Function of Adenylyl Cyclase in Heart: the AKAP Connection

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Adenylyl Cyclases (ACs) and Their Role in Cardiac Function: Knockout Phenotypes
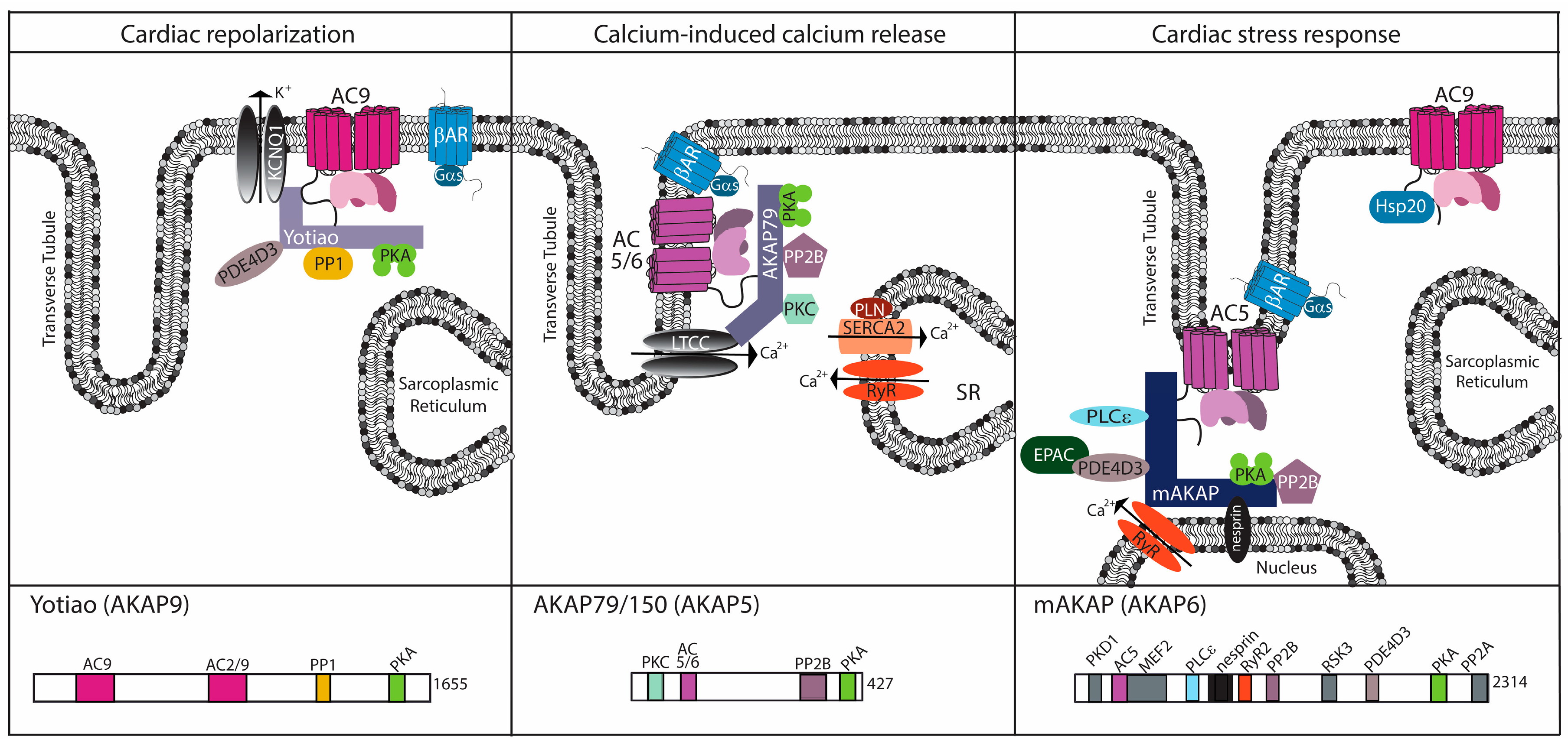
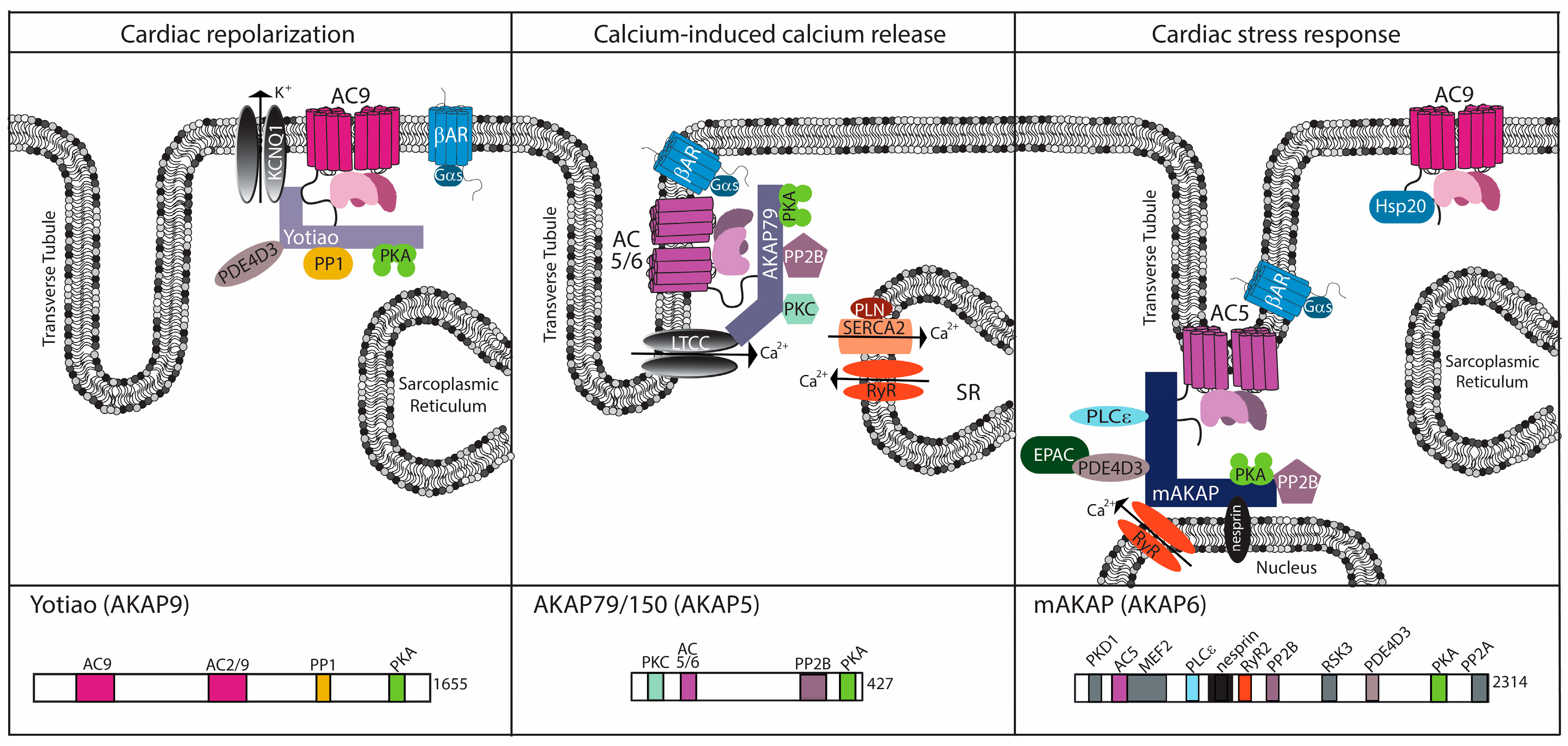
3. The AKAP Connection: Generating Specificity for AC Function
3.1. AKAP5
3.2. mAKAP (AKAP6)
3.3. Yotiao (AKAP9)
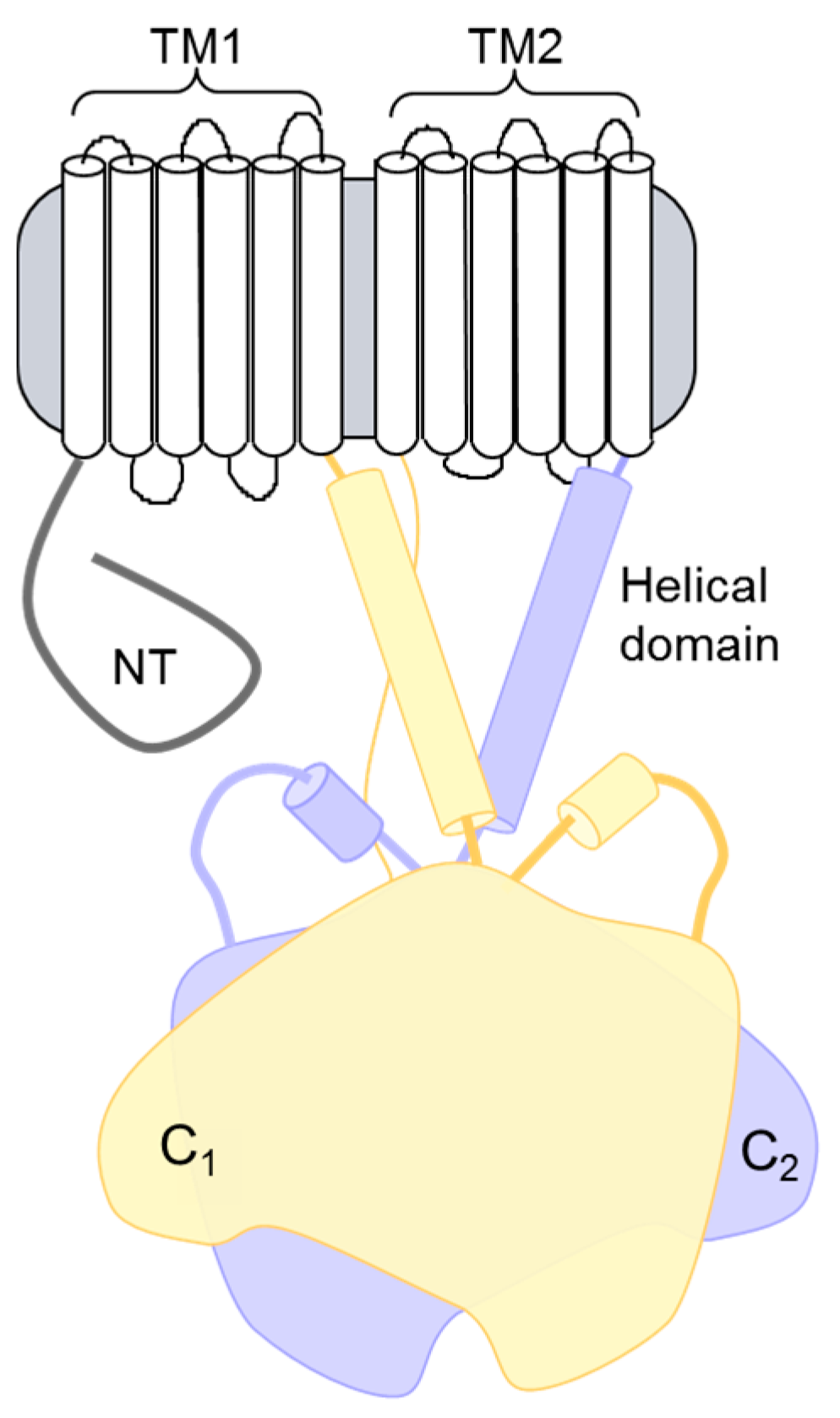
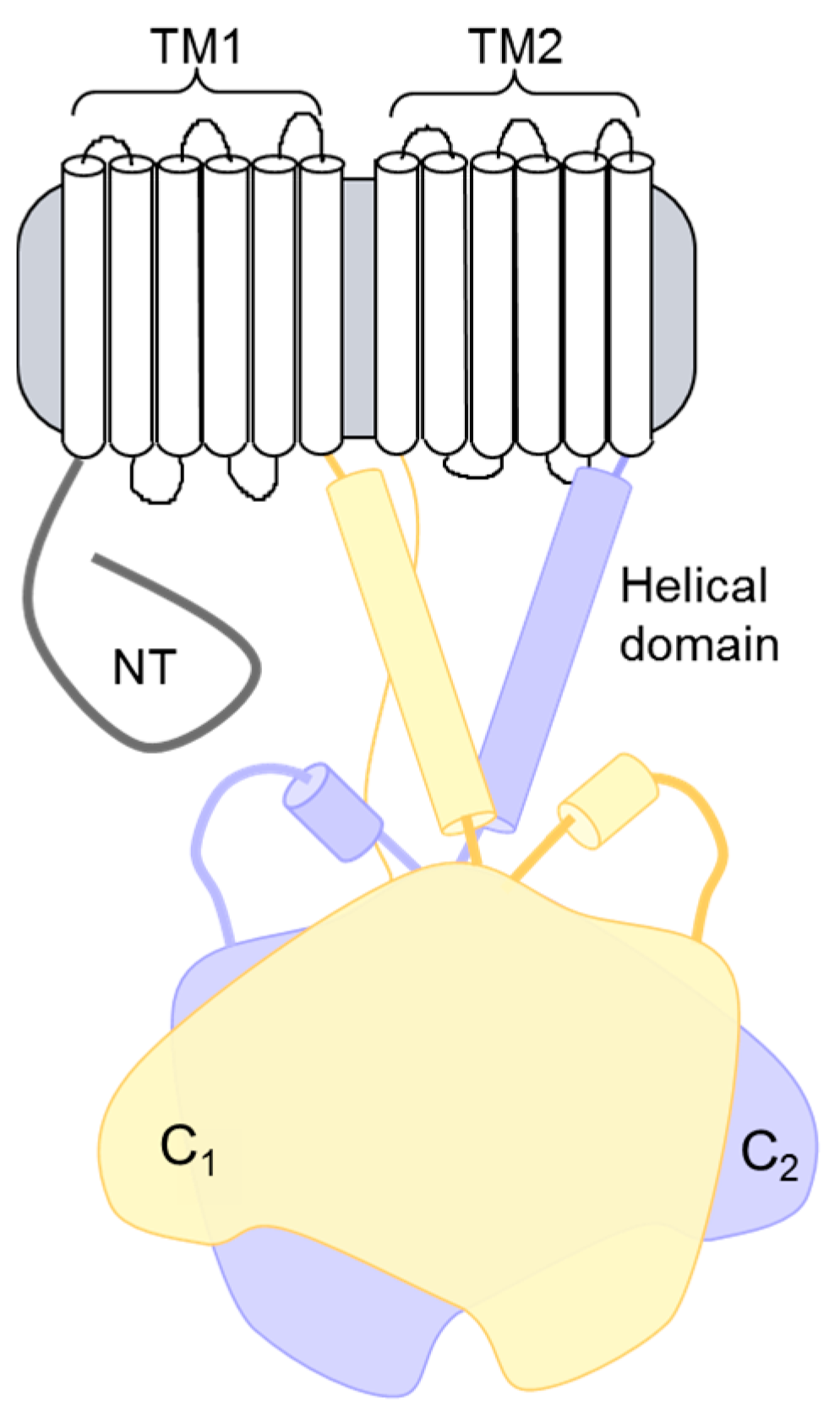
4. Newly Appreciated ACs in Heart
4.1. AC9 Knockout Phenotype
4.2. Complexes and Signaling Alterations in AC9−/− Heart
4.3. AC9 Regulation
4.3.1. G-Protein Regulation
4.3.2. Kinase and Phosphatase Regulation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Antos, C.L.; Frey, N.; Marx, S.O.; Reiken, S.; Gaburjakova, M.; Richardson, J.A.; Marks, A.R.; Olson, E.N. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase A. Circ. Res. 2001, 89, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.A.; Zakhary, D.R.; Mackey, J.A.; Desnoyer, R.W.; Apperson-Hansen, C.; Damron, D.S.; Bond, M. AKAP-mediated targeting of protein kinase A regulates contractility in cardiac myocytes. Circ. Res. 2001, 88, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhao, J.; Mandveno, A.; Potter, J.D. Cardiac troponin i phosphorylation increases the rate of cardiac muscle relaxation. Circ. Res. 1995, 76, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Alig, J.; Marger, L.; Mesirca, P.; Ehmke, H.; Mangoni, M.E.; Isbrandt, D. Control of heart rate by cAMP sensitivity of hcn channels. Proc. Natl. Acad. Sci. USA 2009, 106, 12189–12194. [Google Scholar] [CrossRef] [PubMed]
- Metrich, M.; Laurent, A.C.; Breckler, M.; Duquesnes, N.; Hmitou, I.; Courillau, D.; Blondeau, J.P.; Crozatier, B.; Lezoualc’h, F.; Morel, E. Epac activation induces histone deacetylase nuclear export via a ras-dependent signalling pathway. Cell Signal 2010, 22, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Schindler, R.F.; Brand, T. The popeye domain containing protein family—A novel class of cAMP effectors with important functions in multiple tissues. Prog. Biophys. Mol. Biol. 2016, 120, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, D.; Cooper, D.M. Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol. Rev. 2007, 87, 965–1010. [Google Scholar] [CrossRef] [PubMed]
- Sadana, R.; Dessauer, C.W. Physiological roles for g protein-regulated adenylyl cyclase isoforms: Insights from knockout and overexpression studies. Neurosignals 2009, 17, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, R.S.; Naugle, J.E.; Hase, M.; Gregorian, C.; Swaney, J.S.; Insel, P.A.; Brunton, L.L.; Meszaros, J.G. Angiotensin ii enhances adenylyl cyclase signaling via Ca2+/calmodulin. Gq-Gs cross-talk regulates collagen production in cardiac fibroblasts. J. Biol. Chem. 2003, 278, 24461–24468. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Kawabe, J.; Yatani, A.; Takagi, G.; Lee, M.C.; Hong, C.; Liu, J.; Takagi, I.; Sadoshima, J.; Vatner, D.E.; et al. Type 5 adenylyl cyclase disruption alters not only sympathetic but also parasympathetic and calcium-mediated cardiac regulation. Circ. Res. 2003, 93, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, K.; Minamisawa, S.; Tsunematsu, T.; Nakagome, M.; Toya, Y.; Tomlinson, J.E.; Umemura, S.; Scarborough, R.M.; Levy, D.E.; Ishikawa, Y. Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J. Biol. Chem. 2004, 279, 40938–40945. [Google Scholar] [CrossRef] [PubMed]
- Ping, P.; Anzai, T.; Gao, M.; Hammond, H.K. Adenylyl cyclase and g protein receptor kinase expression during development of heart failure. Am. J. Physiol. Heart Circ. Physiol. 1997, 273, H707–H717. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Kass, R.S.; Dessauer, C.W. The A-kinase anchoring protein Yotiao facilitates complex formation between type 9 adenylyl cyclase and the IKs potassium channel in heart. J. Biol. Chem. 2012, 287, 29815–29824. [Google Scholar] [CrossRef] [PubMed]
- Efendiev, R.; Dessauer, C.W. A kinase-anchoring proteins and adenylyl cyclase in cardiovascular physiology and pathology. J. Cardiovasc. Pharmacol. 2011, 58, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Dessauer, C.W.; Watts, V.J.; Ostrom, R.S.; Conti, M.; Dove, S.; Seifert, R. International union of basic and clinical pharmacology. Ci. Structures and small molecule modulators of mammalian adenylyl cyclases. Pharmacol. Rev. 2017, 69, 93–139. [Google Scholar] [CrossRef] [PubMed]
- Scarpace, P.J.; Matheny, M.; Tumer, N. Myocardial adenylyl cyclase type V and Vi mRNA: Differential regulation with age. J. Cardiovasc. Pharmacol. 1996, 27, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.L.; Chandra, R.; Ge, H.; Pain, J.; Yan, L.; Babu, G.; Depre, C.; Iwatsubo, K.; Ishikawa, Y.; Sadoshima, J.; et al. Adenylyl cyclase type 5 protein expression during cardiac development and stress. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1776–H1782. [Google Scholar] [CrossRef] [PubMed]
- Timofeyev, V.; Myers, R.E.; Kim, H.J.; Woltz, R.L.; Sirish, P.; Heiserman, J.P.; Li, N.; Singapuri, A.; Tang, T.; Yarov-Yarovoy, V.; et al. Adenylyl cyclase subtype-specific compartmentalization: Differential regulation of l-type Ca2+ current in ventricular myocytes. Circ. Res. 2013, 112, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Takagi, G.; Kawabe, J.; Yang, G.; Lee, M.C.; Hong, C.; Liu, J.; Vatner, D.E.; Sadoshima, J.; Vatner, S.F.; et al. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc. Natl. Acad. Sci. USA 2003, 100, 9986–9990. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Lai, N.C.; Roth, D.M.; Drumm, J.; Guo, T.; Lee, K.W.; Han, P.L.; Dalton, N.; Gao, M.H. Adenylyl cyclase type V deletion increases basal left ventricular function and reduces left ventricular contractile responsiveness to β-adrenergic stimulation. Basic Res. Cardiol. 2006, 101, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Gao, M.H.; Lai, N.C.; Firth, A.L.; Takahashi, T.; Guo, T.; Yuan, J.X.; Roth, D.M.; Hammond, H.K. Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation 2008, 117, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Ping, P.; Post, S.; Insel, P.A.; Tang, R.; Hammond, H.K. Increased expression of adenylylcyclase type vi proportionately increases beta-adrenergic receptor-stimulated production of cAMP in neonatal rat cardiac myocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Tepe, N.M.; Lorenz, J.N.; Yatani, A.; Dash, R.; Kranias, E.G.; Dorn, G.W., II; Liggett, S.B. Altering the receptor-effector ratio by transgenic overexpression of type V adenylyl cyclase: Enhanced basal catalytic activity and function without increased cardiomyocyte β-adrenergic signalling. Biochemistry 1999, 38, 16706–16713. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Vatner, D.E.; Kurotani, R.; Bai, Y.; Gao, S.; Yuan, Z.; Iwatsubo, K.; Ulucan, C.; Kawabe, J.; Ghosh, K.; et al. Disruption of type 5 adenylyl cyclase enhances desensitization of cyclic adenosine monophosphate signal and increases akt signal with chronic catecholamine stress. Circulation 2007, 116, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Vatner, D.E.; O’Connor, J.P.; Ivessa, A.; Ge, H.; Chen, W.; Hirotani, S.; Ishikawa, Y.; Sadoshima, J.; Vatner, S.F. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell 2007, 130, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Timofeyev, V.; Porter, C.A.; Tuteja, D.; Qiu, H.; Li, N.; Tang, T.; Singapuri, A.; Han, P.L.; Lopez, J.E.; Hammond, H.K.; et al. Disruption of adenylyl cyclase type v does not rescue the phenotype of cardiac-specific overexpression of galphaq protein-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1459–H1467. [Google Scholar] [CrossRef] [PubMed]
- Lai, N.C.; Tang, T.; Gao, M.H.; Saito, M.; Takahashi, T.; Roth, D.M.; Hammond, H.K. Activation of cardiac adenylyl cyclase expression increases function of the failing ischemic heart in mice. J. Am. Coll. Cardiol. 2008, 51, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.M.; Bayat, H.; Drumm, J.D.; Gao, M.H.; Swaney, J.S.; Ander, A.; Hammond, H.K. Adenylyl cyclase increases survival in cardiomyopathy. Circulation 2002, 105, 1989–1994. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.M.; Gao, M.H.; Lai, N.C.; Drumm, J.; Dalton, N.; Zhou, J.Y.; Zhu, J.; Entrikin, D.; Hammond, H.K. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation 1999, 99, 3099–3102. [Google Scholar] [CrossRef] [PubMed]
- Guellich, A.; Gao, S.; Hong, C.; Yan, L.; Wagner, T.E.; Dhar, S.K.; Ghaleh, B.; Hittinger, L.; Iwatsubo, K.; Ishikawa, Y.; et al. Effects of cardiac overexpression of type 6 adenylyl cyclase affects on the response to chronic pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H707–H712. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.H.; Lai, N.C.; Giamouridis, D.; Kim, Y.C.; Guo, T.; Hammond, H.K. Cardiac-directed expression of a catalytically inactive adenylyl cyclase 6 protects the heart from sustained beta-adrenergic stimulation. PLoS ONE 2017, 12, e0181282. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.S.; Chen, C.C.; Chien, C.L.; Lai, H.L.; Jiang, S.T.; Chen, Y.C.; Lai, L.P.; Hsiao, W.F.; Chen, W.P.; Chern, Y. The type VI adenylyl cyclase protects cardiomyocytes from β-adrenergic stress by a PKA/STAT3-dependent pathway. J. Biomed. Sci. 2017, 24, 68. [Google Scholar] [CrossRef] [PubMed]
- Hammond, H.K.; Penny, W.F.; Traverse, J.H.; Henry, T.D.; Watkins, M.W.; Yancy, C.W.; Sweis, R.N.; Adler, E.D.; Patel, A.N.; Murray, D.R.; et al. Intracoronary gene transfer of adenylyl cyclase 6 in patients with heart failure: A randomized clinical trial. JAMA Cardiol. 2016, 1, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Zhang, X.; Li, Y.; Guan, Y.; Ai, X.; Szeto, C.; Nakayama, H.; Zhang, H.; Ge, S.; Molkentin, J.D.; et al. Enhanced basal contractility but reduced excitation-contraction coupling efficiency and β-adrenergic reserve of hearts with increased Cav1.2 activity. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H519–H528. [Google Scholar] [CrossRef] [PubMed]
- Bravo, C.A.; Vatner, D.E.; Pachon, R.; Zhang, J.; Vatner, S.F. A food and drug administration-approved antiviral agent that inhibits adenylyl cyclase type 5 protects the ischemic heart even when administered after reperfusion. J. Pharmacol. Exp. Ther. 2016, 357, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Brand, C.S.; Hocker, H.J.; Gorfe, A.A.; Cavasotto, C.N.; Dessauer, C.W. Isoform selectivity of adenylyl cyclase inhibitors: Characterization of known and novel compounds. J. Pharmacol. Exp. Ther. 2013, 347, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Braeunig, J.H.; Schweda, F.; Han, P.L.; Seifert, R. Similarly potent inhibition of adenylyl cyclase by p-site inhibitors in hearts from wild type and ac5 knockout mice. PLoS ONE 2013, 8, e68009. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Lai, N.C.; Wright, A.T.; Gao, M.H.; Lee, P.; Guo, T.; Tang, R.; McCulloch, A.D.; Hammond, H.K. Adenylyl cyclase 6 deletion increases mortality during sustained β-adrenergic receptor stimulation. J. Mol. Cell. Cardiol. 2013, 60, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Ikoma, E.; Tsunematsu, T.; Nakazawa, I.; Shiwa, T.; Hibi, K.; Ebina, T.; Mochida, Y.; Toya, Y.; Hori, H.; Uchino, K.; et al. Polymorphism of the type 6 adenylyl cyclase gene and cardiac hypertrophy. J. Cardiovasc. Pharmacol. 2003, 42 (Suppl. 1), S27–S32. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Friedman, J.R.; Chen, D.H.; Chan, G.C.; Bloss, C.S.; Hisama, F.M.; Topol, S.E.; Carson, A.R.; Pham, P.H.; Bonkowski, E.S.; et al. Gain-of-function adcy5 mutations in familial dyskinesia with facial myokymia. Ann. Neurol. 2014, 75, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Matsushita, M.M.; Robertson, P.; Rieder, M.; Girirajan, S.; Antonacci, F.; Lipe, H.; Eichler, E.E.; Nickerson, D.A.; Bird, T.D.; et al. Autosomal dominant familial dyskinesia and facial myokymia: Single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch. Neurol. 2012, 69, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, I.; Rezabkova, L.; Olieric, V.; Polyhach, Y.; Weinert, T.; Kammerer, R.A.; Jeschke, G.; Korkhov, V.M. Role of the nucleotidyl cyclase helical domain in catalytically active dimer formation. Proc. Natl. Acad. Sci. USA 2017, 114, E9821–E9828. [Google Scholar] [CrossRef] [PubMed]
- Mattick, P.; Parrington, J.; Odia, E.; Simpson, A.; Collins, T.; Terrar, D. Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the i(f) pacemaker current. J. Physiol. 2007, 582, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Lyashkov, A.E.; Graham, D.; Sheydina, A.; Volkova, M.V.; Mitsak, M.; Vinogradova, T.M.; Lukyanenko, Y.O.; Li, Y.; Ruknudin, A.M.; et al. Ca(2+) -stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J. Biol. Chem. 2008, 283, 14461–14468. [Google Scholar] [CrossRef] [PubMed]
- Vedantham, V.; Galang, G.; Evangelista, M.; Deo, R.C.; Srivastava, D. Rna sequencing of mouse sinoatrial node reveals an upstream regulatory role for islet-1 in cardiac pacemaker cells. Circ. Res. 2015, 116, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.J.; Sucharov, J.; van Dusen, R.; Cecil, M.; Nunley, K.; Wickers, S.; Karimpur-Fard, A.; Stauffer, B.L.; Miyamoto, S.D.; Sucharov, C.C. Cardiac adenylyl cyclase and phosphodiesterase expression profiles vary by age, disease, and chronic phosphodiesterase inhibitor treatment. J. Card. Fail. 2017, 23, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Dessauer, C.W.; Tasken, K. Creating order from chaos: Cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Efendiev, R.; Bavencoffe, A.; Hu, H.; Zhu, M.X.; Dessauer, C.W. Scaffolding by A-kinase anchoring protein enhances functional coupling between adenylyl cyclase and TRPV1 channel. J. Biol. Chem. 2013, 288, 3929–3937. [Google Scholar] [CrossRef] [PubMed]
- Bauman, A.L.; Soughayer, J.; Nguyen, B.T.; Willoughby, D.; Carnegie, G.K.; Wong, W.; Hoshi, N.; Langeberg, L.K.; Cooper, D.M.; Dessauer, C.W.; et al. Dynamic regulation of cAMP synthesis through anchored pka-adenylyl cyclase v/vi complexes. Mol. Cell 2006, 23, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, P.K.; Jaggi, A.S. Trpv1 channels in cardiovascular system: A double edged sword? Int. J. Cardiol. 2017, 228, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.B.; Rossow, C.F.; Navedo, M.F.; Westenbroek, R.E.; Catterall, W.A.; Santana, L.F.; McKnight, G.S. Sympathetic stimulation of adult cardiomyocytes requires association of akap5 with a subpopulation of L-type calcium channels. Circ. Res. 2010, 107, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, J.; Motoike, H.K.; Rao, J.; Kass, R.S. Regulatory actions of the A-kinase anchoring protein yotiao on a heart potassium channel downstream of pka phosphorylation (vol 101, pg 16374, 2004). Proc. Natl. Acad. Sci. USA 2004, 101, 16374–16378. [Google Scholar] [CrossRef] [PubMed]
- Efendiev, R.; Samelson, B.K.; Nguyen, B.T.; Phatarpekar, P.V.; Baameur, F.; Scott, J.D.; Dessauer, C.W. AKAP79 interacts with multiple adenylyl cyclase (Ac) isoforms and scaffolds AC5 and -6 to alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J. Biol. Chem. 2010, 285, 14450–14458. [Google Scholar] [CrossRef] [PubMed]
- Navedo, M.F.; Nieves-Cintron, M.; Amberg, G.C.; Yuan, C.; Votaw, V.S.; Lederer, W.J.; McKnight, G.S.; Santana, L.F. Akap150 is required for stuttering persistent Ca2+ sparklets and angiotensin ii-induced hypertension. Circ. Res. 2008, 102, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Tajada, S.; Moreno, C.M.; O’Dwyer, S.; Woods, S.; Sato, D.; Navedo, M.F.; Santana, L.F. Distance constraints on activation of trpv4 channels by akap150-bound pkcalpha in arterial myocytes. J. Gen. Physiol. 2017, 149, 639–659. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nooh, M.M.; Bahouth, S.W. Role of AKAP79/150 protein in β1-adrenergic receptor trafficking and signaling in mammalian cells. J. Biol. Chem. 2013, 288, 33797–33812. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, J.; Drum, B.M.; Chen, Y.; Yin, H.; Guo, X.; Luckey, S.W.; Gilbert, M.L.; McKnight, G.S.; Scott, J.D.; et al. Loss of akap150 promotes pathological remodelling and heart failure propensity by disrupting calcium cycling and contractile reserve. Cardiovasc. Res. 2017, 113, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Nieves-Cintron, M.; Hirenallur-Shanthappa, D.; Nygren, P.J.; Hinke, S.A.; Dell’Acqua, M.L.; Langeberg, L.K.; Navedo, M.; Santana, L.F.; Scott, J.D. Akap150 participates in calcineurin/nfat activation during the down-regulation of voltage-gated k(+) currents in ventricular myocytes following myocardial infarction. Cell Signal 2016, 28, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Matheus, A.S.; Tannus, L.R.; Cobas, R.A.; Palma, C.C.; Negrato, C.A.; Gomes, M.B. Impact of diabetes on cardiovascular disease: An update. Int. J. Hypertens. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Wang, J.; Li, N.; Shen, M.; Wang, D.; Yu, Q.; Wang, H. Akap150 mobilizes cpkc-dependent cardiac glucotoxicity. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E384–E397. [Google Scholar] [CrossRef] [PubMed]
- Kapiloff, M.S.; Schillace, R.V.; Westphal, A.M.; Scott, J.D. Makap: An A-kinase anchoring protein targeted to the nuclear membrane of differentiated myocytes. J. Cell Sci. 1999, 112, 2725–2736. [Google Scholar] [PubMed]
- Kapiloff, M.S.; Piggott, L.A.; Sadana, R.; Li, J.; Heredia, L.A.; Henson, E.; Efendiev, R.; Dessauer, C.W. An adenylyl cyclase-makapbeta signaling complex regulates cAMP levels in cardiac myocytes. J. Biol. Chem. 2009, 284, 23540–23546. [Google Scholar] [CrossRef] [PubMed]
- Pare, G.C.; Easlick, J.L.; Mislow, J.M.; McNally, E.M.; Kapiloff, M.S. Nesprin-1α contributes to the targeting of makap to the cardiac myocyte nuclear envelope. Exp. Cell Res. 2005, 303, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Ruehr, M.L.; Russell, M.A.; Bond, M. A-kinase anchoring protein targeting of protein kinase A in the heart. J. Mol. Cell. Cardiol. 2004, 37, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Puri, T.S.; Gerhardstein, B.L.; Chien, A.J.; Green, R.D.; Hosey, M.M. Identification and subcellular localization of the subunits of l-type calcium channels and adenylyl cyclase in cardiac myocytes. J. Biol. Chem. 1997, 272, 19401–19407. [Google Scholar] [CrossRef] [PubMed]
- Escobar, M.; Cardenas, C.; Colavita, K.; Petrenko, N.B.; Franzini-Armstrong, C. Structural evidence for perinuclear calcium microdomains in cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 50, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.A.; Tirnauer, J.S.; Glidden, N.; Kapiloff, M.S.; Dodge-Kafka, K.L. Myocyte enhancer factor 2 (mef2) tethering to muscle selective a-kinase anchoring protein (makap) is necessary for myogenic differentiation. Cell Signal 2012, 24, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Dodge, K.L.; Khouangsathiene, S.; Kapiloff, M.S.; Mouton, R.; Hill, E.V.; Houslay, M.D.; Langeberg, L.K.; Scott, J.D. Makap assembles a protein kinase a/pde4 phosphodiesterase cAMP signaling module. EMBO J. 2001, 20, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Goehring, A.S.; Kapiloff, M.S.; Langeberg, L.K.; Scott, J.D. Makap compartmentalizes oxygen-dependent control of HIF-1α. Sci. Signal 2008, 1, ra18. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.L.; Bauman, A.; Mayer, N.; Henson, E.; Heredia, L.; Ahn, J.; McAvoy, T.; Nairn, A.C.; Kapiloff, M.S. cAMP-stimulated protein phosphatase 2a activity associated with muscle a kinase-anchoring protein (makap) signaling complexes inhibits the phosphorylation and activity of the cAMP-specific phosphodiesterase pde4d3. J. Biol. Chem. 2010, 285, 11078–11086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Malik, S.; Pang, J.; Wang, H.; Park, K.M.; Yule, D.I.; Blaxall, B.C.; Smrcka, A.V. Phospholipase cepsilon hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 2013, 153, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kritzer, M.D.; Michel, J.J.; Le, A.; Thakur, H.; Gayanilo, M.; Passariello, C.L.; Negro, A.; Danial, J.B.; Oskouei, B.; et al. Anchored p90 ribosomal s6 kinase 3 is required for cardiac myocyte hypertrophy. Circ. Res. 2013, 112, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.L.; Soughayer, J.; Pare, G.C.; Carlisle Michel, J.J.; Langeberg, L.K.; Kapiloff, M.S.; Scott, J.D. The protein kinase A anchoring protein makap coordinates two integrated cAMP effector pathways. Nature 2005, 437, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Ruehr, M.L.; Russell, M.A.; Ferguson, D.G.; Bhat, M.; Ma, J.J.; Damron, D.S.; Scott, J.D.; Bond, M. Targeting of protein kinase A by muscle a kinase-anchoring protein (makap) regulates phosphorylation and function of the skeletal muscle ryanodine receptor. J. Biol. Chem. 2003, 278, 24831–24836. [Google Scholar] [CrossRef] [PubMed]
- Pare, G.C.; Bauman, A.L.; McHenry, M.; Michel, J.J.; Dodge-Kafka, K.L.; Kapiloff, M.S. The makap complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. J. Cell Sci. 2005, 118, 5637–5646. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, M.D.; Li, J.; Passariello, C.L.; Gayanilo, M.; Thakur, H.; Dayan, J.; Dodge-Kafka, K.; Kapiloff, M.S. The scaffold protein muscle A-kinase anchoring protein beta orchestrates cardiac myocyte hypertrophic signaling required for the development of heart failure. Circ. Heart Fail. 2014, 7, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Negro, A.; Lopez, J.; Bauman, A.L.; Henson, E.; Dodge-Kafka, K.; Kapiloff, M.S. The makapbeta scaffold regulates cardiac myocyte hypertrophy via recruitment of activated calcineurin. J. Mol. Cell. Cardiol. 2010, 48, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Malik, S.; Kelley, G.G.; Kapiloff, M.S.; Smrcka, A.V. Phospholipase c epsilon scaffolds to muscle-specific a kinase anchoring protein (makapbeta) and integrates multiple hypertrophic stimuli in cardiac myocytes. J. Biol. Chem. 2011, 286, 23012–23021. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Kurokawa, J.; Reiken, S.; Motoike, H.; D’Armiento, J.; Marks, A.R.; Kass, R.S. Requirement of a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 2002, 295, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Marquardt, M.L.; Tester, D.J.; Sampson, K.J.; Ackerman, M.J.; Kass, R.S. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 20990–20995. [Google Scholar] [CrossRef] [PubMed]
- Duggal, P.; Vesely, M.R.; Wattanasirichaigoon, D.; Villafane, J.; Kaushik, V.; Beggs, A.H. Mutation of the gene for isk associated with both jervell and lange-nielsen and romano-ward forms of long-QT syndrome. Circulation 1998, 97, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Westphal, R.S.; Tavalin, S.J.; Lin, J.W.; Alto, N.M.; Fraser, I.D.; Langeberg, L.K.; Sheng, M.; Scott, J.D. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science 1999, 285, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.W.; Wyszynski, M.; Madhavan, R.; Sealock, R.; Kim, J.U.; Sheng, M. Yotiao, a novel protein of neuromuscular junction and brain that interacts with specific splice variants of NMDA receptor subunit nr1. J. Neurosci. 1998, 18, 2017–2027. [Google Scholar] [PubMed]
- Terrenoire, C.; Houslay, M.D.; Baillie, G.S.; Kass, R.S. The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J. Biol. Chem. 2009, 284, 9140–9146. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Spatjens, R.L.; Seyen, S.R.; Lentink, V.; Kuijpers, H.J.; Boulet, I.R.; de Windt, L.J.; David, M.; Volders, P.G. Dominant-negative control of cAMP-dependent IKs upregulation in human long-QT syndrome type 1. Circ. Res. 2012, 110, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Piggott, L.A.; Bauman, A.L.; Scott, J.D.; Dessauer, C.W. The A-kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. Proc. Natl. Acad. Sci. USA 2008, 105, 13835–13840. [Google Scholar] [CrossRef] [PubMed]
- Antoni, F.A. Adenylyl cyclase type 9. UCSD Nat. Mol. Pages 2006. [Google Scholar] [CrossRef]
- Li, Y.; Baldwin, T.A.; Wang, Y.; Subramaniam, J.; Carbajal, A.G.; Brand, C.S.; Cunha, S.R.; Dessauer, C.W. Loss of type 9 adenylyl cyclase triggers reduced phosphorylation of hsp20 and diastolic dysfunction. Sci. Rep. 2017, 7, 5522. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.; Zhao, X.; Gao, S.; Hong, C.; Vatner, D.E.; Vatner, S.F. Heart rate and electrocardiography monitoring in mice. Curr. Protoc. Mouse Biol. 2011, 1, 123–139. [Google Scholar] [PubMed]
- Honore, E.; Attali, B.; Romey, G.; Heurteaux, C.; Ricard, P.; Lesage, F.; Lazdunski, M.; Barhanin, J. Cloning, expression, pharmacology and regulation of a delayed rectifier k+ channel in mouse heart. EMBO J. 1991, 10, 2805–2811. [Google Scholar] [PubMed]
- Salama, G.; Baker, L.; Wolk, R.; Barhanin, J.; London, B. Arrhythmia phenotype in mouse models of human long QT. J. Interv. Card. Electrophysiol. 2009, 24, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C.; Yuan, Q.; Song, G.; Wang, Y.; Chen, G.; Qian, J.; Zhou, X.; Lee, Y.J.; Ashraf, M.; Kranias, E.G. Small heat-shock protein hsp20 attenuates β-agonist-mediated cardiac remodeling through apoptosis signal-regulating kinase 1. Circ. Res. 2006, 99, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C.; Zhou, X.; Wang, X.; Song, G.; Qian, J.; Nicolaou, P.; Chen, G.; Ren, X.; Kranias, E.G. Heat shock protein 20 interacting with phosphorylated akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ. Res. 2008, 103, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.P.; Hortigon-Vinagre, M.P.; Findlay, J.E.; Elliott, C.; Currie, S.; Baillie, G.S. Targeted disruption of the heat shock protein 20-phosphodiesterase 4D (PDE4D) interaction protects against pathological cardiac remodelling in a mouse model of hypertrophy. FEBS Open Bio 2014, 4, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Baskerville, S.; Bartel, D.P. Microarray profiling of micrornas reveals frequent coexpression with neighboring mirnas and host genes. RNA 2005, 11, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Voellenkle, C.; van Rooij, J.; Cappuzzello, C.; Greco, S.; Arcelli, D.; Di Vito, L.; Melillo, G.; Rigolini, R.; Costa, E.; Crea, F.; et al. Microrna signatures in peripheral blood mononuclear cells of chronic heart failure patients. Physiol. Genom. 2010, 42, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, R.D.; Tsoutsman, T.; Shephard, R.E.; Ritchie, W.; Semsarian, C. Global microrna profiling of the mouse ventricles during development of severe hypertrophic cardiomyopathy and heart failure. PLoS ONE 2012, 7, e44744. [Google Scholar] [CrossRef] [PubMed]
- Tijsen, A.J.; Pinto, Y.M.; Creemers, E.E. Circulating micrornas as diagnostic biomarkers for cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1085–H1095. [Google Scholar] [CrossRef] [PubMed]
- Antoni, F.A.; Barnard, R.J.O.; Shipston, M.J.; Smith, S.M.; Simpson, J.; Paterson, J.M. Calcineurin feedback inhibition of agonist-evoked cAMP formation. J. Biol. Chem. 1995, 270, 28055–28061. [Google Scholar] [PubMed]
- Hacker, B.M.; Tomlinson, J.E.; Wayman, G.A.; Sultana, R.; Chan, G.; Villacres, E.; Disteche, C.; Storm, D.R. Cloning, chromosomal mapping, and regulatory properties of the human type 9 adenylyl cyclase (ADCY9). Genomics 1998, 50, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Paterson, J.M.; Smith, S.M.; Simpson, J.; Grace, O.C.; Sosunov, A.A.; Bell, J.E.; Antoni, F.A. Characterisation of human adenylyl cyclase ix reveals inhibition by Ca2+/calcineurin and differential mRNA plyadenylation. J. Neurochem. 2000, 75, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Sosunov, S.A.; Kemaikin, S.P.; Kurnikova, I.A.; Antoni, F.A.; Sosunov, A.A. Expression of adenylyl cyclase type ix and calcineurin in synapses of the central nervous system. Bull. Exp. Biol. Med. 2001, 131, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gritz, D.; Parent, C.A. PkcβII acts downstream of chemoattractant receptors and MTORC2 to regulate cAMP production and myosin ii activity in neutrophils. Mol. Biol. Cell. 2014, 25, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Cumbay, M.G.; Watts, V.J. Galphaq potentiation of adenylate cyclase type 9 activity through a Ca2+/calmodulin-dependent pathway. Biochem. Pharmacol. 2005, 69, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Cumbay, M.G.; Watts, V.J. Novel regulatory properties of human type 9 adenylate cyclase. J. Pharmacol. Exp. Ther. 2004, 310, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Dessauer, C.W.; Tesmer, J.J.; Sprang, S.R.; Gilman, A.G. Identification of a gialpha binding site on type v adenylyl cyclase. J. Biol. Chem. 1998, 273, 25831–25839. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Das, S.; Losert, W.; Parent, C.A. mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev. Cell 2010, 19, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Brand, C.S.; Sadana, R.; Malik, S.; Smrcka, A.V.; Dessauer, C.W. Adenylyl cyclase 5 regulation by gbetagamma involves isoform-specific use of multiple interaction sites. Mol. Pharmacol. 2015, 88, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Paterson, J.M.; Smith, S.M.; Harmar, A.J.; Antoni, F.A. Control of a novel adenylyl cyclase by calcineurin. Biochem. Biophys. Res. Commun. 1995, 214, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldwin, T.A.; Dessauer, C.W. Function of Adenylyl Cyclase in Heart: the AKAP Connection. J. Cardiovasc. Dev. Dis. 2018, 5, 2. https://doi.org/10.3390/jcdd5010002
Baldwin TA, Dessauer CW. Function of Adenylyl Cyclase in Heart: the AKAP Connection. Journal of Cardiovascular Development and Disease. 2018; 5(1):2. https://doi.org/10.3390/jcdd5010002
Chicago/Turabian StyleBaldwin, Tanya A., and Carmen W. Dessauer. 2018. "Function of Adenylyl Cyclase in Heart: the AKAP Connection" Journal of Cardiovascular Development and Disease 5, no. 1: 2. https://doi.org/10.3390/jcdd5010002
APA StyleBaldwin, T. A., & Dessauer, C. W. (2018). Function of Adenylyl Cyclase in Heart: the AKAP Connection. Journal of Cardiovascular Development and Disease, 5(1), 2. https://doi.org/10.3390/jcdd5010002