Diagnostic Yield of Whole Exome Sequencing in Pediatric Dilated Cardiomyopathy

Abstract
1. Introduction
2. Materials and Methods
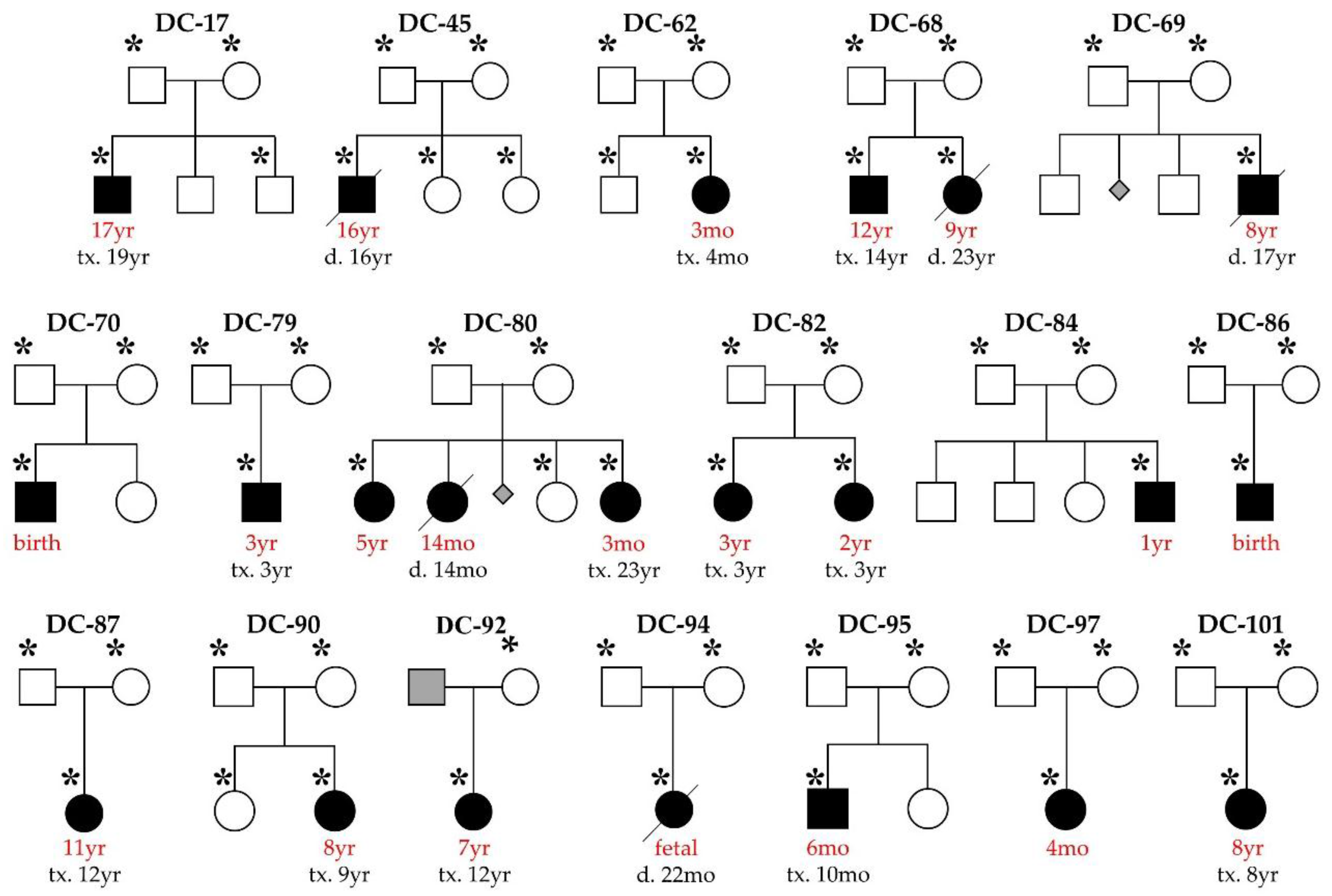
2.1. Study Subjects
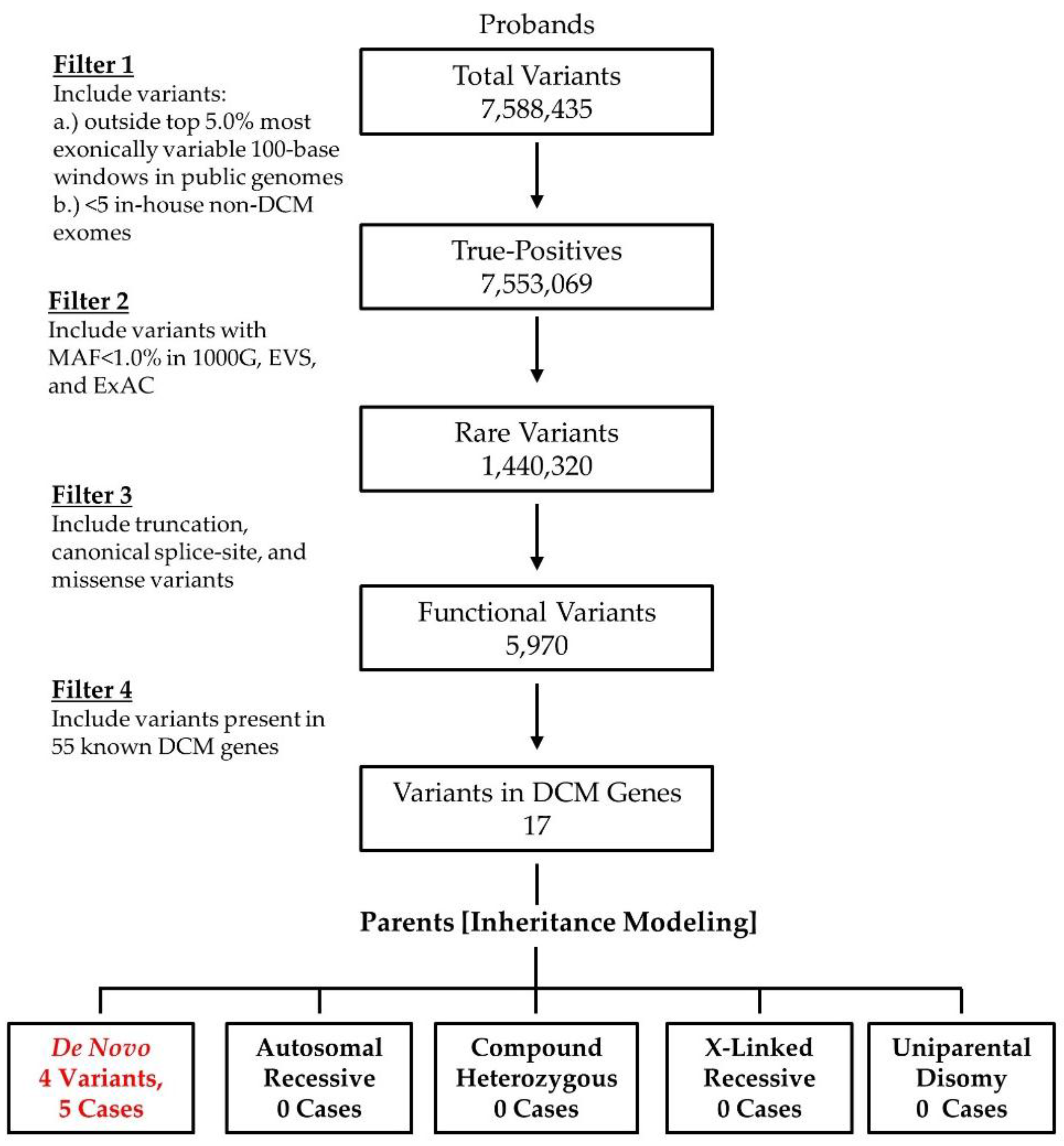
2.2. WES and Bioinformatics Analysis
2.3. Transmission Electron Microscopy
3. Results
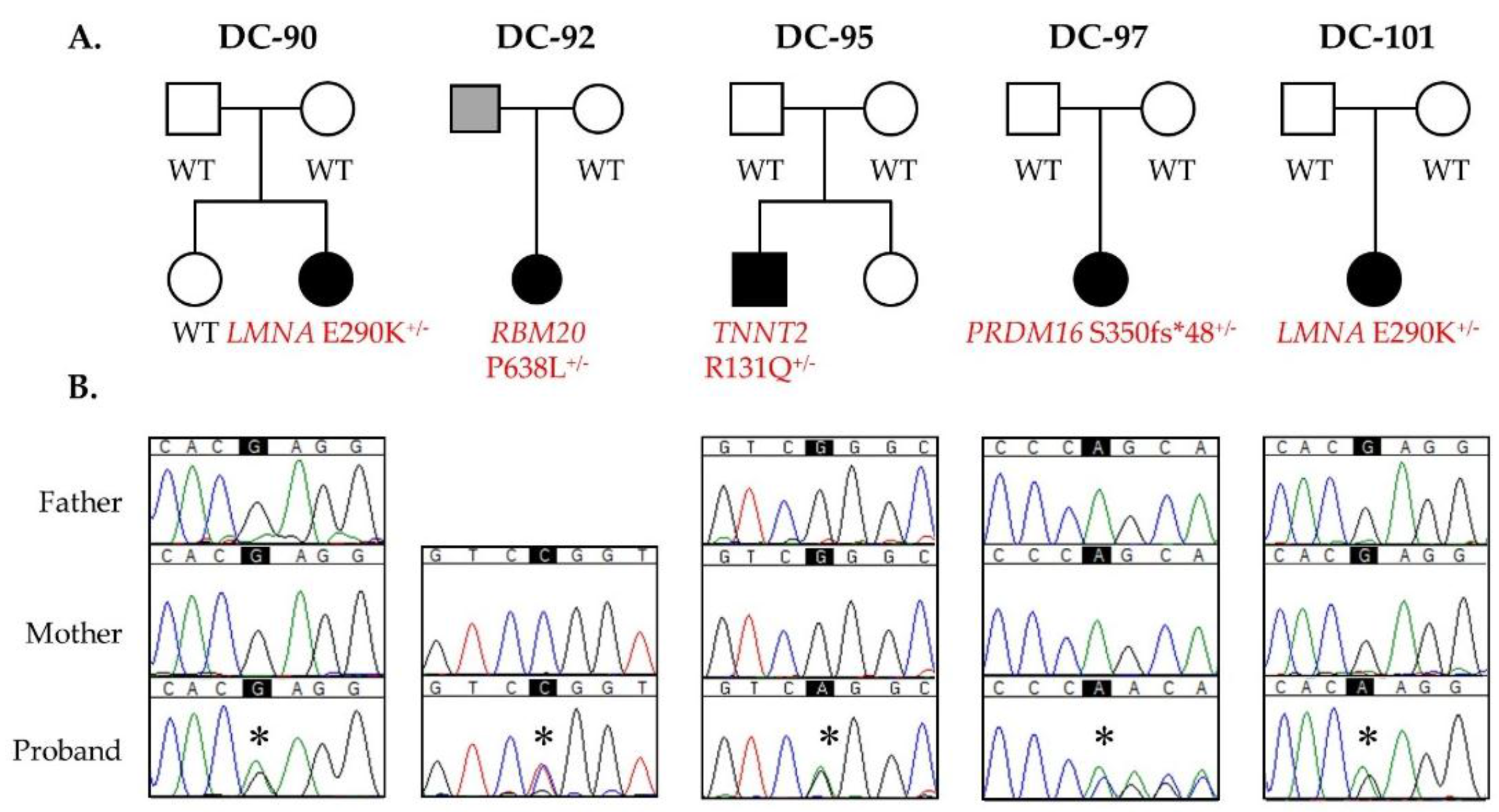
3.1. WES Uncovers De Novo Mutations as a Cause for Early-Onset DCM
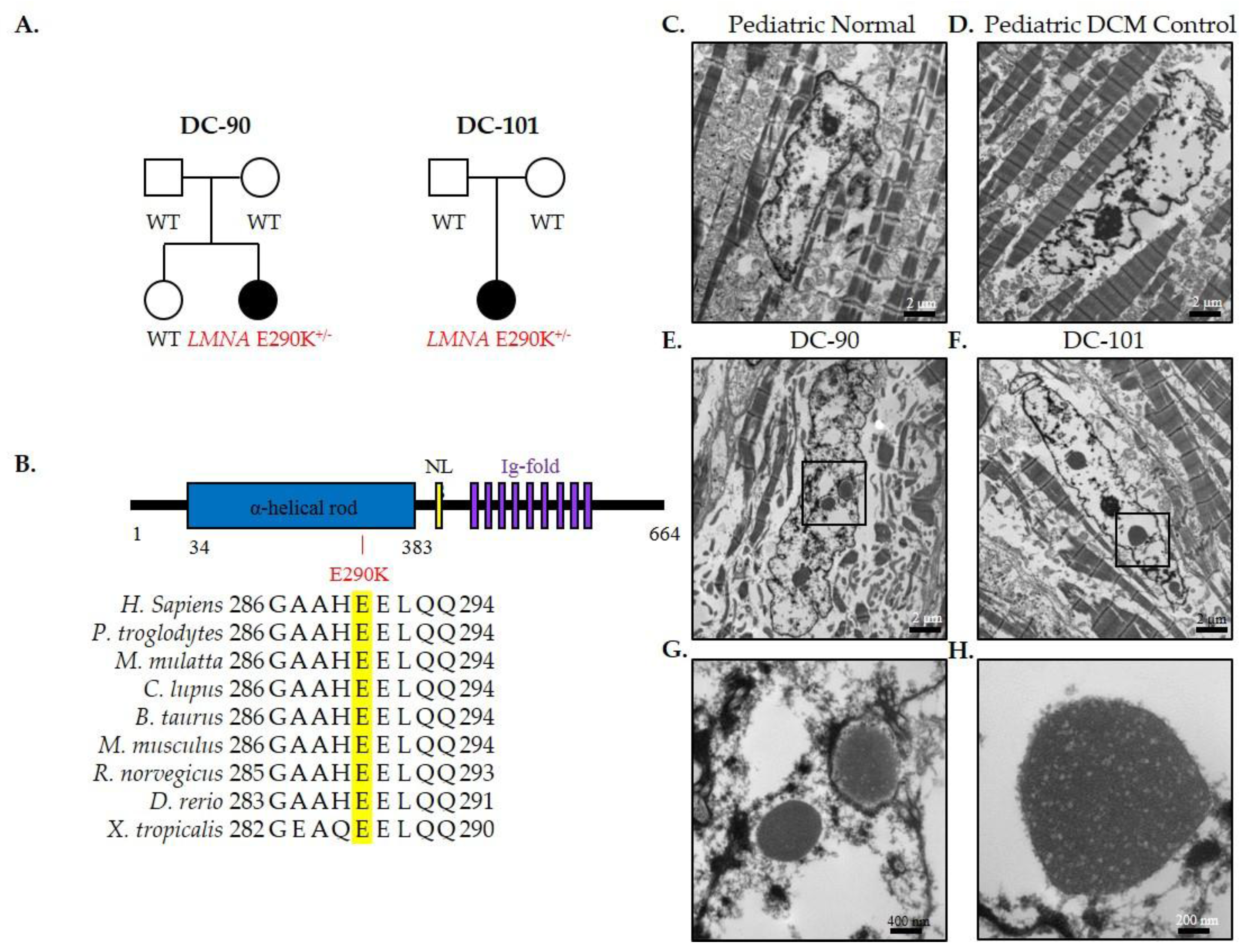
3.2. Recurrent LMNA Mutation Associated with Nuclear Inclusions
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Michels, V.V.; Moll, P.P.; Miller, F.A.; Tajik, A.J.; Chu, J.S.; Driscoll, D.J.; Burnett, J.C.; Rodeheffer, R.J.; Chesebro, J.H.; Tazelaar, H.D. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N. Engl. J. Med. 1992, 326, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Dipchand, A.I.; Kirk, R.; Edwards, L.B.; Kucheryavaya, A.Y.; Benden, C.; Christie, J.D.; Dobbels, F.; Lund, L.H.; Rahmel, A.O.; Yusen, R.D.; et al. International Society for Heart and Lung Transplantation. The registry of the international society for heart and lung transplantation: Sixteenth official pediatric heart transplantation report—2013; focus theme: Age. J. Heart Lung Transplant. 2013, 32, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.H.; Edwards, L.B.; Kucheryavaya, A.Y.; Dipchand, A.I.; Benden, C.; Christie, J.D.; Dobbels, F.; Kirk, R.; Rahmel, A.O.; Yusen, R.D.; et al. The registry of the international society for heart and lung transplantation: Thirtieth official adult heart transplant report—2013; focus theme: Age. J. Heart Lung Transplant. 2013, 32, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Chan, D.P. Dilated cardiomyopathy. In Moss and Adams’ Heart Disease in Infants, Children, and Adolescents: Including the Fetus and Young Adult, 8th ed.; Allen, H.D., Driscoll, D.J., Shaddy, R.E., Feltes, T.F., Eds.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1235–1246. [Google Scholar]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.; Seidman, M.A.; Baxter, S.M.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014, 16, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Long, P.A.; Evans, J.M.; Olson, T.M. Exome sequencing establishes diagnosis of Alström syndrome in an infant presenting with non-syndromic dilated cardiomyopathy. Am. J. Med. Genet. A 2015, 167A, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Long, P.A.; Larsen, B.T.; Evans, J.M.; Olson, T.M. Exome sequencing identifies pathogenic and modifier mutations in a child with sporadic dilated cardiomyopathy. J. Am. Heart Assoc. 2015, 4, e002443. [Google Scholar] [CrossRef] [PubMed]
- Long, P.A.; Zimmermann, M.T.; Kim, M.; Evans, J.M.; Xu, X.; Olson, T.M. De novo RRAGC mutation activates mTORC1 signaling in syndromic fetal dilated cardiomyopathy. Hum. Genet. 2016, 135, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Long, P.A.; Theis, J.L.; Shih, Y.H.; Maleszewski, J.J.; Abell Aleff, P.C.; Evans, J.M.; Xu, X.; Olson, T.M. Recessive TAF1A mutations reveal ribosomopathy in siblings with end-stage pediatric dilated cardiomyopathy. Hum. Mol. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maquire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- 1000 genomes project consortium; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [PubMed]
- Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 23 May 2017).
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. Available online: http://exac.broadinstitute.org (accessed on 23 May 2017). [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Pinto, J.R.; Parks, S.B.; Kushner, J.D.; Li, D.; Ludwigsen, S.; Cowan, J.; Morales, A.; Parvatiyar, M.S.; Potter, J.D. Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.W.; Morimoto, S.; Harada, K.; Du, C.K.; Takahashi-Yanaga, F.; Miwa, Y.; Sasaguri, T.; Ohtsuki, I. Cardiac troponin T mutation R141W found in dilated cardiomyopathy stabilizes the troponin T-tropomyosin interaction and causes a Ca2+ desensitization. J. Mol. Cell. Cardiol. 2003, 35, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.; Marston, S.; Willott, R.; Ashley, C.; Mogensen, J.; McKenna, W.; Robinson, P.; Redwood, C.; Watkins, H. Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J. Biol. Chem. 2005, 280, 28498–28506. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, G.; Gomes, A.V.; Kerrick, W.G.; Potter, J.D. Characterization of troponin T dilated cardiomyopathy mutations in the fetal troponin isoform. J. Biol. Chem. 2005, 280, 17584–17592. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Griffiths, P.J.; Watkins, H.; Redwood, C.S. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 2007, 101, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tikunova, S.B.; Kline, K.P.; Siddiqui, J.K.; Davis, J.P. Disease-related cardiac troponins alter thin filament Ca2+ association and dissociation rates. PLoS ONE 2012, 7, e38259. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Maatz, H.; Jens, M.; Liss, M.; Schafer, S.; Heinig, M.; Kirchner, M.; Adami, E.; Rintisch, C.; Dauksaite, V.; Radke, M.H.; et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Investig. 2014, 124, 3419–3430. [Google Scholar] [CrossRef] [PubMed]
- Bjork, B.C.; Turbe-Doan, A.; Prysak, M.; Herron, B.J.; Beier, D.R. Prdm16 is required for normal palatogenesis in mice. Hum. Mol. Genet. 2010, 19, 774–789. [Google Scholar] [CrossRef] [PubMed]
- Kinameri, E.; Inoue, T.; Aruga, J.; Imayoshi, I.; Kageyama, R.; Shimogori, T.; Moore, A.W. Prdm proto-oncogene transcription factor family expression and interaction with the Notch-Hes pathway in mouse neurogenesis. PLoS ONE 2008, 3, e3859. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, N.; Shimizu, S.; Nagasawa, T.; Tanaka, H.; Taniwaki, M.; Yokota, J.; Morishita, K. A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t(1;3)(p36;q21)-positive leukemia cells. Blood 2000, 96, 3209–3214. [Google Scholar] [PubMed]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scimè, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Arndt, A.K.; Schafer, S.; Drenckhahn, J.D.; Sabeh, M.K.; Plovie, E.R.; Caliebe, A.; Klopocki, E.; Musso, G.; Werdich, A.A.; Kalwa, H.; et al. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am. J. Hum. Genet. 2013, 93, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.Z.; Chaudhary, N.; Blobel, G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 6450–6454. [Google Scholar] [CrossRef] [PubMed]
- McKeon, F.D.; Kirschner, M.W.; Caput, D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 1986, 319, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Capell, B.C.; Collins, F.S. Human laminopathies: Nuclei gone genetically awry. Nat. Rev. Genet. 2006, 7, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.H.; Kennedy, B.K. When lamins go bad: Nuclear structure and disease. Cell 2013, 152, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Kishimoto, N.Y.; Whitby, F.G.; Michels, V.V. Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2001, 33, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Illenberger, S.; Kishimoto, N.Y.; Huttelmaier, S.; Keating, M.T.; Jockusch, B.M. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation 2002, 105, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Beraldi, R.; Li, X.; Martinez Fernandez, A.; Reyes, S.; Secreto, F.; Terzic, A.; Olson, T.M.; Nelson, T.J. Rbm20-deficient cardiogenesis reveals early disruption of RNA processing and sarcomere remodeling establishing a developmental etiology for dilated cardiomyopathy. Hum. Mol. Genet. 2014, 23, 3779–3791. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Gene Symbol | Inheritance | Transcript Variant | Protein Variant | CADD or pLI Score (%ile) | ExAC MAF (Eur) | Gene Ontology |
|---|---|---|---|---|---|---|---|
| DC-82 * | TAF1A | CH | c.251T>C c.1021G>A NM_005681.3 | p.L84S p.G341R | 27.2 (85) 34.0 (99) | 0.007% 0.004% | rRNA transcription |
| DC-86 * | ALMS1 | CH | c.4156dupA c.6436C>T NM_015120.4 | p.T1386fs*15 p.R2146* | pLI = n/a | 0.001% 0.001% | Formation/maintenance of primary cilia |
| DC-87 * | TNNT2 | De novo | c.421C>T NM_001001430.2 | p.R141W | 33.0 (>95) | -- | Sarcomeric force generation |
| DC-90 | LMNA | De novo | c.868G>A NM_170707.3 | p.E290K | 35.0 (>99) | -- | Nuclear lamina |
| DC-92 | RBM20 | Presumed de novo | c.1913C>T NM_001134363.2 | p.P638L | 28.0 (90) | -- | Spliceosome |
| DC-94 * | RRAGC | De novo | c.224C>A NM_022157.3 | p.S75Y | 27.2 (85) | -- | mTORC1 activation |
| DC-95 | TNNT2 | De novo | c.392G>A NM_001001430.2 | p.R131Q | 33.0 (>95) | -- | Sarcomeric force generation |
| DC-97 | PRDM16 | De novo | c.1047dupC NM_022114.3 | p.S350fs*48 | pLI = 1.00 (100) | -- | Transcriptional regulation |
| DC-101 | LMNA | De novo | c.868G>A NM_170707.3 | p.E290K | 35.0 (>99) | -- | Nuclear lamina |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, P.A.; Evans, J.M.; Olson, T.M. Diagnostic Yield of Whole Exome Sequencing in Pediatric Dilated Cardiomyopathy. J. Cardiovasc. Dev. Dis. 2017, 4, 11. https://doi.org/10.3390/jcdd4030011
Long PA, Evans JM, Olson TM. Diagnostic Yield of Whole Exome Sequencing in Pediatric Dilated Cardiomyopathy. Journal of Cardiovascular Development and Disease. 2017; 4(3):11. https://doi.org/10.3390/jcdd4030011
Chicago/Turabian StyleLong, Pamela A., Jared M. Evans, and Timothy M. Olson. 2017. "Diagnostic Yield of Whole Exome Sequencing in Pediatric Dilated Cardiomyopathy" Journal of Cardiovascular Development and Disease 4, no. 3: 11. https://doi.org/10.3390/jcdd4030011
APA StyleLong, P. A., Evans, J. M., & Olson, T. M. (2017). Diagnostic Yield of Whole Exome Sequencing in Pediatric Dilated Cardiomyopathy. Journal of Cardiovascular Development and Disease, 4(3), 11. https://doi.org/10.3390/jcdd4030011