Functions of PDE3 Isoforms in Cardiac Muscle


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The PDE3 Family of Cyclic Nucleotide Phosphodiesterases
3. Intracellular Localisation of PDE3A and PDE3B in Cardiac Myocytes and Their Protein–Protein Interactions
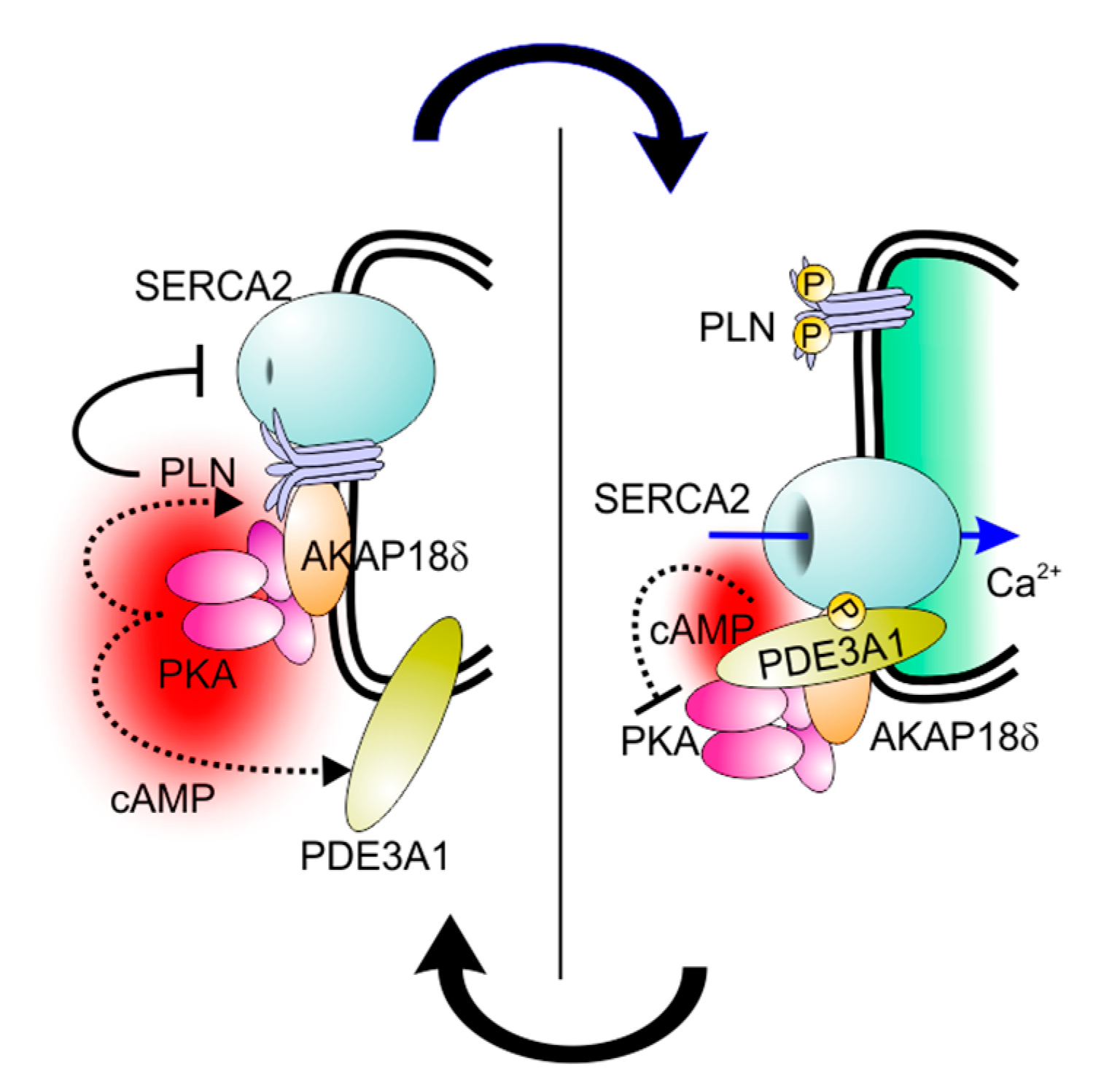
3.1. PDE3A
3.2. PDE3B
4. Inotropic Actions of PDE3A Inhibition
5. Pro-Apoptotic Actions of PDE3A Inhibition
6. Pro-Hypertrophic Actions of PDE3A Inhibition
7. Clinical Experience with PDE3 Inhibition in the Treatment of Heart Disease
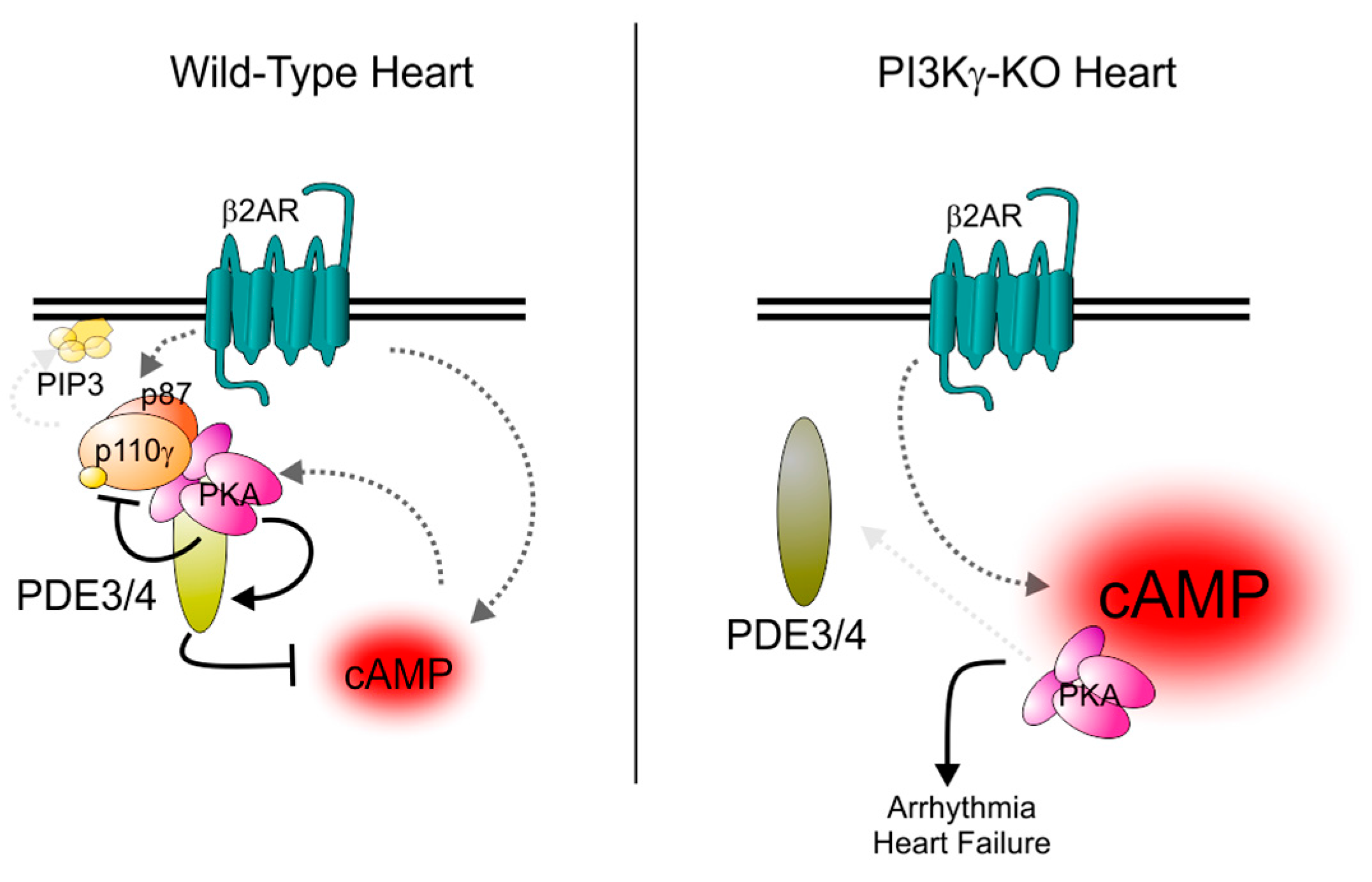
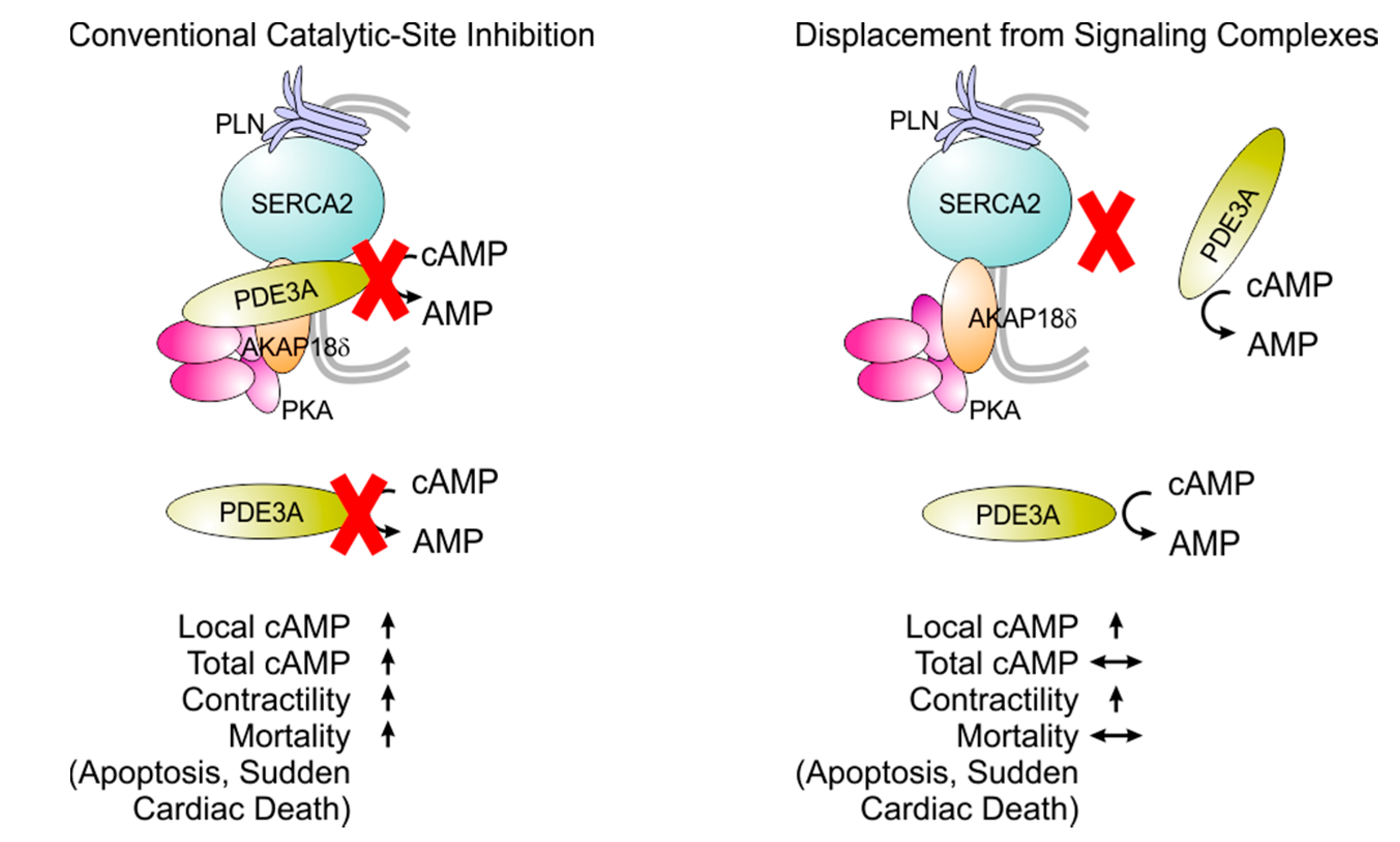
8. Selective Targeting of PDE3 Isoforms
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| aa | amino acid |
| AKAP | A-kinase-anchoring protein |
| β2-AR | β2-adrenergic receptor |
| cAMP | Cyclic adenosine monophosphate |
| IRS | Insulin-receptor substrate |
| NHR | N-terminal hydrophobic region |
| PLN | Phospholamban |
| PKA | cAMP-dependent protein kinase |
| PKA-RII | cAMP-dependent protein kinase regulatory subunit II |
| PKB (AKT) | Protein kinase B |
| PKC | Protein kinase C |
| PI3Kγ | Phosphoinositide 3-kinase γ |
| PIP3 | Phosphatidylinositol (3,4,5)-triphosphate |
| p110γ | Phosphoinositide 3-kinase γ (PI3Kγ) catalytic subunit |
| p87 | PI3Kγ regulatory subunit (also known as p87PIKAP p87-PI3K adapter protein) |
| SERCA2 | Sarcoplasmic/endoplasmic reticulum calcium ATPase-2 |
References
- Grant, P.G.; Colman, R.W. Purification and characterization of a human platelet cyclic nucleotide phosphodiesterase. Biochemistry 1984, 23, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Boyes, S.; Loten, E.G. Purification of an insulin-sensitive cyclic AMP phosphodiesterase from rat liver. Eur. J. Biochem. 1988, 174, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Degerman, E.; Belfrage, P.; Manganiello, V.C. Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3). J. Biol. Chem. 1997, 272, 6823–6826. [Google Scholar] [CrossRef] [PubMed]
- DiBianco, R.; Shabetai, R.; Kostuk, W.; Moran, J.; Schlant, R.C.; Wright, R. A comparison of oral milrinone, digoxin, and their combination in the treatment of patients with chronic heart failure. N. Engl. J. Med. 1989, 320, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Uretsky, B.F.; Jessup, M.; Konstam, M.A.; Dec, G.W.; Leier, C.V.; Benotti, J.; Murali, S.; Herrmann, H.C.; Sandberg, J.A. Multicenter trial of oral enoximone in patients with moderate to moderately severe congestive heart failure. Lack of benefit compared with placebo. Enoximone Multicenter Trial Group. Circulation 1990, 82, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Narahara, K.A. Oral enoximone therapy in chronic heart failure: A placebo-controlled randomized trial. The Western Enoximone Study Group. Am. Heart J. 1991, 121, 1471–1479. [Google Scholar] [CrossRef]
- Packer, M.; Carver, J.R.; Rodeheffer, R.J.; Ivanhoe, R.J.; DiBianco, R.; Zeldis, S.M.; Hendrix, G.H.; Bommer, W.J.; Elkayam, U.; Kukin, M.L.; et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N. Engl. J. Med. 1991, 325, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Narahara, K.A.; Elkayam, U.; Sullivan, J.M.; Pearle, D.L.; Massie, B.M.; Creager, M.A. Double-blind, placebo-controlled study of the efficacy of flosequinan in patients with chronic heart failure. Principal Investigators of the REFLECT Study. J. Am. Coll. Cardiol. 1993, 22, 65–72. [Google Scholar] [CrossRef]
- Cohn, J.N.; Goldstein, S.O.; Greenberg, B.H.; Lorell, B.H.; Bourge, R.C.; Jaski, B.E.; Gottlieb, S.O.; McGrew, F., 3rd; DeMets, D.L.; White, B.G. A dose-dependent increase in mortality with vesnarinone among patients with severe heart failure. Vesnarinone Trial Investigators. N. Engl. J. Med. 1998, 339, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Amsallem, E.; Kasparian, C.; Haddour, G.; Boissel, J.P.; Nony, P. Phosphodiesterase III inhibitors for heart failure. Cochrane Database Syst. Rev. 2005. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterases (PDE) and peptide motifs. Curr. Pharm. Des. 2010, 16, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Meacci, E.; Taira, M.; Moos, M., Jr.; Smith, C.J.; Movsesian, M.A.; Degerman, E.; Belfrage, P.; Manganiello, V. Molecular cloning and expression of human myocardial cGMP-inhibited cAMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 1992, 89, 3721–3725. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Taira, M.; Hockman, S.; Shimada, F.; Lieman, J.; Napolitano, M.; Ward, D.; Taira, M.; Makino, H.; Manganiello, V.C. Characterization of the cDNA and gene encoding human PDE3B, the cGIP1 isoform of the human cyclic GMP-inhibited cyclic nucleotide phosphodiesterase family. Genomics 1996, 36, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, J.; Choi, Y.H.; Krall, J.; Ahmad, F.; Manganiello, V.C.; Movsesian, M.A. Isoforms of cyclic nucleotide phosphodiesterase PDE3A in cardiac myocytes. J. Biol. Chem. 2002, 277, 38072–38078. [Google Scholar] [CrossRef] [PubMed]
- Kenan, Y.; Murata, T.; Shakur, Y.; Degerman, E.; Manganiello, V.C. Functions of the N-terminal region of cyclic nucleotide phosphodiesterase 3 (PDE 3) isoforms. J. Biol. Chem. 2000, 275, 12331–12338. [Google Scholar] [CrossRef] [PubMed]
- Shakur, Y.; Takeda, K.; Kenan, Y.; Yu, Z.X.; Rena, G.; Brandt, D.; Houslay, M.D.; Degerman, E.; Ferrans, V.J.; Manganiello, V.C. Membrane localization of cyclic nucleotide phosphodiesterase 3 (PDE3). Two N-terminal domains are required for the efficient targeting to, and association of, PDE3 with endoplasmic reticulum. J. Biol. Chem. 2000, 275, 38749–38761. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Vaccari, S.; Nedachi, T.; Andersen, C.B.; Kovacina, K.S.; Roth, R.A.; Conti, M. Protein kinase B/Akt phosphorylation of PDE3A and its role in mammalian oocyte maturation. EMBO J. 2006, 25, 5716–5725. [Google Scholar] [CrossRef] [PubMed]
- Pozuelo Rubio, M.; Campbell, D.G.; Morrice, N.A.; Mackintosh, C. Phosphodiesterase 3A binds to 14-3-3 proteins in response to PMA-induced phosphorylation of Ser428. Biochem. J. 2005, 392, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.W.; Mackintosh, C.; Hers, I. Protein kinase C-mediated phosphorylation and activation of PDE3A regulate cAMP levels in human platelets. J. Biol. Chem. 2009, 284, 12339–12348. [Google Scholar] [CrossRef] [PubMed]
- Hambleton, R.; Krall, J.; Tikishvili, E.; Honeggar, M.; Ahmad, F.; Manganiello, V.C.; Movsesian, M.A. Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to cAMP hydrolytic activity in subcellular fractions of human myocardium. J. Biol. Chem. 2005, 280, 39168–39174. [Google Scholar] [CrossRef] [PubMed]
- Leroy, M.J.; Degerman, E.; Taira, M.; Murata, T.; Wang, L.H.; Movsesian, M.A.; Meacci, E.; Manganiello, V.C. Characterization of two recombinant PDE3 (cGMP-inhibited cyclic nucleotide phosphodiesterase) isoforms, RcGIP1 and HcGIP2, expressed in NIH 3006 murine fibroblasts and Sf9 insect cells. Biochemistry 1996, 35, 10194–10202. [Google Scholar] [CrossRef] [PubMed]
- Taira, M.; Hockman, S.C.; Calvo, J.C.; Taira, M.; Belfrage, P.; Manganiello, V.C. Molecular cloning of the rat adipocyte hormone-sensitive cyclic GMP-inhibited cyclic nucleotide phosphodiesterase. J. Biol. Chem. 1993, 268, 18573–18579. [Google Scholar] [PubMed]
- Reinhardt, R.R.; Chin, E.; Zhou, J.; Taira, M.; Murata, T.; Manganiello, V.C.; Bondy, C.A. Distinctive anatomical patterns of gene expression for cGMP-inhibited cyclic nucleotide phosphodiesterases. J. Clin. Investig. 1995, 95, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.W.; Lagranha, C.; Chen, Y.; Sun, J.; Tong, G.; Hockman, S.C.; Ahmad, F.; Esfahani, S.G.; Bae, D.H.; Polidovitch, N.; et al. Targeted disruption of PDE3B, but not PDE3A, protects murine heart from ischemia/reperfusion injury. Proc. Natl. Acad. Sci. USA 2015, 112, E2253–E2262. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C.; Keravis, T.; Le Bec, A.; Pauvert, O.; Proteau, S.; Rousseau, E. Characterization of cyclic nucleotide phosphodiesterase isoforms associated to isolated cardiac nuclei. Biochim. Biophys. Acta 1999, 1472, 431–446. [Google Scholar] [CrossRef]
- Zaccolo, M. cAMP signal transduction in the heart: Understanding spatial control for the development of novel therapeutic strategies. Br. J. Pharmacol. 2009, 158, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci. 2010, 35, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Raymond, D.R.; Wilson, L.S.; Carter, R.L.; Maurice, D.H. Numerous distinct PKA-, or EPAC-based, signalling complexes allow selective phosphodiesterase 3 and phosphodiesterase 4 coordination of cell adhesion. Cell Signal. 2007, 19, 2507–2518. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Santana, L.F. A-kinase anchoring proteins: Getting to the heart of the matter. Circulation 2010, 121, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Stangherlin, A.; Zaccolo, M. Local termination of 3′-5′-cyclic adenosine monophosphate signals: The role of A kinase anchoring protein-tethered phosphodiesterases. J. Cardiovasc. Pharmacol. 2011, 58, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, M.D.; Li, J.; Dodge-Kafka, K.; Kapiloff, M.S. AKAPs: The architectural underpinnings of local cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Ghigo, A.; Scott, J.D.; Hirsch, E. Anchoring proteins as regulators of signaling pathways. Circ. Res. 2012, 111, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Movsesian, M.A.; Smith, C.J.; Krall, J.; Bristow, M.R.; Manganiello, V.C. Sarcoplasmic reticulum-associated cyclic adenosine 5′-monophosphate phosphodiesterase activity in normal and failing human hearts. J. Clin. Investig. 1991, 88, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C.; Muller, B.; Le Bec, A.; Beaudry, C.; Rousseau, E. Characterization of indolidan- and rolipram-sensitive cyclic nucleotide phosphodiesterases in canine and human cardiac microsomal fractions. J. Pharmacol. Exp. Ther. 1993, 265, 1142–1151. [Google Scholar] [PubMed]
- Beca, S.; Ahmad, F.; Shen, W.; Liu, J.; Makary, S.; Polidovitch, N.; Sun, J.; Hockman, S.; Chung, Y.W.; Movsesian, M.; et al. Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ. Res. 2013, 112, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Shen, W.; Vandeput, F.; Szabo-Fresnais, N.; Krall, J.; Degerman, E.; Goetz, F.; Klussmann, E.; Movsesian, M.; Manganiello, V. Regulation of sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2) activity by phosphodiesterase 3A (PDE3A) in human myocardium: Phosphorylation-dependent interaction of PDE3A1 with SERCA2. J. Biol. Chem. 2015, 290, 6763–6776. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, A.; Laffargue, M.; Li, M.; Hirsch, E. PI3K and Calcium Signaling in Cardiovascular Disease. Circ. Res. 2017, 121, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Xie, M.; Scheitrum, C.; Krall, J.; Movsesian, M.A.; Conti, M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res. Cardiol. 2011, 106, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.L.; Conti, M.; Marks, A.R. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Kerfant, B.G.; Zhao, D.; Lorenzen-Schmidt, I.; Wilson, L.S.; Cai, S.; Chen, S.R.; Maurice, D.H.; Backx, P.H. PI3Kgamma is required for PDE4, not PDE3, activity in subcellular microdomains containing the sarcoplasmic reticular calcium ATPase in cardiomyocytes. Circ. Res. 2007, 101, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Richter, W.; Mika, D.; Castro, L.R.; Abi-Gerges, A.; Xie, M.; Scheitrum, C.; Lefebvre, F.; Schittl, J.; Mateo, P.; et al. Phosphodiesterase 4B in the cardiac l-type Ca(2)(+) channel complex regulates Ca(2)(+) current and protects against ventricular arrhythmias in mice. J. Clin. Investig. 2011, 121, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Macphee, C.H.; Reifsnyder, D.H.; Moore, T.A.; Lerea, K.M.; Beavo, J.A. Phosphorylation results in activation of a cAMP phosphodiesterase in human platelets. J. Biol. Chem. 1988, 263, 10353–10358. [Google Scholar] [PubMed]
- Grant, P.G.; Mannarino, A.F.; Colman, R.W. cAMP-mediated phosphorylation of the low-Km cAMP phosphodiesterase markedly stimulates its catalytic activity. Proc. Natl. Acad. Sci. USA 1988, 85, 9071–9075. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Lindh, R.; Tang, Y.; Ruishalme, I.; Ost, A.; Sahachartsiri, B.; Stralfors, P.; Degerman, E.; Manganiello, V.C. Differential regulation of adipocyte PDE3B in distinct membrane compartments by insulin and the beta3-adrenergic receptor agonist CL316243: Effects of caveolin-1 knockdown on formation/maintenance of macromolecular signalling complexes. Biochem. J. 2009, 424, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Lindh, R.; Tang, Y.; Weston, M.; Degerman, E.; Manganiello, V.C. Insulin-induced formation of macromolecular complexes involved in activation of cyclic nucleotide phosphodiesterase 3B (PDE3B) and its interaction with PKB. Biochem. J. 2007, 404, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Onuma, H.; Osawa, H.; Yamada, K.; Ogura, T.; Tanabe, F.; Granner, D.K.; Makino, H. Identification of the insulin-regulated interaction of phosphodiesterase 3B with 14-3-3 beta protein. Diabetes 2002, 51, 3362–3367. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Palmer, D.; Jimmo, S.L.; Raymond, D.R.; Wilson, L.S.; Carter, R.L.; Maurice, D.H. Protein kinase A phosphorylation of human phosphodiesterase 3B promotes 14-3-3 protein binding and inhibits phosphatase-catalyzed inactivation. J. Biol. Chem. 2007, 282, 9411–9419. [Google Scholar] [CrossRef] [PubMed]
- Vandeput, F.; Szabo-Fresnais, N.; Ahmad, F.; Kho, C.; Lee, A.; Krall, J.; Dunlop, A.; Hazel, M.W.; Wohlschlegel, J.A.; Hajjar, R.J.; et al. Selective regulation of cyclic nucleotide phosphodiesterase PDE3A isoforms. Proc. Natl. Acad. Sci. USA 2013, 110, 19778–19783. [Google Scholar] [CrossRef] [PubMed]
- Weninger, S.; Van Craenenbroeck, K.; Cameron, R.T.; Vandeput, F.; Movsesian, M.A.; Baillie, G.S.; Lefebvre, R.A. Phosphodiesterase 4 interacts with the 5-HT4(b) receptor to regulate cAMP signaling. Cell Signal. 2014, 26, 2573–2582. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, A.; Perino, A.; Mehel, H.; Zahradnikova, A., Jr.; Morello, F.; Leroy, J.; Nikolaev, V.O.; Damilano, F.; Cimino, J.; De Luca, E.; et al. Phosphoinositide 3-kinase gamma protects against catecholamine-induced ventricular arrhythmia through protein kinase A-mediated regulation of distinct phosphodiesterases. Circulation 2012, 126, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol. Cell 2011, 42, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Marcantoni, A.; Levi, R.C.; Gallo, M.P.; Hirsch, E.; Alloatti, G. Phosphoinositide 3-kinasegamma (PI3Kgamma) controls l-type calcium current (ICa,L) through its positive modulation of type-3 phosphodiesterase (PDE3). J. Cell. Physiol. 2006, 206, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Rondinone, C.M.; Carvalho, E.; Rahn, T.; Manganiello, V.C.; Degerman, E.; Smith, U.P. Phosphorylation of PDE3B by phosphatidylinositol 3-kinase associated with the insulin receptor. J. Biol. Chem. 2000, 275, 10093–10098. [Google Scholar] [CrossRef] [PubMed]
- DiPilato, L.M.; Ahmad, F.; Harms, M.; Seale, P.; Manganiello, V.; Birnbaum, M.J. The Role of PDE3B Phosphorylation in the Inhibition of Lipolysis by Insulin. Mol. Cell. Biol. 2015, 35, 2752–2760. [Google Scholar] [CrossRef] [PubMed]
- Odagiri, K.; Katoh, H.; Kawashima, H.; Tanaka, T.; Ohtani, H.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J. Mol. Cell. Cardiol. 2009, 46, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Kitamura, Y.; Kuroda, S.; Hino, Y.; Ando, M.; Kotani, K.; Konishi, H.; Matsuzaki, H.; Kikkawa, U.; Ogawa, W.; et al. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Mol. Cell. Biol. 1999, 19, 6286–6296. [Google Scholar] [CrossRef] [PubMed]
- Onuma, H.; Osawa, H.; Ogura, T.; Tanabe, F.; Nishida, W.; Makino, H. A newly identified 50 kDa protein, which is associated with phosphodiesterase 3B, is phosphorylated by insulin in rat adipocytes. Biochem. Biophys. Res. Commun. 2005, 337, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.; Ahmad, F.; Sward, K.; Andersson, U.; Weston, M.; Manganiello, V.; Degerman, E. Plasma membrane cyclic nucleotide phosphodiesterase 3B (PDE3B) is associated with caveolae in primary adipocytes. Cell Signal. 2006, 18, 1713–1721. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patrucco, E.; Notte, A.; Barberis, L.; Selvetella, G.; Maffei, A.; Brancaccio, M.; Marengo, S.; Russo, G.; Azzolino, O.; Rybalkin, S.D.; et al. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 2004, 118, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Beretta, M.; Kilic, A.; Ghigo, A.; Carnevale, D.; Repetto, I.E.; Braccini, L.; Longo, D.; Liebig-Gonglach, M.; Zaglia, T.; et al. Combined inhibition of PI3Kbeta and PI3Kgamma reduces fat mass by enhancing alpha-MSH-dependent sympathetic drive. Sci. Signal. 2014, 7, ra110. [Google Scholar] [CrossRef] [PubMed]
- Damilano, F.; Perino, A.; Hirsch, E. PI3K kinase and scaffold functions in heart. Ann. N. Y. Acad. Sci. 2010, 1188, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, M.; Wu, M.; Lim, S.; Knight, W.E.; Miller, C.L.; Cai, Y.; Lu, Y.; Blaxall, B.C.; Takeishi, Y.; Abe, J.; et al. Cyclic nucleotide phosphodiesterase 3A1 protects the heart against ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2013, 64, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Abe, J.; Wei, H.; Xu, H.; Che, W.; Aizawa, T.; Liu, W.; Molina, C.A.; Sadoshima, J.; Blaxall, B.C.; et al. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 14771–14776. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Abe, J.; Wei, H.; Huang, Q.; Walsh, R.A.; Molina, C.A.; Zhao, A.; Sadoshima, J.; Blaxall, B.C.; Berk, B.C.; et al. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: Implication in heart failure. Circulation 2005, 111, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Zoccarato, A.; Surdo, N.C.; Aronsen, J.M.; Fields, L.A.; Mancuso, L.; Dodoni, G.; Stangherlin, A.; Livie, C.; Jiang, H.; Sin, Y.Y.; et al. Cardiac Hypertrophy Is Inhibited by a Local Pool of cAMP Regulated by Phosphodiesterase 2. Circ. Res. 2015, 117, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Maass, P.G.; Aydin, A.; Luft, F.C.; Schachterle, C.; Weise, A.; Stricker, S.; Lindschau, C.; Vaegler, M.; Qadri, F.; Toka, H.R.; et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat. Genet. 2015, 47, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Boda, H.; Uchida, H.; Takaiso, N.; Ouchi, Y.; Fujita, N.; Kuno, A.; Hata, T.; Nagatani, A.; Funamoto, Y.; Miyata, M.; et al. A PDE3A mutation in familial hypertension and brachydactyly syndrome. J. Hum. Genet. 2016, 61, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Toka, O.; Tank, J.; Schachterle, C.; Aydin, A.; Maass, P.G.; Elitok, S.; Bartels-Klein, E.; Hollfinger, I.; Lindschau, C.; Mai, K.; et al. Clinical Effects of Phosphodiesterase 3A Mutations in Inherited Hypertension With Brachydactyly. Hypertension 2015, 66, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Ginsburg, R.; Minobe, W.; Cubicciotti, R.S.; Sageman, W.S.; Lurie, K.; Billingham, M.E.; Harrison, D.C.; Stinson, E.B. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N. Engl. J. Med. 1982, 307, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Ginsburg, R.; Umans, V.; Fowler, M.; Minobe, W.; Rasmussen, R.; Zera, P.; Menlove, R.; Shah, P.; Jamieson, S.; et al. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: Coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ. Res. 1986, 59, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.; Parruti, G.; Bohm, M.; Puzicha, M.; DeBlasi, A.; Erdmann, E.; Lohse, M.J. Expression of beta-arrestins and beta-adrenergic receptor kinases in the failing human heart. Circ. Res. 1994, 74, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.; Bohm, M.; Elce, J.S.; Erdmann, E.; Lohse, M.J. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation 1993, 87, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Schmitz, W.; Scholz, H.; von Meyerinck, L.; Doring, V.; Kalmar, P. Increase in myocardial Gi-proteins in heart failure. Lancet 1988, 2, 936–937. [Google Scholar] [CrossRef]
- Feldman, A.M.; Cates, A.E.; Veazey, W.B.; Hershberger, R.E.; Bristow, M.R.; Baughman, K.L.; Baumgartner, W.A.; Van Dop, C. Increase of the 40,000-mol wt pertussis toxin substrate (G protein) in the failing human heart. J. Clin. Investig. 1988, 82, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.D.; Copelas, L.; Gwathmey, J.K.; Phillips, P.; Warren, S.E.; Schoen, F.J.; Grossman, W.; Morgan, J.P. Deficient production of cyclic AMP: Pharmacologic evidence of an important cause of contractile dysfunction in patients with end-stage heart failure. Circulation 1987, 75, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, W.; v der Leyen, H.; Meyer, W.; Neumann, J.; Schmitz, W.; Scholz, H.; Starbatty, J.; Stein, B.; Doring, V.; Kalmar, P. Basal and isoprenaline-stimulated cAMP content in failing versus nonfailing human cardiac preparations. J. Cardiovasc. Pharmacol. 1989, 14, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Bohm, M.; Reiger, B.; Schwinger, R.H.; Erdmann, E. cAMP concentrations, cAMP dependent protein kinase activity, and phospholamban in non-failing and failing myocardium. Cardiovasc. Res. 1994, 28, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Beuckelmann, D.J.; Nabauer, M.; Erdmann, E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 1992, 85, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Baim, D.S.; McDowell, A.V.; Cherniles, J.; Monrad, E.S.; Parker, J.A.; Edelson, J.; Braunwald, E.; Grossman, W. Evaluation of a new bipyridine inotropic agent—Milrinone—In patients with severe congestive heart failure. N. Engl. J. Med. 1983, 309, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Benotti, J.R.; Grossman, W.; Braunwald, E.; Davolos, D.D.; Alousi, A.A. Hemodynamic assessment of amrinone. A new inotropic agent. N. Engl. J. Med. 1978, 299, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Jaski, B.E.; Fifer, M.A.; Wright, R.F.; Braunwald, E.; Colucci, W.S. Positive inotropic and vasodilator actions of milrinone in patients with severe congestive heart failure. Dose-response relationships and comparison to nitroprusside. J. Clin. Investig. 1985, 75, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Sinoway, L.S.; Maskin, C.S.; Chadwick, B.; Forman, R.; Sonnenblick, E.H.; Le Jemtel, T.H. Long-term therapy with a new cardiotonic agent, WIN 47203: Drug-dependent improvement in cardiac performance and progression of the underlying disease. J. Am. Coll. Cardiol. 1983, 2, 327–331. [Google Scholar] [CrossRef]
- Maskin, C.S.; Sinoway, L.; Chadwick, B.; Sonnenblick, E.H.; Le Jemtel, T.H. Sustained hemodynamic and clinical effects of a new cardiotonic agent, WIN 47203, in patients with severe congestive heart failure. Circulation 1983, 67, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L. Hemodynamic and clinical benefits with intravenous milrinone in severe chronic heart failure: Results of a multicenter study in the United States. Am. Heart J. 1991, 121, 1956–1964. [Google Scholar] [CrossRef]
- Monrad, E.S.; McKay, R.G.; Baim, D.S.; Colucci, W.S.; Fifer, M.A.; Heller, G.V.; Royal, H.D.; Grossman, W. Improvement in indexes of diastolic performance in patients with congestive heart failure treated with milrinone. Circulation 1984, 70, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Pande, R.L.; Hiatt, W.R.; Zhang, P.; Hittel, N.; Creager, M.A. A pooled analysis of the durability and predictors of treatment response of cilostazol in patients with intermittent claudication. Vasc. Med. 2010, 15, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Del Monte, F.; Lebeche, D.; Guerrero, J.L.; Tsuji, T.; Doye, A.A.; Gwathmey, J.K.; Hajjar, R.J. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc. Natl. Acad. Sci. USA 2004, 101, 5622–5627. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; Bannister, M.L.; Collins, T.; Pearce, E.; Sepehripour, A.H.; Dubb, S.S.; Garcia, E.; O’Gara, P.; Liang, L.; Kohlbrenner, E.; et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ. Arrhythm. Electrophysiol. 2011, 4, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.M.; Yu, H.Y.; Li, Y.L.; Jia, C.J. Design and discovery of 2-(4-(1H-tetrazol-5-yl)-1H-pyrazol-1-yl)-4-(4-phenyl)thiazole derivatives as cardiotonic agents via inhibition of PDE3. Bioorg. Med. Chem. 2015, 23, 6111–6117. [Google Scholar] [CrossRef] [PubMed]
- Lygren, B.; Carlson, C.R.; Santamaria, K.; Lissandron, V.; McSorley, T.; Litzenberg, J.; Lorenz, D.; Wiesner, B.; Rosenthal, W.; Zaccolo, M.; et al. AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007, 8, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.S.; Baillie, G.S.; Pritchard, L.M.; Umana, B.; Terrin, A.; Zaccolo, M.; Houslay, M.D.; Maurice, D.H. A phosphodiesterase 3B-based signaling complex integrates exchange protein activated by cAMP 1 and phosphatidylinositol 3-kinase signals in human arterial endothelial cells. J. Biol. Chem. 2011, 286, 16285–16296. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Movsesian, M.; Ahmad, F.; Hirsch, E. Functions of PDE3 Isoforms in Cardiac Muscle. J. Cardiovasc. Dev. Dis. 2018, 5, 10. https://doi.org/10.3390/jcdd5010010
Movsesian M, Ahmad F, Hirsch E. Functions of PDE3 Isoforms in Cardiac Muscle. Journal of Cardiovascular Development and Disease. 2018; 5(1):10. https://doi.org/10.3390/jcdd5010010
Chicago/Turabian StyleMovsesian, Matthew, Faiyaz Ahmad, and Emilio Hirsch. 2018. "Functions of PDE3 Isoforms in Cardiac Muscle" Journal of Cardiovascular Development and Disease 5, no. 1: 10. https://doi.org/10.3390/jcdd5010010
APA StyleMovsesian, M., Ahmad, F., & Hirsch, E. (2018). Functions of PDE3 Isoforms in Cardiac Muscle. Journal of Cardiovascular Development and Disease, 5(1), 10. https://doi.org/10.3390/jcdd5010010