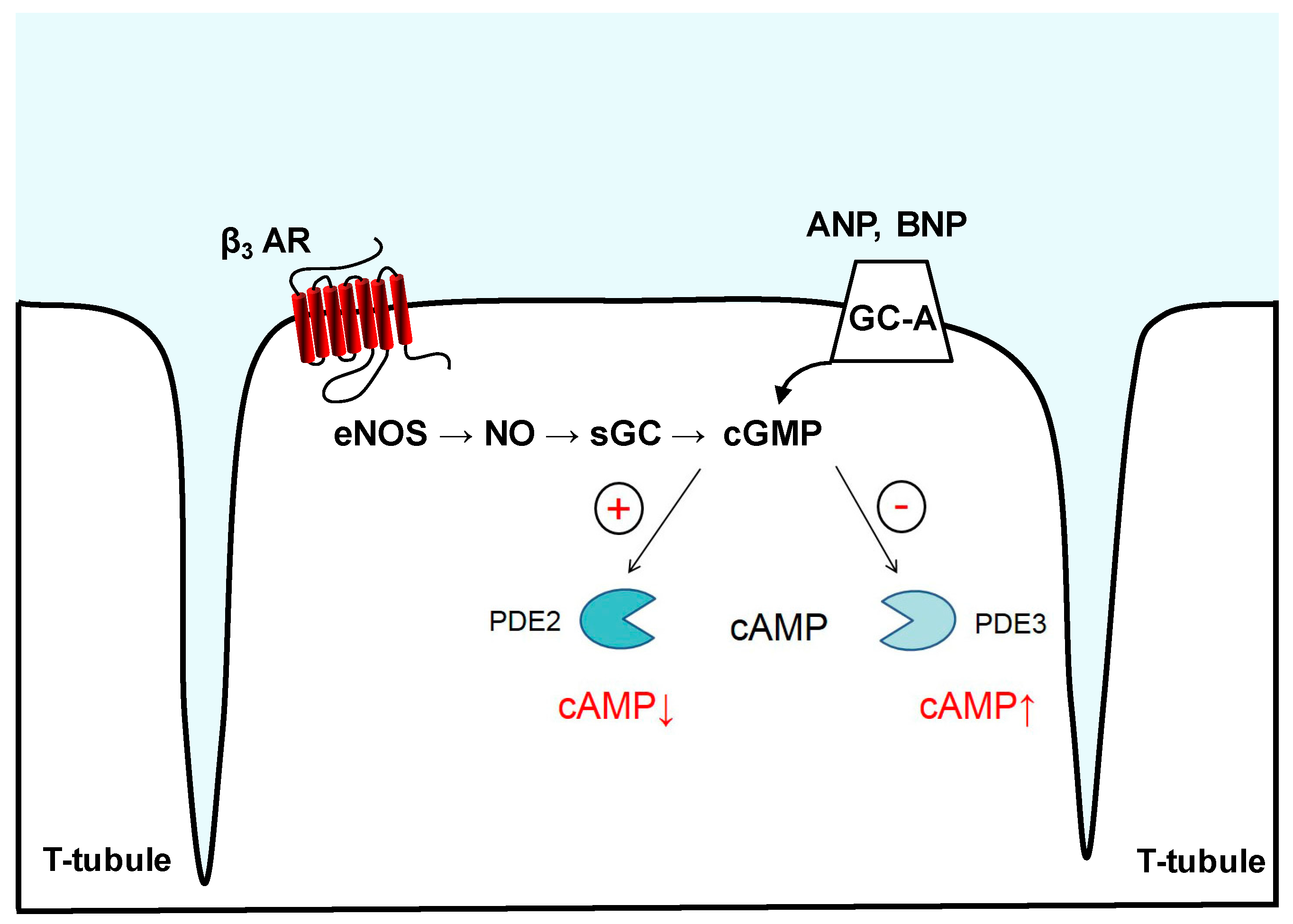
3.1. Phosphodiesterase 2
3.2. Phosphodiesterase 3
4. Visualization of Compartmentalized cAMP and cGMP
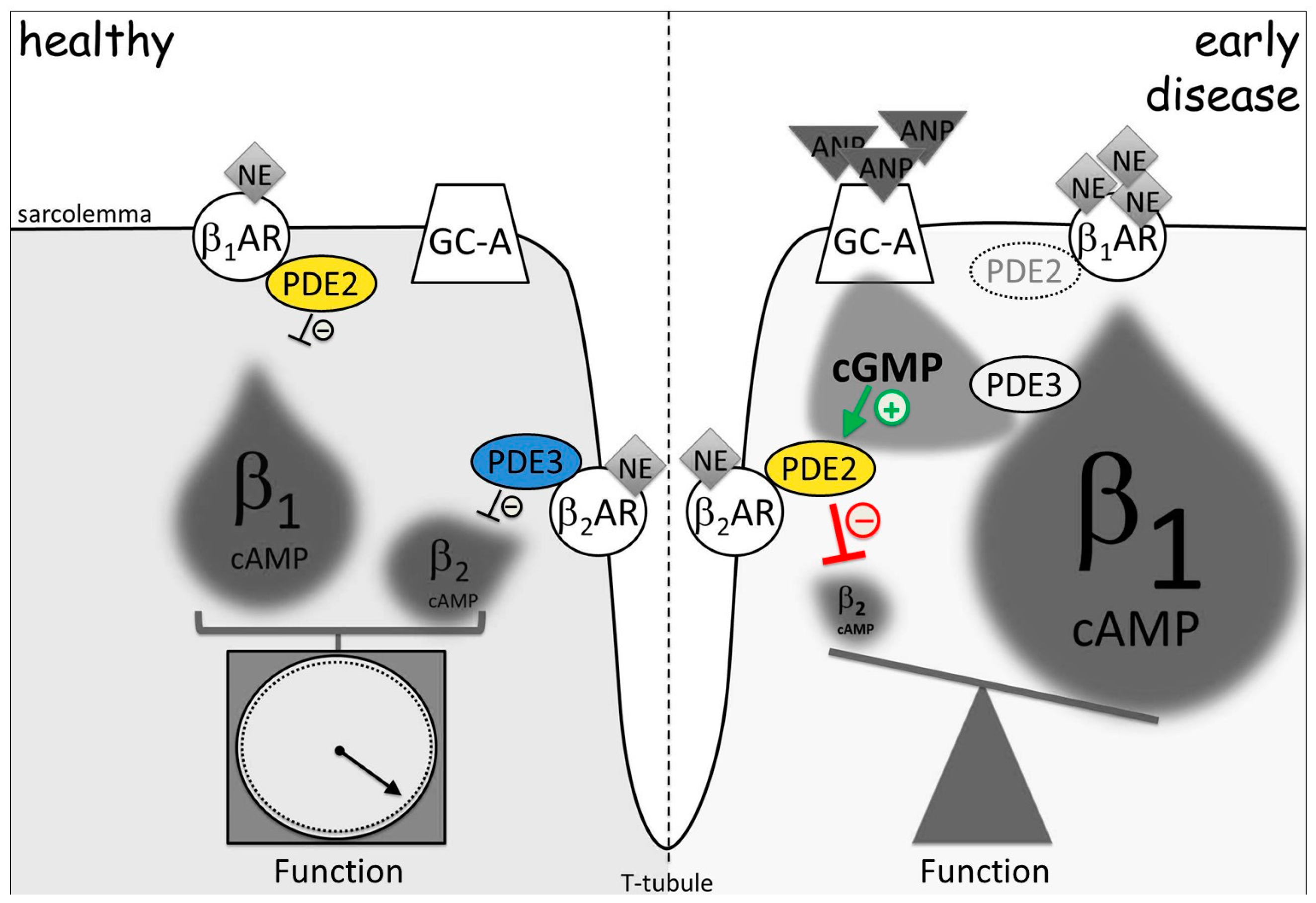
5. Imaging of cGMP-to-cAMP Crosstalk via PDE2 and PDE3
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Defer, N.; Best-Belpomme, M.; Hanoune, J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am. J. Physiol. Ren. Physiol. 2000, 279, F400–F416. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Marcantoni, A.; Gastineau, M.; Birkedal, R.; Rochais, F.; Garnier, A.; Lompre, A.M.; Vandecasteele, G.; Lezoualc’h, F. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ. Res. 2005, 97, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Métrich, M.; Lucas, A.; Gastineau, M.; Samuel, J.L.; Heymes, C.; Morel, E.; Lezoualc’h, F. Epac mediates β-adrenergic receptor–induced cardiomyocyte hypertrophy. Circ. Res. 2008, 102, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Wit, A.L.; Rosen, M.R. Pathophysiologic mechanisms of cardiac arrhythmias. Am. Heart J. 1983, 106, 798–811. [Google Scholar] [CrossRef]
- Larsson, H.P. How is the heart rate regulated in the sinoatrial node? Another piece to the puzzle. J. Gen. Physiol. 2010, 88, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Zoccarato, A.; Zaccolo, M. cAMP compartmentalisation and hypertrophy of the heart: “Good” pools of cAMP and “Bad” pools of cAMP coexist in the same cardiac myocyte. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer Nature: Cham, Switzerland, 2017. [Google Scholar]
- Froese, A.; Breher, S.S.; Waldeyer, C.; Schindler, R.F.; Nikolaev, V.O.; Rinné, S.; Vauti, F. Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J. Clin. Investig. 2012, 122. [Google Scholar] [CrossRef] [PubMed]
- Schindler, R.F.R.; Brand, T. The Popeye domain containing protein family-A novel class of cAMP effectors with important functions in multiple tissues. Prog. Biophys. Mol. Biol. 2016, 120, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Cherry, P.D.; Furchgott, R.F.; Zawadski, J.V.; Jothianandan, D. Role of endothelial cells in relaxation of isolated arteries by bradykinin. Proc. Natl. Acad. Sci. USA 1982, 79, 2106–2110. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, R.M.; Draznin, M.B.; Murad, F. Endothelium-dependent relaxation in rat aorta may be mediated through cyclic GMP-dependent protein phosphorylation. Nature 1983, 306, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, R.M.; Murad, F. Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ. Res. 1983, 52, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Carvalho, M.H.; Khan, M.T.; Matsunaga, K. Evidence for endothelium-dependent vasodilation of resistance vessels by acetylcholine. J. Vasc. Res. 1987, 24, 145–149. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Byrns, R.E.; Buga, G.M.; Wood, K.S. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ. Res. 1987, 61, 866–879. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Koide, M.; Kawahara, Y.; Nakayama, I.; Tsuda, T.; Yokoyama, M. Cyclic AMP-elevating agents induce an inducible type of nitric oxide synthase in cultured vascular smooth muscle cells. Synergism with the induction elicited by inflammatory cytokines. J. Biol. Chem. 1993, 268, 24959–24966. [Google Scholar] [PubMed]
- Balligand, J.L.; Kobzik, L.; Han, X.; Kaye, D.M.; Belhassen, L.; O’Hara, D.S.; Kelly, R.A.; Smith, T.W.; Michel, T. Nitric oxide-dependent parasympathetic signaling is due to activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes. J. Biol. Chem. 1995, 270, 14582–14586. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, N.; Alfie, M.E.; Sigmon, D.H.; Rhaleb, N.E.; Shesely, E.G.; Carretero, O.A. Role of nNOS in blood pressure regulation in eNOS null mutant mice. Hypertension 1998, 32, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Gyurko, R.; Kuhlencordt, P.; Fishman, M.C.; Huang, P.L. Modulation of mouse cardiac function in vivo by eNOS and ANP. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H971–H981. [Google Scholar] [CrossRef] [PubMed]
- MacNaul, K.L.; Hutchinson, N.I. Differential expression of iNOS and cNOS mRNA in human vascular smooth muscle cells and endothelial cells under normal and inflammatory conditions. Biochem. Biophys. Res. Commun. 1993, 196, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef] [PubMed]
- Yuen, P.S.; Potter, L.R.; Garbers, D.L. A new form of guanylyl cyclase is preferentially expressed in rat kidney. Biochemistry 1990, 29, 10872–10878. [Google Scholar] [CrossRef] [PubMed]
- Harteneck, C.; Wedel, B.; Koesling, D.; Bo, E. Molecular cloning and expression of a new α-subunit of soluble guanylyl cyclase. Interchangeability of the α-subunits of the enzyme. FEBS Lett. 1991, 292, 217–222. [Google Scholar] [PubMed]
- Behrends, S.; Harteneck, C.; Schultz, G.; Koesling, D. A variant of the α2 subunit of soluble guanylyl cyclase contains an insert homologous to a region within adenylyl cyclases and functions as a dominant negative protein. J. Biol. Chem. 1995, 270, 21109–21113. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Kass, D.A. Phosphodiesterases and cyclic GMP regulation in heart muscle. Physiology 2012, 27, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Y.; Greenstein, J.L.; Winslow, R.L. Interaction between phosphodiesterases in the regulation of the cardiac β-adrenergic pathway. J. Mol. Cell. Cardiol. 2015, 88, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Y.; Greenstein, J.L.; Winslow, R.L. Roles of phosphodiesterases in the regulation of the cardiac cyclic nucleotide cross-talk signaling network. J. Mol. Cell. Cardiol. 2016, 91, 215–227. [Google Scholar] [CrossRef] [PubMed]
- D’souza, S.P.; Davis, M.; Baxter, G.F. Autocrine and paracrine actions of natriuretic peptides in the heart. Pharmacol. Ther. 2004, 101, 113–129. [Google Scholar] [CrossRef] [PubMed]
- De Bold, A.J.; Ma, K.K.Y.; Zhang, Y.; de Bold, M.L.K.; Bensimon, M.; Khoshbaten, A. The physiological and pathophysiological modulation of the endocrine function of the heart. Can. J. Physiol. Pharmacol. 2001, 79, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Molecular physiology of membrane guanylyl cyclase receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef] [PubMed]
- Kambayashi, Y.; Nakao, K.; Mukoyama, M.; Saito, Y.; Ogawa, Y.; Shiono, S.; Inouye, K.; Yoshida, N.; Imura, H. Isolation and sequence determination of human brain natriuretic peptide in human atrium. FEBS Lett. 1990, 259, 341–345. [Google Scholar] [CrossRef]
- Mukoyama, M.; Nakao, K.; Saito, Y.; Ogawa, Y.; Hosoda, K.; Suga, S.I.; Shirakami, G.; Jougasaki, M.; Imura, H. Human brain natriuretic peptide, a novel cardiac hormone. Lancet 1990, 335, 801–802. [Google Scholar] [CrossRef]
- Nakao, K.; Itoh, H.; Kambayashi, Y.; Hosoda, K.; Saito, Y.; Yamada, T.; Mukoyama, M.; Arai, H.; Shirakami, G.; Suga, S.I. Rat brain natriuretic peptide. Isolation from rat heart and tissue distribution. Hypertension 1990, 15, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Doyle, D.D.; Upshaw-Earley, J.; Bell, E.L.; Palfrey, H.C. Natriuretic peptide receptor-B in adult rat ventricle is predominantly confined to the nonmyocyte population. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2117–H2123. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, J.; Shanks, J.; Li, D.; Hao, G.; Athwal, A.; Langenickel, T.H.; Wright, H.; da Costa Goncalves, A.C.; Monti, J.; Plehm, R.; et al. C-type natriuretic peptide and natriuretic peptide receptor B signalling inhibits cardiac sympathetic neurotransmission and autonomic function. Cardiovasc. Res. 2016, 112, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, H.G.; Trindade, P.T.; Cunanan, D.B.; Wu, C.F.; Pratt, R.E. Mechanisms of natriuretic-peptide-induced growth inhibition of vascular smooth muscle cells. Cardiovasc. Res. 1997, 35, 158–167. [Google Scholar] [CrossRef]
- Rosenkranz, A.C.; Woods, R.L.; Dusting, G.J.; Ritchie, R.H. Antihypertrophic actions of the natriuretic peptides in adult rat cardiomyocytes: Importance of cyclic GMP. Cardiovasc. Res. 2003, 57, 515–522. [Google Scholar] [CrossRef]
- Tokudome, T.; Horio, T.; Soeki, T.; Mori, K.; Kishimoto, I.; Suga, S.I.; Yoshihara, F.; Kawano, Y.; Kohno, M.; Kangawa, K. Inhibitory effect of C-type natriuretic peptide (CNP) on cultured cardiac myocyte hypertrophy: Interference between CNP and endothelin-1 signaling pathways. Endocrinology 2004, 145, 2131–2140. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R. cCMP and cUMP: Emerging second messengers. Trends Biochem. Sci. 2015, 40, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R.; Schneider, E.H.; Bähre, H. From canonical to non-canonical cyclic nucleotides as second messengers: Pharmacological implications. Pharmacol. Ther. 2015, 148, 154–184. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M. Spatial control of cAMP signalling in health and disease. Curr. Opin. Pharmacol. 2011, 11, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Saucerman, J.J.; Greenwald, E.C.; Polanowska-Grabowska, R. Mechanisms of cyclic AMP compartmentation revealed by computational models. J. Gen. Physiol. 2014, 143, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Mika, D.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. PDEs create local domains of cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Ziolo, M.T.; Kohr, M.J.; Wang, H. Nitric oxide signaling and the regulation of myocardial function. J. Mol. Cell. Cardiol. 2008, 45, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Fischmeister, R.; Castro, L.R.; Abi-Gerges, A.; Rochais, F.; Jurevičius, J.; Leroy, J.; Vandecasteele, G. Compartmentation of cyclic nucleotide signaling in the heart. Circ. Res. 2006, 99, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Vandecasteele, G.; Rochais, F.; Abi-Gerges, A.; Fischmeister, R. Functional localization of cAMP signalling in cardiac myocytes. Biochem. Soc. Trans. 2006, 34, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [PubMed]
- Balijepalli, R.C.; Foell, J.D.; Hall, D.D.; Hell, J.W.; Kamp, T.J. Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 7500–7505. [Google Scholar] [CrossRef] [PubMed]
- Pani, B.; Singh, B.B. Lipid rafts/caveolae as microdomains of calcium signalling. Cell Calcium 2009, 45, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Kamp, T.J.; Hell, J.W. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 2000, 87, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.R.; Ostrom, R.S.; Harvey, R.D. Membrane Microdomains and cAMP Compartmentation in Cardiac Myocytes. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 17–35. [Google Scholar]
- Guellich, A.; Mehel, H.; Fischmeister, R. Cyclic AMP synthesis and hydrolysis in the normal and failing heart. Pflügers Arch. 2014, 466, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Timofeyev, V.; Myers, R.E.; Kim, H.J.; Woltz, R.L.; Sirish, P.; Heiserman, J.P.; Li, N.; Singapuri, A.; Tang, T.; Yarov-Yarovoy, V.; et al. Adenylyl cyclase subtype-specific compartmentalization: Differential regulation of L-type Ca2+ current in ventricular myocytes. Circ. Res. 2013, 112, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Redden, J.M.; Dodge-Kafka, K.L.; Kapiloff, M.S. Function to Failure: Compartmentalization of Cardiomyocyte Signaling by A-Kinase-Anchoring Proteins. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 37–57. [Google Scholar]
- Schrade, K.; Klussmann, E. Pharmacological approaches for delineating functions of AKAP-based signalling complexes and finding therapeutic targets. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 59–83. [Google Scholar]
- Ghigo, A.; Pirozzi, F.; Li, M.; Hirsch, E. Chatting Second Messengers: PIP3 and cAMP. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 85–95. [Google Scholar]
- Dodge-Kafka, K.L.; Langeberg, L.; Scott, J.D. Compartmentation of cyclic nucleotide signaling in the heart: The role of A-kinase anchoring proteins. Circ. Res. 2006, 98, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Balijepalli, R.C.; Kamp, T.J. Caveolae, ion channels and cardiac arrhythmias. Prog. Biophys. Mol. Biol. 2008, 98, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, G.K.; Means, C.K.; Scott, J.D. A-kinase anchoring proteins: From protein complexes to physiology and disease. IUBMB Life 2009, 61, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.B.; Rossow, C.F.; Navedo, M.F.; Westenbroek, R.E.; Catterall, W.A.; Santana, L.F.; McKnight, G.S. Sympathetic stimulation of adult cardiomyocytes requires association of akap5 with a subpopulation of l-type calcium channels novelty and significance. Circ. Res. 2010, 107, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Cheepala, S.; Hulot, J.S.; Morgan, J.A.; Sassi, Y.; Zhang, W.; Naren, A.P.; Schuetz, J.D. Cyclic nucleotide compartmentalization: Contributions of phosphodiesterases and ATP-binding cassette transporters. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 231–253. [Google Scholar] [CrossRef] [PubMed]
- Sassi, Y.; Abi-Gerges, A.; Fauconnier, J.; Mougenot, N.; Reiken, S.; Haghighi, K.; Kranias, E.G.; Marks, A.R.; Lacampagne, A.; Engelhardt, S.; et al. Regulation of cAMP homeostasis by the efflux protein MRP4 in cardiac myocytes. FASEB J. 2012, 26, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci. 2010, 35, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Mika, D.; Richter, W. Cyclic AMP compartments and signaling specificity: Role of cyclic nucleotide phosphodiesterases. J. Gen. Physiol. 2014, 143, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Movsesian, M.A. cAMP and cGMP signaling cross-talk: Role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res. 2007, 100, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kass, D.A. Phosphodiesterases and cardiac cGMP: Evolving roles and controversies. Trends Pharmacol. Sci. 2011, 32, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Brescia, M.; Zaccolo, M. Modulation of compartmentalised cyclic nucleotide signalling via local inhibition of phosphodiesterase activity. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Muratal, T.; Shimizul, K.; Degerma, E.; Maurice, D.; Manganiello, V. Cyclic nucleotide phosphodiesterases: Important signaling modulators and therapeutic targets. Oral Dis. 2015, 21, e25–e50. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Y. Computational modeling of cyclic nucleotide signaling mechanisms in cardiac myocytes. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer Nature: Cham, Switzerland, 2017; pp. 175–213. [Google Scholar]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Bledsoe, R.; Chai, J.; Daka, P.; Deng, H.; Ding, Y.; Harris-Gurley, S.; Kryn, L.H.; Nartey, E.; Nichols, J.; et al. The role of phosphodiesterase 12 (PDE12) as a negative regulator of the innate immune response and the discovery of antiviral inhibitors. J. Biol. Chem. 2015, 290, 19681–19696. [Google Scholar] [CrossRef] [PubMed]
- Vandeput, F.; Wolda, S.L.; Krall, J.; Hambleton, R.; Uher, L.; McCaw, K.N.; Radwanski, P.B.; Florio, V.; Movsesian, M.A. Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes. J. Biol. Chem. 2007, 282, 32749–32757. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; Tocchetti, C.G.; Terrin, A.; Lissandron, V.; Cheung, Y.F.; Dostmann, W.R.; Pozzan, T.; Kass, D.A.; Paolocci, N.; Houslay, M.D.; et al. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ. Res. 2006, 98, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Xie, M.; Scheitrum, C.; Krall, J.; Movsesian, M.A.; Conti, M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res. Cardiol. 2011, 106, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Gamanuma, M.; Yuasa, K.; Sasaki, T.; Sakurai, N.; Kotera, J.; Omori, K. Comparison of enzymatic characterization and gene organization of cyclic nucleotide phosphodiesterase 8 family in humans. Cell Signal. Technol. 2003, 15, 565–574. [Google Scholar] [CrossRef]
- Houslay, M.D.; Baillie, G.S.; Maurice, D.H. cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: A molecular toolbox for generating compartmentalized cAMP signaling. Circ. Res. 2007, 100, 950–966. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.J.; Mumby, M.C.; Beavo, J.A. Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J. Biol. Chem. 1982, 257, 1973–1979. [Google Scholar] [PubMed]
- Mumby, M.C.; Martins, T.J.; Chang, M.L.; Beavo, J.A. Identification of cGMP-stimulated cyclic nucleotide phosphodiesterase in lung tissue with monoclonal antibodies. J. Biol. Chem. 1982, 257, 13283–13290. [Google Scholar] [PubMed]
- Martinez, S.E.; Wu, A.Y.; Glavas, N.A.; Tang, X.B.; Turley, S.; Hol, W.G.; Beavo, J.A. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc. Natl. Acad. Sci. USA 2002, 99, 13260–13265. [Google Scholar] [CrossRef] [PubMed]
- Rosman, G.J.; Martins, T.J.; Sonnenburg, W.K.; Beavo, J.A.; Ferguson, K.; Loughney, K. Isolation and characterization of human cDNAs encoding a cGMP-stimulated 3′, 5′-cyclic nucleotide phosphodiesterase. Gene 1997, 191, 89–95. [Google Scholar] [CrossRef]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Paskind, M.; Bolger, G.; Thompson, W.J.; Repaske, D.R.; Cutler, L.S.; Epstein, P.M. A novel cyclic GMP stimulated phosphodiesterase from rat brain. Biochem. Biophys. Res. Commun. 1994, 205, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Manganiello, V.C.; Vaughan, M. Purification and characterization of cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from calf liver. Effects of divalent cations on activity. J. Biol. Chem. 1983, 258, 12526–12533. [Google Scholar] [PubMed]
- Bender, A.T.; Beavo, J.A. Specific localized expression of cGMP PDEs in Purkinje neurons and macrophages. Neurochem. Int. 2004, 45, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Juilfs, D.M.; Soderling, S.; Burns, F.; Beavo, J.A. Cyclic GMP as substrate and regulator of cyclic nucleotide phosphodiesterases (PDEs). In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 1999. [Google Scholar]
- Dickinson, N.T.; Haslam, R.J. Activation of cGMP-stimulated phosphodiesterase by nitroprusside limits cAMP accumulation in human platelets: Effects on platelet aggregation. Biochem. J. 1997, 323, 371–377. [Google Scholar] [CrossRef] [PubMed]
- MacFarland, R.T.; Zelus, B.D.; Beavo, J.A. High concentrations of a cGMP-stimulated phosphodiesterase mediate ANP-induced decreases in cAMP and steroidogenesis in adrenal glomerulosa cells. J. Biol. Chem. 1991, 266, 136–142. [Google Scholar] [PubMed]
- Simmons, M.A.; Hartzell, H.C. Role of phosphodiesterase in regulation of calcium current in isolated cardiac myocytes. Mol. Pharmacol. 1988, 33, 664–671. [Google Scholar] [PubMed]
- Acin-Perez, R.; Gatti, D.L.; Bai, Y.; Manfredi, G. Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: Coupled mechanisms of energy metabolism regulation. Cell Metab. 2011, 13, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.R.; Verde, I.; Cooper, D.M.; Fischmeister, R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation 2006, 113, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Zeller, M.; Guan, K.; Wunder, F.; Wagner, M.; El-Armouche, A. PDE2 at the crossway between cAMP and cGMP signalling in the heart. Cell Signal. 2017, 38, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Shakur, Y.; Holst, L.S.; Landstrom, T.R.; Movsesian, M.; Degerman, E.; Manganiello, V. Regulation and function of the cyclic nucleotide phosphodiesterase (PDE3) gene family. Prog. Nucleic Acid Res. Mol. Biol. 2001, 66, 241–277. [Google Scholar] [PubMed]
- Degerman, E.; Belfrage, P.; Manganiello, V.C. Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3). J. Biol. Chem. 1997, 272, 6823–6826. [Google Scholar] [CrossRef] [PubMed]
- Beca, S.; Ahmad, F.; Shen, W.; Liu, J.; Makary, S.; Polidovitch, N.; Sun, J.; Hockman, S.; Chung, Y.W.; Movsenian, M.; et al. Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart novelty and significance. Circ. Res. 2013, 112, 289–297. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Böhm, M.; Dickstein, K.; Jaarsma, T. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012. Eur. J. Heart Fail. 2012, 14, 803–869. [Google Scholar] [CrossRef] [PubMed]
- Landry, Y.; Gies, J.P. Drugs and their molecular targets: An updated overview. Fundam. Clin. Pharmacol. 2008, 22, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Carver, J.R.; Rodeheffer, R.J.; Ivanhoe, R.J.; DiBianco, R.; Zeldis, S.M.; Mallis, G.I. Effect of oral milrinone on mortality in severe chronic heart failure. N. Engl. J. Med. 1991, 325, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Park, S.; Hockman, S.; Zmuda-Trzebiatowska, E.; Svennelid, F.; Haluzik, M.; Gavrilova, O.; Ahmad, F.; Pepin, L.; Napolitano, M.; et al. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B–null mice. J. Clin. Investig. 2006, 116, 3240–3251. [Google Scholar] [CrossRef] [PubMed]
- Degerman, E.; Ahmad, F.; Chung, Y.W.; Guirguis, E.; Omar, B.; Stenson, L.; Manganiello, V. From PDE3B to the regulation of energy homeostasis. Curr. Opin. Pharmacol. 2011, 11, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.W.; Lagranha, C.; Chen, Y.; Sun, J.; Tong, G.; Hockman, S.C.; Ahmad, F.; Esfahani, S.G.; Bae, D.H.; Polidovitch, N.; et al. Targeted disruption of PDE3B, but not PDE3A, protects murine heart from ischemia/reperfusion injury. Proc. Natl. Acad. Sci. USA 2015, 112, E2253–E2262. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Li, H.; Shakur, Y.; Hensley, J.; Hockman, S.; Kambayashi, J.; Manganiello, V.M.; Liu, Y. Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell Signal. 2007, 19, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Abe, J.I.; Wei, H.; Xu, H.; Che, W.; Aizawa, T.; Liu, W.; Molina, C.A.; Sadoshima, J.; Blaxall, B.C.; et al. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 14771–14776. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, M.; Wu, M.; Lim, S.; Knight, W.E.; Miller, C.L.; Cai, Y.; Lu, C.; Blaxall, B.C.; Takeishi, Y.; Abe, J.I.; et al. Cyclic nucleotide phosphodiesterase 3A1 protects the heart against ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2013, 64, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, E.; Notte, A.; Barberis, L.; Selvetella, G.; Maffei, A.; Brancaccio, M.; Marengo, S.; Russo, G.; Azzolino, O.; Rybalkin, S.D.; et al. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 2004, 118, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating cardiac PIP 3 and cAMP signaling through a PKA anchoring function of p110γ. Mol. Cell 2011, 42, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, A.; Perino, A.; Mehel, H.; Zahradníková, A.; Morello, F.; Leroy, J.; Nikolaev, V.O.; Damilano, F.; Cimino, J.; De Luca, E.; et al. Phosphoinositide 3-kinase γ protects against catecholamine-induced ventricular arrhythmia through protein kinase a–mediated regulation of distinct phosphodiesterases. Circulation 2012, 126, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Götz, K.R.; Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Kuhn, M.; Gorelik, J.; Balligand, J.L.; Nikolaev, V.O. Transgenic mice for real-time visualization of cGMP in intact adult cardiomyocytes. Circ. Res. 2014, 114, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.F.; Cui, X.; Jin, J.Y.; Kim, S.M.; Kim, S.Z.; Kim, S.H.; Kim, S.H.; Lee, H.S.; Cho, K.W. High and low gain switches for regulation of camp efflux concentration. Circ. Res. 2004, 94, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.S.; Brunton, L.L. Functional compartments in cyclic nucleotide action. J. Cycl. Nucleotide Res. 1982, 8, 1–16. [Google Scholar]
- Corbin, J.D.; Sugden, P.H.; Lincoln, T.M.; Keely, S.L. Compartmentalization of adenosine 3′: 5′-monophosphate and adenosine 3′: 5′-monophosphate-dependent protein kinase in heart tissue. J. Biol. Chem. 1977, 252, 3854–3861. [Google Scholar] [PubMed]
- Hayes, J.S.; Brunton, L.L.; Brown, J.H.; Reese, J.B.; Mayer, S.E. Hormonally specific expression of cardiac protein kinase activity. Proc. Natl. Acad. Sci. USA 1979, 76, 1570–1574. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.S.; Brunton, L.L.; Mayer, S.E. Selective activation of particulate cAMP-dependent protein kinase by isoproterenol and prostaglandin E1. J. Biol. Chem. 1980, 255, 5113–5119. [Google Scholar] [PubMed]
- Keely, S.L. Activation of cAMP-dependent protein kinase without a corresponding increase in phosphorylase activity. Res. Commun. Chem. Pathol. Pharmacol. 1977, 18, 283–290. [Google Scholar] [PubMed]
- Buxton, I.L.; Brunton, L.L. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem. 1983, 258, 10233–10239. [Google Scholar] [PubMed]
- Jurevicius, J.; Fischmeister, R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc. Natl. Acad. Sci. USA 1996, 93, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Nikolaev, V.O. Biophysical techniques for detection of cAMP and cGMP in Living Cells. Int. J. Mol. Sci. 2013, 14, 8025–8046. [Google Scholar] [CrossRef] [PubMed]
- Förster, T. Zwischenmolekulare energiewanderung und fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Lohse, M.J. Novel techniques for real-time monitoring of cGMP in living cells. Handb. Exp. Pharmacol. 2009, 191, 229–243. [Google Scholar]
- Honda, A.; Adams, S.R.; Sawyer, C.L.; Lev-Ram, V.; Tsien, R.Y.; Dostmann, W.R. Spatiotemporal dynamics of guanosine 3′, 5′-cyclic monophosphate revealed by a genetically encoded, fluorescent indicator. Proc. Natl. Acad. Sci. USA 2001, 98, 2437–2442. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Hida, N.; Ozawa, T.; Umezawa, Y. Fluorescent indicators for cyclic GMP based on cyclic GMP-dependent protein kinase I alpha and green fluorescent proteins. Anal. Chem. 2000, 72, 5918–5924. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Gambaryan, S.; Lohse, M.J. Fluorescent sensors for rapid monitoring of intracellular cGMP. Nat. Methods 2006, 3, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Niino, Y.; Hotta, K.; Oka, K. Simultaneous live cell imaging using dual FRET sensors with a single excitation light. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.R.; Harootunian, A.T.; Buechler, Y.J.; Taylor, S.S.; Tsien, R.Y. Fluorescence ratio imaging of cyclic AMP in single cells. Nature 1991, 349, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Bacskai, B.J.; Hochner, B.; Mahaut-Smith, M.; Adams, S.R.; Kaang, B.K.; Kandel, E.R.; Tsien, R.Y. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science 1993, 260, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; De Giorgi, F.; Cho, C.Y.; Feng, L.; Knapp, T.; Negulescu, P.A.; Taylor, S.S.; Tsien, R.Y.; Pozzan, T. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell. Biol. 2000, 2, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.L.; Conti, M.; Marks, A.R. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bünemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 2004, 279, 37215–37218. [Google Scholar] [CrossRef] [PubMed]
- DiPilato, L.M.; Cheng, X.; Zhang, J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci. USA 2004, 101, 16513–16518. [Google Scholar] [CrossRef] [PubMed]
- Ponsioen, B.; Zhao, J.; Riedl, J.; Zwartkruis, F.; van der Krogt, G.; Zaccolo, M.; Moolenaar, W.H.; Bos, J.L.; Jalink, K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004, 5, 1176–1180. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Gambaryan, S.; Engelhardt, S.; Walter, U.; Lohse, M.J. Real-time monitoring of the PDE2 activity of live cells: Hormone-stimulated cAMP hydrolysis is faster than hormone-stimulated cAMP synthesis. J. Biol. Chem. 2005, 280, 1716–1719. [Google Scholar] [CrossRef] [PubMed]
- Calebiro, D.; Nikolaev, V.O.; Gagliani, M.C.; de Filippis, T.; Dees, C.; Tacchetti, C.; Persani, L.; Lohse, M.J. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 2009, 7. [Google Scholar] [CrossRef] [PubMed]
- Klarenbeek, J.; Goedhart, J.; van Batenburg, A.; Groenewald, D.; Jalink, K. Fourth-generation epac-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: Characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Rich, T.C.; Fagan, K.A.; Nakata, H.; Schaack, J.; Cooper, D.M.; Karpen, J.W. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J. Gen. Physiol. 2000, 116, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Rich, T.C.; Tse, T.E.; Rohan, J.G.; Schaack, J.; Karpen, J.W. In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J. Gen. Physiol. 2001, 118, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bünemann, M.; Schmitteckert, E.; Lohse, M.J.; Engelhardt, S. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ. Res. 2006, 99, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.K.; Nikolaev, V.O. Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol. 2013, 207, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Bork, N.I.; Nikolaev, V.O. Receptor-cyclic nucleotide microdomains in the heart. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer Nature: Cham, Switzerland, 2017; pp. 3–15. [Google Scholar]
- Hansma, P.K.; Drake, B.; Marti, O.; Gould, S.A.; Prater, C.B. The scanning ion-conductance microscope. Science 1989, 243, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Korchev, Y.E.; Bashford, C.L.; Milovanovic, M.; Vodyanoy, I.; Lab, M.J. Scanning ion conductance microscopy of living cells. Biophys. J. 1997, 73, 653–658. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Moshkov, A.; Novak, P.; Shevchuk, A.; Nikolaev, V.O.; El-Hamamsy, I.; Potter, C.M.; Wright, P.; Kadir, S.H.; Lyon, A.R.; et al. Scanning ion conductance microscopy: A convergent high-resolution technology for multi-parametric analysis of living cardiovascular cells. J. R. Soc. Interface 2011, 8, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.; Nikolaev, V.O. Imaging alterations of cardiomyocyte cAMP microdomains in disease. Front. Pharmacol. 2015, 6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Berisha, F.; Nikolaev, V.O. Cyclic nucleotide imaging and cardiovascular disease. Pharmacol. Ther. 2017, 175, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Fischmeister, R.; Castro, L.; Abi-Gerges, A.; Rochais, F.; Vandecasteele, G. Species-and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2005, 142, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Vandecasteele, G.; Verde, I.; Rücker-Martin, C.; Donzeau-Gouge, P.; Fischmeister, R. Cyclic GMP regulation of the L-type Ca2+ channel current in human atrial myocytes. J. Physiol. 2001, 533, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Mehel, H.; Emons, J.; Vettel, C.; Wittkopper, K.; Seppelt, D.; Dewenter, M.; Lutz, S.; Sossalla, S.; Maier, L.S.; Lechene, P.; et al. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts beta adrenergic responses in cardiomyocytes. J. Am. Coll. Cardiol. 2013, 62, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Zoccarato, A.; Surdo, N.C.; Aronsen, J.M.; Fields, L.A.; Mancuso, L.; Dodoni, G.; Stangherlin, A.; Livie, C.; Jiang, H.; Sin, Y.Y.; et al. Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2 novelty and significance. Circ. Res. 2015, 117, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V.O. In vivo model with targeted cAMP biosensor reveals changes in receptor microdomain communication in cardiac disease. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.K.; Sprenger, J.U.; Steinbrecher, J.H.; Hübscher, D.; Lehnart, S.E.; Abesser, M.; Schuh, K.; El-Armouche, A.; Nikolaev, V.O. Microdomain switch of cGMP-regulated phosphodiesterases leads to ANP-induced augmentation of beta-adrenoceptor-stimulated contractility in early cardiac hypertrophy. Circ. Res. 2015, 116, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Vettel, C.; Lindner, M.; Dewenter, M.; Lorenz, K.; Schanbacher, C.; Riedel, M.; Lämmle, S. Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ. Res. 2017, 120, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Zoccarato, A.; Lissandron, V.; Terrin, A.; Li, X.; Houslay, M.D.; Baillie, G.S.; Zaccolo, M. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ. Res. 2008, 103, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Stangherlin, A.; Gesellchen, F.; Zoccarato, A.; Terrin, A.; Fields, L.A.; Berrera, M.; Surdo, N.C.; Craig, M.A.; Smith, G.; Hamilton, G.; et al. cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ. Res. 2011, 108, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Surapisitchat, J.; Jeon, K.I.; Yan, C.; Beavo, J.A. Differential regulation of endothelial cell permeability by cGMP via phosphodiesterases 2 and 3. Circ. Res. 2007, 101, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Stangherlin, A.; Zaccolo, M. Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H379–H390. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lu, C.J.; Hao, G.; Wright, H.; Woodward, L.; Liu, K.; Vergari, E.; Surdo, N.C.; Herring, N.; Zaccolo, M.; et al. Efficacy of B-type natriuretic peptide is coupled to phosphodiesterase 2A in cardiac sympathetic neurons novelty and significance. Hypertension 2015, 66, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.; Andressen, K.W.; Aronsen, J.M.; Sjaastad, I.; Hougen, K.; Skomedal, T.; Osnes, J.B.; Qvigstad, E.; Levy, F.O.; Moltzau, L.R. PDE3 inhibition by C-type natriuretic peptide-induced cGMP enhances cAMP-mediated signaling in both non-failing and failing hearts. Eur. J. Pharmacol. 2017, 812, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; McSorley, T.; Evellin, S.; Sood, A.; Lissandron, V.; Terrin, A.; Huston, E.; Hannawacker, A.; Lohse, M.J.; Pozzan, T.; et al. Fluorescence resonance energy transfer–based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ. Res. 2004, 95, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Rochais, F.; Abi-Gerges, A.; Horner, K.; Lefebvre, F.; Cooper, D.M.; Conti, M.; Fischmeister, R.; Vandecasteele, G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ. Res. 2006, 98, 1081–1088. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlaki, N.; Nikolaev, V.O. Imaging of PDE2- and PDE3-Mediated cGMP-to-cAMP Cross-Talk in Cardiomyocytes. J. Cardiovasc. Dev. Dis. 2018, 5, 4. https://doi.org/10.3390/jcdd5010004
Pavlaki N, Nikolaev VO. Imaging of PDE2- and PDE3-Mediated cGMP-to-cAMP Cross-Talk in Cardiomyocytes. Journal of Cardiovascular Development and Disease. 2018; 5(1):4. https://doi.org/10.3390/jcdd5010004
Chicago/Turabian StylePavlaki, Nikoleta, and Viacheslav O. Nikolaev. 2018. "Imaging of PDE2- and PDE3-Mediated cGMP-to-cAMP Cross-Talk in Cardiomyocytes" Journal of Cardiovascular Development and Disease 5, no. 1: 4. https://doi.org/10.3390/jcdd5010004
APA StylePavlaki, N., & Nikolaev, V. O. (2018). Imaging of PDE2- and PDE3-Mediated cGMP-to-cAMP Cross-Talk in Cardiomyocytes. Journal of Cardiovascular Development and Disease, 5(1), 4. https://doi.org/10.3390/jcdd5010004