A-Kinase Anchoring Protein-Lbc: A Molecular Scaffold Involved in Cardiac Protection

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Role of the cAMP/PKA Pathways in Cardiac Protection
3. AKAP-Lbc Signaling and Cardiac Protection
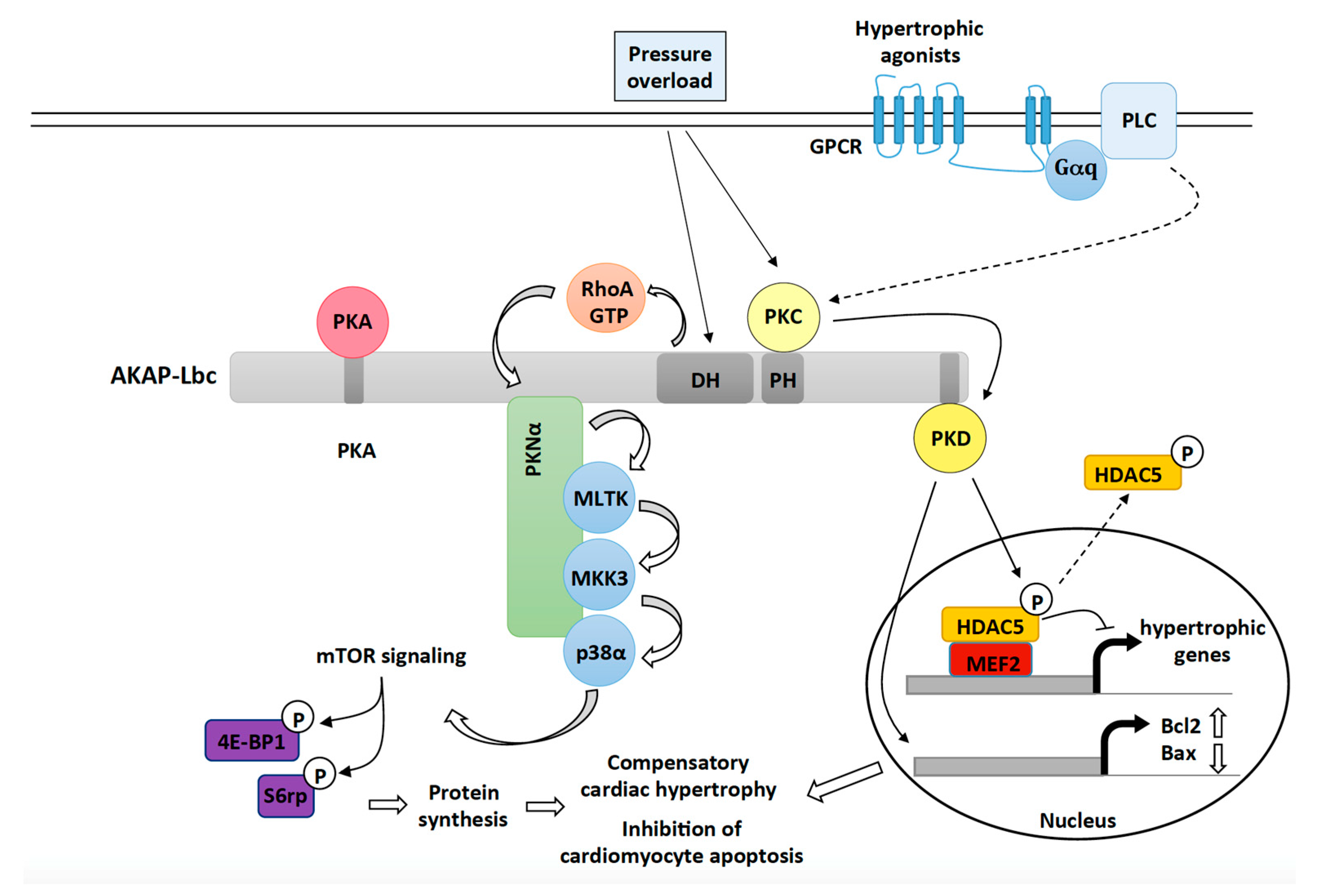
3.1. AKAP-Lbc Mediates Protection against Pressure Overload-Induced Cardiac Dysfunction
3.1.1. The Role of AKAP-Lbc-Mediated Regulation of p38α
3.1.2. The Role of AKAP-Lbc-Mediated Regulation of PKD1
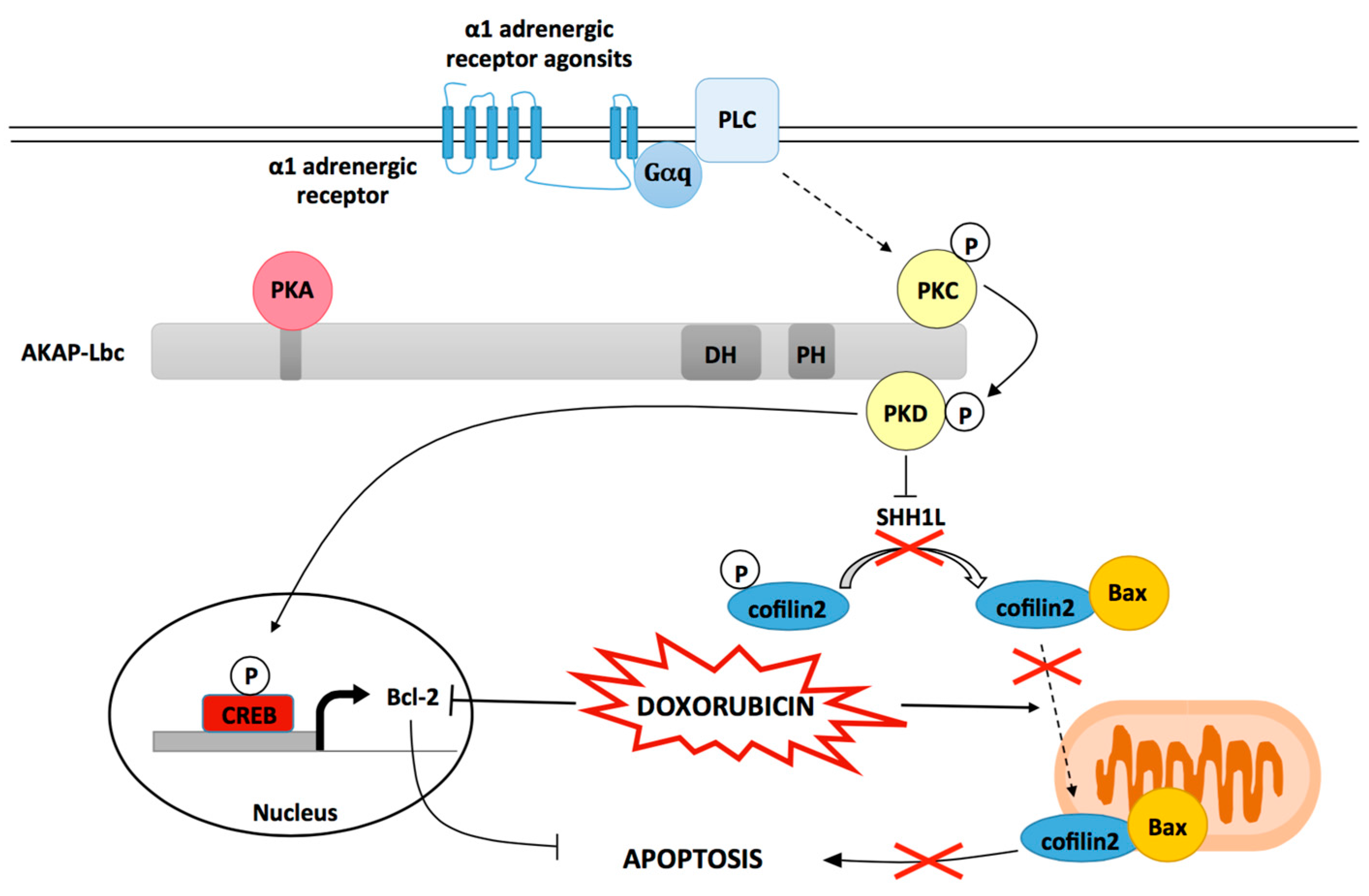
3.2. AKAP-Lbc Mediates Protection against Doxorubicin-Induced Cardiomyocyte Toxicity
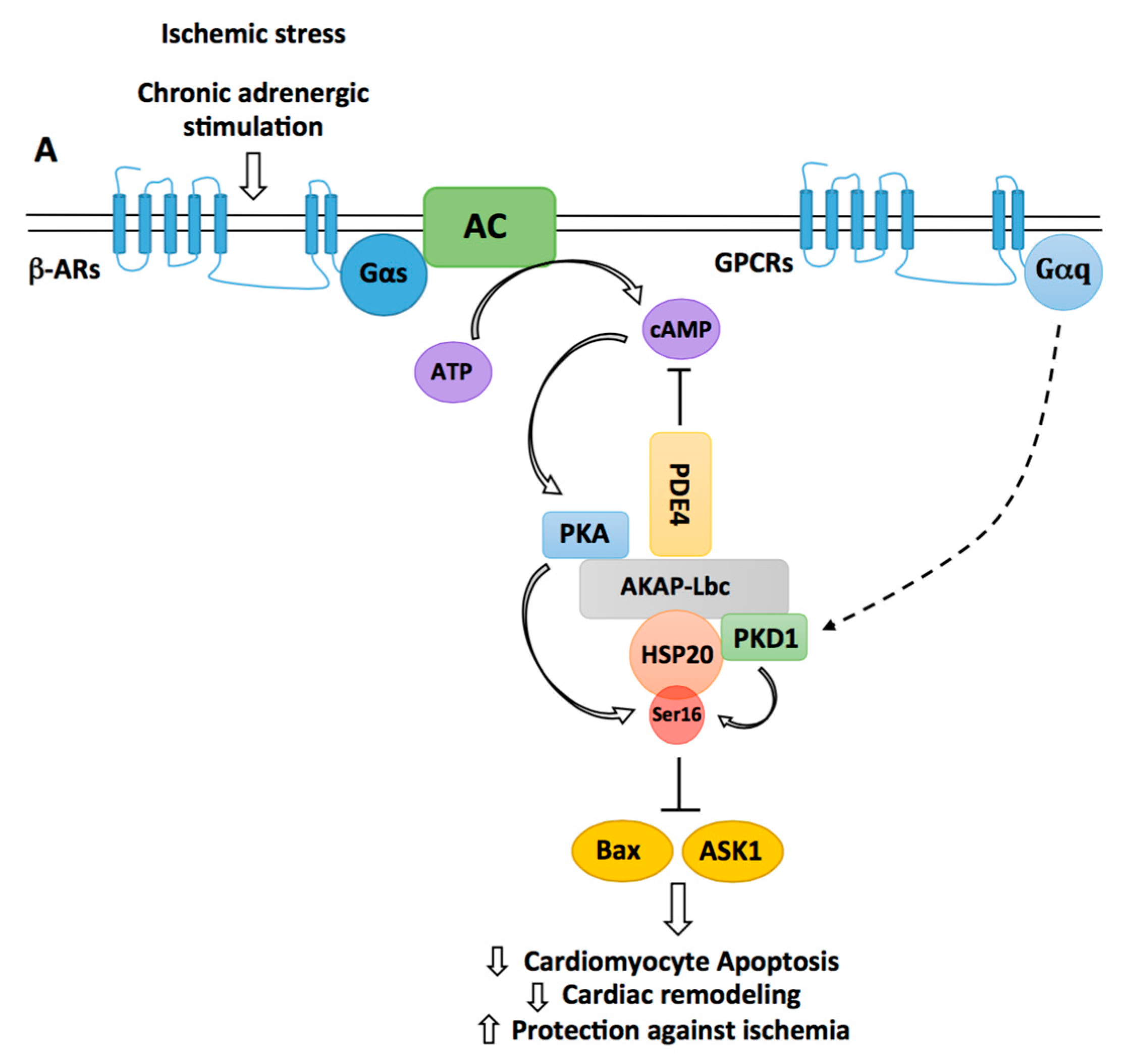
3.3. The Cardioprotective Role of the AKAP-Lbc/HSP20 Complex
4. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Towbin, J.A.; Bowles, N.E. The failing heart. Nature 2002, 415, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Olson, E.N.; Bassel-Duby, R. Mending broken hearts: Cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 2013, 14, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Sun, Y.; Diez, J. Fibrosis: A living tissue and the infarcted heart. J. Am. Coll. Cardiol. 2008, 52, 2029–2031. [Google Scholar] [CrossRef] [PubMed]
- Burchfield, J.S.; Xie, M.; Hill, J.A. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Maric, D.; Lopez, I.P.; Cavin, S.; del Vescovo, C.D. A-kinase anchoring proteins: Molecular regulators of the cardiac stress response. Biochim. Biophys. Acta 2013, 1833, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.R.; Rosenzweig, A. Targeting survival signaling in heart failure. Curr. Opin. Pharmacol. 2005, 5, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Esseltine, J.L.; Scott, J.D. AKAP signaling complexes: Pointing towards the next generation of therapeutic targets? Trends Pharmacol. Sci. 2013, 34, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Langeberg, L.K.; Scott, J.D. Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 2015, 16, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Dema, A.; Perets, E.; Schulz, M.S.; Deak, V.A.; Klussmann, E. Pharmacological targeting of AKAP-directed compartmentalized cAMP signalling. Cell. Signal. 2015, 27, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Cavin, S.; Maric, D.; Diviani, D. A-kinase anchoring protein-Lbc promotes pro-fibrotic signaling in cardiac fibroblasts. Biochim. Biophys. Acta 2014, 1843, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.G.; Lygren, B.; Dokurno, P.; Hoshi, N.; McConnachie, G.; Tasken, K.; Carlson, C.R.; Scott, J.D.; Barford, D. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol. Cell 2006, 24, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Kinderman, F.S.; Kim, C.; von Daake, S.; Ma, Y.; Pham, B.Q.; Spraggon, G.; Xuong, N.H.; Jennings, P.A.; Taylor, S.S. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol. Cell 2006, 24, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, E. Protein-protein interactions of PDE4 family members—Functions, interactions and therapeutic value. Cell. Signal 2016, 28, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Dessauer, C.W. Adenylyl cyclase—A-kinase anchoring protein complexes: The next dimension in cAMP signaling. Mol. Pharmacol. 2009, 76, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Kapiloff, M.S.; Piggott, L.A.; Sadana, R.; Li, J.; Heredia, L.A.; Henson, E.; Efendiev, R.; Dessauer, C.W. An adenylyl cyclase-mAKAPβ signaling complex regulates cAMP levels in cardiac myocytes. J. Biol. Chem. 2009, 284, 23540–23546. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Soderling, J.; Scott, J.D. AKAP-Lbc anchors protein kinase A and nucleates Gα 12-selective Rho-mediated stress fiber formation. J. Biol. Chem. 2001, 276, 44247–44257. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Dessauer, C.W.; Tasken, K. Creating order from chaos: Cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Redden, J.M.; Dodge-Kafka, K.L. AKAP phosphatase complexes in the heart. J. Cardiovasc. Pharmacol. 2011, 58, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Wild, A.R.; Dell’Acqua, M.L. Potential for therapeutic targeting of AKAP signaling complexes in nervous system disorders. Pharmacol. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Aye, T.T.; Mohammed, S.; van den Toorn, H.W.; van Veen, T.A.; van der Heyden, M.A.; Scholten, A.; Heck, A.J. Selectivity in enrichment of cAMP-dependent protein kinase regulatory subunits type I and type II and their interactors using modified cAMP affinity resins. Mol. Cell. Proteom. 2009, 8, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Aye, T.T.; Soni, S.; van Veen, T.A.; van der Heyden, M.A.; Cappadona, S.; Varro, A.; de Weger, R.A.; de Jonge, N.; Vos, M.A.; Heck, A.J.; et al. Reorganized PKA-AKAP associations in the failing human heart. J. Mol. Cell. Cardiol. 2012, 52, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Reggi, E.; Arambasic, M.; Caso, S.; Maric, D. Emerging roles of A-kinase anchoring proteins in cardiovascular pathophysiology. Biochim. Biophys. Acta 2016, 1863, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Santana, L.F. A-kinase anchoring proteins: Getting to the heart of the matter. Circulation 2010, 121, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, M.D.; Li, J.; Dodge-Kafka, K.; Kapiloff, M.S. AKAPs: The architectural underpinnings of local cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol. Cell 2011, 42, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Lopez, I.P.; Cariolato, L.; Maric, D.; Gillet, L.; Abriel, H.; Diviani, D. A-kinase anchoring protein Lbc coordinates a p38 activating signaling complex controlling compensatory cardiac hypertrophy. Mol. Cell. Biol. 2013, 33, 2903–2917. [Google Scholar] [CrossRef] [PubMed]
- Taglieri, D.M.; Johnson, K.R.; Burmeister, B.T.; Monasky, M.M.; Spindler, M.J.; DeSantiago, J.; Banach, K.; Conklin, B.R.; Carnegie, G.K. The C-terminus of the long AKAP13 isoform (AKAP-Lbc) is critical for development of compensatory cardiac hypertrophy. J. Mol. Cell. Cardiol. 2014, 66, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Caso, S.; Maric, D.; Arambasic, M.; Cotecchia, S.; Diviani, D. AKAP-Lbc mediates protection against doxorubicin-induced cardiomyocyte toxicity. Biochim. Biophys. Acta 2017, 1864, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Goehring, A.S.; Kapiloff, M.S.; Langeberg, L.K.; Scott, J.D. mAKAP compartmentalizes oxygen-dependent control of HIF-1α, Science Signaling. Sci. Signal. 2008, 1, ra18. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Matta, S.M.; Sullivan, R.D.; Bahouth, S.W. Carvedilol reverses cardiac insufficiency in AKAP5 knockout mice by normalizing the activities of calcineurin and CaMKII. Cardiovasc. Res. 2014, 104, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; Feliciello, A.; Schiattarella, G.G.; Esposito, G.; Guerriero, R.; Zaccaro, L.; del Gatto, A.; Saviano, M.; Garbi, C.; Carangi, R.; et al. AKAP121 downregulation impairs protective cAMP signals, promotes mitochondrial dysfunction, and increases oxidative stress. Cardiovasc. Res. 2010, 88, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Scimia, M.C.; Wilkinson, D.; Trelles, R.D.; Wood, M.R.; Bowtell, D.; Dillin, A.; Mercola, M.; Ronai, Z.A. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell. 2011, 44, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, V.; Suresh, S.C.; Thirunavukkarasu, M.; Mannu, J.; Foye, J.L.C.; Mathur, P.P.; Palesty, J.A.; Sanchez, J.A.; McFadden, D.W.; Maulik, N. Regulation of A-Kinase-Anchoring Protein 12 by Heat Shock Protein A12B to Prevent Ventricular Dysfunction Following Acute Myocardial Infarction in Diabetic Rats. J. Cardiovasc. Transl. Res. 2017, 10, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, J.; Drum, B.M.; Chen, Y.; Yin, H.; Guo, X.; Luckey, S.W.; Gilbert, M.L.; McKnight, G.S.; Scott, J.D.; et al. Loss of AKAP150 promotes pathological remodelling and heart failure propensity by disrupting calcium cycling and contractile reserve. Cardiovasc. Res. 2017, 113, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Cattaneo, F.; Pironti, G.; Magliulo, F.; Carotenuto, G.; Pirozzi, M.; Polishchuk, R.; Borzacchiello, D.; Paolillo, R.; Oliveti, M.; et al. Akap1 Deficiency Promotes Mitochondrial Aberrations and Exacerbates Cardiac Injury Following Permanent Coronary Ligation via Enhanced Mitophagy and Apoptosis. PLoS ONE 2016, 11, e0154076. [Google Scholar] [CrossRef] [PubMed]
- Lochner, A.; Genade, S.; Tromp, E.; Podzuweit, T.; Moolman, J.A. Ischemic preconditioning and the β-adrenergic signal transduction pathway. Circulation 1999, 100, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Garcia-Dorado, D.; Ruiz-Meana, M.; Agullo, L.; Pina, P.; Soler-Soler, J. Ischemic preconditioning attenuates calpain-mediated degradation of structural proteins through a protein kinase A-dependent mechanism. Cardiovasc. Res. 2004, 64, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Salie, R.; Moolman, J.A.; Lochner, A. The role of β-adrenergic receptors in the cardioprotective effects of β-preconditioning (βPC). Cardiovasc. Drugs Ther. 2011, 25, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Keyes, K.T.; Zhang, C.; Perez-Polo, J.R.; Lin, Y.; Birnbaum, Y. The myocardial infarct size-limiting effect of sitagliptin is PKA-dependent, whereas the protective effect of pioglitazone is partially dependent on PKA. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1454–H1465. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Sato, T.; Miyazaki, M.; Nakaya, H. Infarct size limitation by adrenomedullin: Protein kinase A but not PI3-kinase is linked to mitochondrial KCa channels. Cardiovasc. Res. 2008, 77, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, A.K.; Wergeland, A.; Helgeland, E.; Mjos, O.D.; Brar, B.K. Activation of corticotropin releasing factor receptor type 2 in the heart by corticotropin releasing factor offers cytoprotection against ischemic injury via PKA and PKC dependent signaling. Regul. Pept. 2012, 174, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.L.; Zhao, J.; Wang, Y.J.; Lau, W.B.; Yuan, Y.X.; Gao, E.H.; Koch, W.J.; Ma, X.L. Adiponectin inhibits oxidative/nitrative stress during myocardial ischemia and reperfusion via PKA signaling. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1436–E1443. [Google Scholar] [CrossRef] [PubMed]
- Younce, C.W.; Burmeister, M.A.; Ayala, J.E. Exendin-4 attenuates high glucose-induced cardiomyocyte apoptosis via inhibition of endoplasmic reticulum stress and activation of SERCA2a. Am. J. Physiol. Cell Physiol. 2013, 304, C508–C518. [Google Scholar] [CrossRef] [PubMed]
- Kobashigawa, L.C.; Xu, Y.C.; Padbury, J.F.; Tseng, Y.T.; Yano, N. Metformin protects cardiomyocyte from doxorubicin induced cytotoxicity through an AMP-activated protein kinase dependent signaling pathway: An in vitro study. PLoS ONE 2014, 9, e104888. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, X.; Tong, M.; Wu, D.; Wu, S.; Chen, J.; Wang, X.; Wang, X.; Kang, Y.; Tang, H.; et al. Intermedin suppresses pressure overload cardiac hypertrophy through activation of autophagy. PLoS ONE 2013, 8, e64757. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C.; Chu, G.; Mitton, B.; Song, Q.; Yuan, Q.; Kranias, E.G. Small heat-shock protein Hsp20 phosphorylation inhibits β-agonist-induced cardiac apoptosis. Circ. Res. 2004, 94, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C.; Yuan, Q.; Song, G.; Wang, Y.; Chen, G.; Qian, J.; Zhou, X.; Lee, Y.J.; Ashraf, M.; Kranias, E.G. Small heat-shock protein Hsp20 attenuates β-agonist-mediated cardiac remodeling through apoptosis signal-regulating kinase 1. Circ. Res. 2006, 99, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, C.S.; He, W.; Foote, K.; Bradley, A.; McGlynn, K.; Vidler, F.; Nixon, C.; Nather, K.; Fattah, C.; Riddell, A.; et al. Runx1 Deficiency Protects Against Adverse Cardiac Remodeling After Myocardial Infarction. Circulation 2018, 137, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Backs, J.; Worst, B.C.; Lehmann, L.H.; Patrick, D.M.; Jebessa, Z.; Kreusser, M.M.; Sun, Q.; Chen, L.; Heft, C.; Katus, H.A.; et al. Selective repression of MEF2 activity by PKA-dependent proteolysis of HDAC4. J. Cell Biol. 2011, 195, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.H.; Kim, J.Y.; Zhao, J.; Wang, W.; Jhun, B.S.; Wong, C.; Jin, Z.G. PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 15467–15472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.L.; McKinsey, T.A.; Chang, S.; Antos, C.L.; Hill, J.A.; Olson, E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002, 110, 479–488. [Google Scholar] [CrossRef]
- Wang, Q.; Oka, T.; Yamagami, K.; Lee, J.K.; Akazawa, H.; Naito, A.T.; Yasui, T.; Ishizu, T.; Nakaoka, Y.; Sakata, Y.; et al. An EP4 Receptor Agonist Inhibits Cardiac Fibrosis Through Activation of PKA Signaling in Hypertrophied Heart. Int. Heart J. 2017, 58, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Azeez, K.R.A.; Knapp, S.; Fernandes, J.M.; Klussmann, E.; Elkins, J.M. The crystal structure of the RhoA-AKAP-Lbc DH-PH domain complex. Biochem. J. 2014, 464, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Olson, M.F.; Hall, A.; Cerione, R.A.; Toksoz, D. Direct involvement of the small GTP-binding protein Rho in lbc oncogene function. J. Biol. Chem. 1995, 270, 9031–9034. [Google Scholar] [CrossRef] [PubMed]
- Toksoz, D.; Williams, D.A. Novel human oncogene lbc detected by transfection with distinct homology regions to signal transduction products. Oncogene 1994, 9, 621–628. [Google Scholar] [PubMed]
- Appert-Collin, A.; Cotecchia, S.; Nenniger-Tosato, M.; Pedrazzini, T.; Diviani, D. The A-kinase anchoring protein (AKAP)-Lbc-signaling complex mediates α1 adrenergic receptor-induced cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 10140–10145. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.W.; Cavagnini, K.S.; Brawley, D.N.; Berkley, C.Y.; Smolski, W.; Garcia, R.G.; Towne, A.L.; Sims, J.R.; Meigs, T.E. A Gα12-specific Binding Domain in AKAP-Lbc and p114RhoGEF. J. Mol. Signal. 2016, 11. [Google Scholar] [CrossRef]
- Carnegie, G.K.; Soughayer, J.; Smith, F.D.; Pedroja, B.S.; Zhang, F.; Diviani, D.; Bristow, M.R.; Kunkel, M.T.; Newton, A.C.; Langeberg, L.K.; et al. AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell 2008, 32, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Cariolato, L.; Cavin, S.; Diviani, D. A-Kinase Anchoring Protein (AKAP)-Lbc Anchors a PKN-based Signaling Complex Involved in α1-Adrenergic Receptor-induced p38 Activation. J. Biol. Chem. 2011, 286, 7925–7937. [Google Scholar] [CrossRef] [PubMed]
- Edwards, H.V.; Scott, J.D.; Baillie, G.S. The A-kinase-anchoring protein AKAP-Lbc facilitates cardioprotective PKA phosphorylation of Hsp20 on Ser(16). Biochem. J. 2012, 446, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Georgakopoulos, D.; Kovacs, A.; Zheng, M.; Lerner, D.; Pu, H.; Saffitz, J.; Chien, K.; Xiao, R.P.; Kass, D.A.; et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc. Natl. Acad. Sci. USA 2001, 98, 12283–12288. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Yamaguchi, O.; Hirotani, S.; Hikoso, S.; Higuchi, Y.; Watanabe, T.; Takeda, T.; Osuka, S.; Morita, T.; Kondoh, G.; et al. p38α mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol. 2004, 24, 10611–10620. [Google Scholar] [CrossRef] [PubMed]
- Braz, J.C.; Bueno, O.F.; Liang, Q.; Wilkins, B.J.; Dai, Y.S.; Parsons, S.; Braunwart, J.; Glascock, B.J.; Klevitsky, R.; Kimball, T.F.; et al. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J. Clin. Investig. 2003, 111, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Weinheimer, C.; Courtois, M.; Kovacs, A.; Zhang, C.E.; Cheng, A.M.; Wang, Y.; Muslin, A.J. The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J. Clin. Investig. 2003, 111, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.J.; Wall, J.A.; Martinez-Longoria, D.M.; Aryal, P.; Rockman, H.A.; Guo, Y.; Bolli, R.; Glembotski, C.C. Overexpression of mitogen-activated protein kinase kinase 6 in the heart improves functional recovery from ischemia in vitro and protects against myocardial infarction in vivo. J. Biol. Chem. 2005, 280, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Streicher, J.M.; Ren, S.; Herschman, H.; Wang, Y. MAPK-Activated Protein Kinase-2 in Cardiac Hypertrophy and Cyclooxygenase-2 Regulation in Heart. Circ. Res. 2010, 106, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Marber, M.S.; Rose, B.; Wang, Y. The p38 mitogen-activated protein kinase pathway—A potential target for intervention in infarction, hypertrophy, and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, G.; Lal, H.; Fidalgo, M.; Guerrero, A.; Zalvide, J.; Force, T.; Pombo, C.M. A novel cardioprotective p38-MAPK/mTOR pathway. Exp. Cell Res. 2011, 317, 2938–2949. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, G.K.; Smith, F.D.; McConnachie, G.; Langeberg, L.K.; Scott, J.D. AKAP-Lbc nucleates a protein kinase D activation scaffold. Mol. Cell 2004, 15, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Nicodemus-Johnson, J.; Spindler, M.J.; Carnegie, G.K. Genome-Wide Gene Expression Analysis Shows AKAP13-Mediated PKD1 Signaling Regulates the Transcriptional Response to Cardiac Hypertrophy. PLoS ONE 2015, 10, e0132474. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Sandhu, N.; Herrmann, J. Systems biology approaches to adverse drug effects: The example of cardio-oncology. Nat. Rev. Clin. Oncol. 2015, 12, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, C.M.; Serrano, J.; Kuehl, D.W.; Wallace, K.B. Preferential oxidation of cardiac mitochondrial DNA following acute intoxication with doxorubicin. Biochim. Biophys. Acta 1997, 1321, 101–106. [Google Scholar] [CrossRef]
- Fajardo, G.; Zhao, M.; Berry, G.; Wong, L.J.; Mochly-Rosen, D.; Bernstein, D. β2-adrenergic receptors mediate cardioprotection through crosstalk with mitochondrial cell death pathways. J. Mol. Cell. Cardiol. 2011, 51, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Kobe, B.; Heierhorst, J.; Feil, S.C.; Parker, M.W.; Benian, G.M.; Weiss, K.R.; Kemp, B.E. Giant protein kinases: Domain interactions and structural basis of autoregulation. EMBO J. 1996, 15, 6810–6821. [Google Scholar] [PubMed]
- De Francesco, E.M.; Rocca, C.; Scavello, F.; Amelio, D.; Pasqua, T.; Rigiracciolo, D.C.; Scarpelli, A.; Avino, S.; Cirillo, F.; Amodio, N.; et al. Protective Role of GPER Agonist G-1 on Cardiotoxicity Induced by Doxorubicin. J. Cell. Physiol. 2017, 232, 1640–1649. [Google Scholar] [CrossRef] [PubMed]
- Aries, A.; Paradis, P.; Lefebvre, C.; Schwartz, R.J.; Nemer, M. Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc. Natl. Acad. Sci. USA 2004, 101, 6975–6980. [Google Scholar] [CrossRef] [PubMed]
- Lebrecht, D.; Geist, A.; Ketelsen, U.P.; Haberstroh, J.; Setzer, B.; Walker, U.A. Dexrazoxane prevents doxorubicin-induced long-term cardiotoxicity and protects myocardial mitochondria from genetic and functional lesions in rats. Br. J. Pharmacol. 2007, 151, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, A.; Li, M.; Hirsch, E. New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochim. Biophys. Acta 2016, 1863, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wright, C.D.; Merkwan, C.L.; Baye, N.L.; Liang, Q.; Simpson, P.C.; O’Connell, T.D. An α1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation 2007, 115, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Beak, J.; Huang, W.; Parker, J.S.; Hicks, S.T.; Patterson, C.; Simpson, P.C.; Ma, A.; Jin, J.; Jensen, B.C. An Oral Selective α-1A Adrenergic Receptor Agonist Prevents Doxorubicin Cardiotoxicity. JACC Basic Transl. Sci. 2017, 2, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.Y.; Ouyang, K.; Yung, B.S.; Miyamoto, S.; Smrcka, A.V.; Chen, J.; Brown, J.H. PLCepsilon, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci. Signal. 2013, 6, ra108. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C.; Ren, X.; Qian, J.; Yuan, Q.; Nicolaou, P.; Wang, Y.; Jones, W.K.; Chu, G.; Kranias, E.G. Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury. Circulation 2005, 111, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C.; Zhou, X.; Wang, X.; Song, G.; Qian, J.; Nicolaou, P.; Chen, G.; Ren, X.; Kranias, E.G. Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ. Res. 2008, 103, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Edwards, H.V.; Scott, J.D.; Baillie, G.S. PKA phosphorylation of the small heat-shock protein Hsp20 enhances its cardioprotective effects. Biochem. Soc. Trans. 2012, 40, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, P.; Knoll, R.; Haghighi, K.; Fan, G.C.; Dorn, G.W., 2nd; Hasenfub, G.; Kranias, E.G. Human mutation in the anti-apoptotic heat shock protein 20 abrogates its cardioprotective effects. J. Biol. Chem. 2008, 283, 33465–33471. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, M.; Kim, E.; Sheng, M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated granylate kinases. J. Neurosci. 1996, 16, 2157–2163. [Google Scholar] [PubMed]
- Sin, Y.Y.; Edwards, H.V.; Li, X.; Day, J.P.; Christian, F.; Dunlop, A.J.; Adams, D.R.; Zaccolo, M.; Houslay, M.D.; Baillie, G.S. Disruption of the cyclic AMP phosphodiesterase-4 (PDE4)-HSP20 complex attenuates the β-agonist induced hypertrophic response in cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 50, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Burmeister, B.T.; Johnson, K.R.; Baillie, G.S.; Karginov, A.V.; Skidgel, R.A.; O’Bryan, J.P.; Carnegie, G.K. UCR1C is a novel activator of phosphodiesterase 4 (PDE4) long isoforms and attenuates cardiomyocyte hypertrophy. Cell. Signal. 2015, 27, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Sin, Y.Y.; Baillie, G.S. Heat shock protein 20 (HSP20) is a novel substrate for protein kinase D1 (PKD1). Cell Biochem. Funct. 2015, 33, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Eng. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Shende, P.; Plaisance, I.; Morandi, C.; Pellieux, C.; Berthonneche, C.; Zorzato, F.; Krishnan, J.; Lerch, R.; Hall, M.N.; Ruegg, M.A.; et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation 2011, 123, 1073. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Raimondi, F.; Del Vescovo, C.D.; Dreyer, E.; Reggi, E.; Osman, H.; Ruggieri, L.; Gonano, C.; Cavin, S.; Box, C.L.; et al. Small-Molecule Protein-Protein Interaction Inhibitor of Oncogenic Rho Signaling. Cell Chem. Biol. 2016, 23, 1135–1146. [Google Scholar]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diviani, D.; Osman, H.; Reggi, E. A-Kinase Anchoring Protein-Lbc: A Molecular Scaffold Involved in Cardiac Protection. J. Cardiovasc. Dev. Dis. 2018, 5, 12. https://doi.org/10.3390/jcdd5010012
Diviani D, Osman H, Reggi E. A-Kinase Anchoring Protein-Lbc: A Molecular Scaffold Involved in Cardiac Protection. Journal of Cardiovascular Development and Disease. 2018; 5(1):12. https://doi.org/10.3390/jcdd5010012
Chicago/Turabian StyleDiviani, Dario, Halima Osman, and Erica Reggi. 2018. "A-Kinase Anchoring Protein-Lbc: A Molecular Scaffold Involved in Cardiac Protection" Journal of Cardiovascular Development and Disease 5, no. 1: 12. https://doi.org/10.3390/jcdd5010012
APA StyleDiviani, D., Osman, H., & Reggi, E. (2018). A-Kinase Anchoring Protein-Lbc: A Molecular Scaffold Involved in Cardiac Protection. Journal of Cardiovascular Development and Disease, 5(1), 12. https://doi.org/10.3390/jcdd5010012