Phosphodiesterases 3 and 4 Differentially Regulate the Funny Current, If, in Mouse Sinoatrial Node Myocytes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Sinoatrial Myocyte Isolation
2.3. Sinoatrial Myocyte Electrophysiology
2.4. Statistical Analysis
3. Results
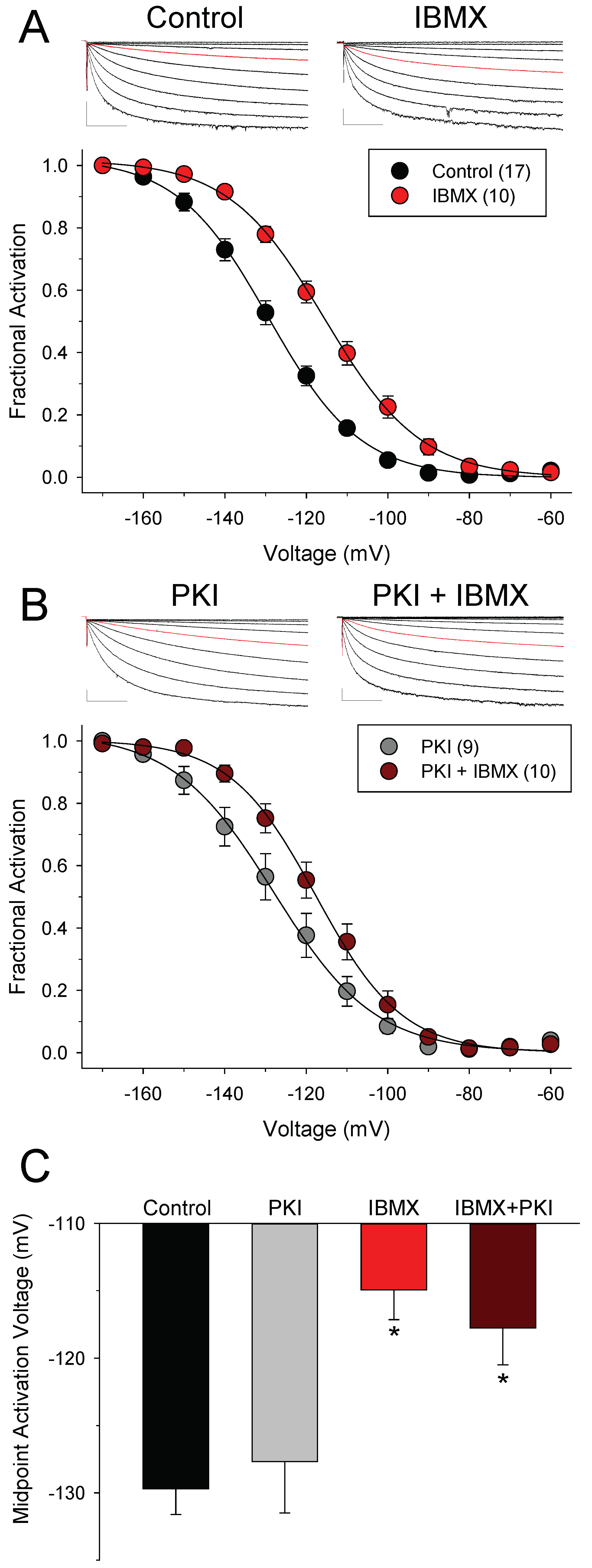
3.1. Phosphodiesterases Restrict Access of cAMP to HCN Channels at Rest
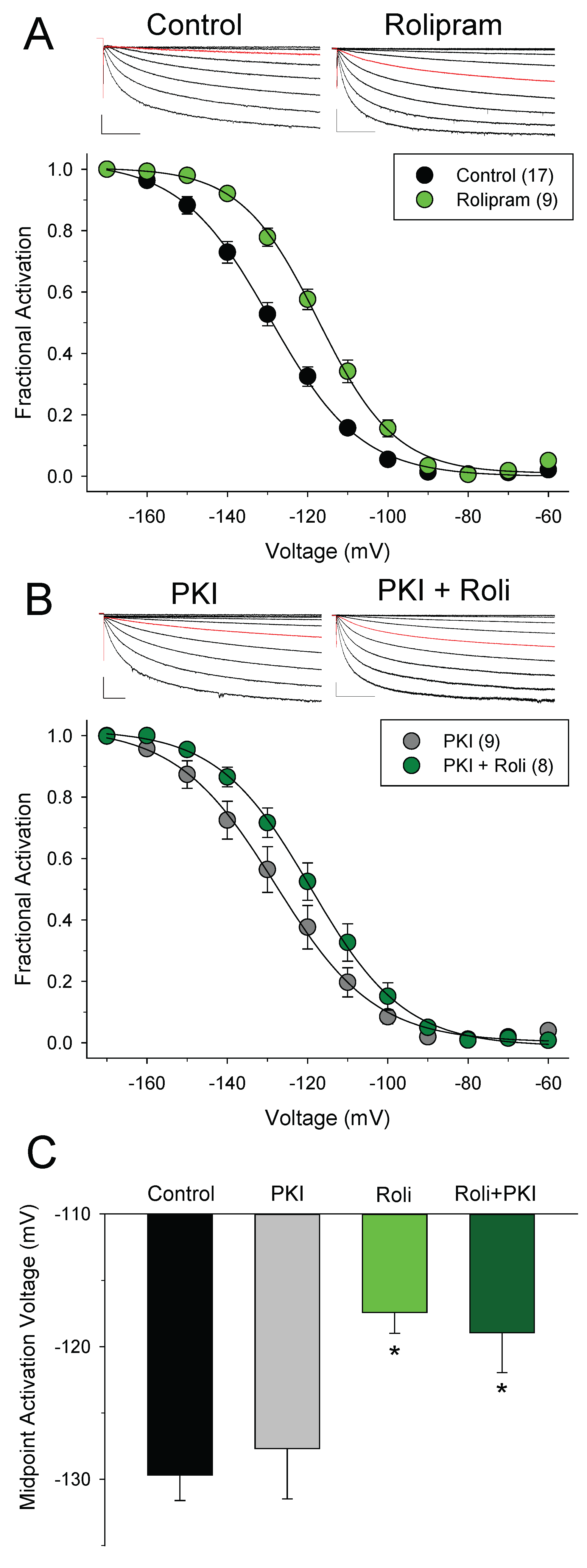
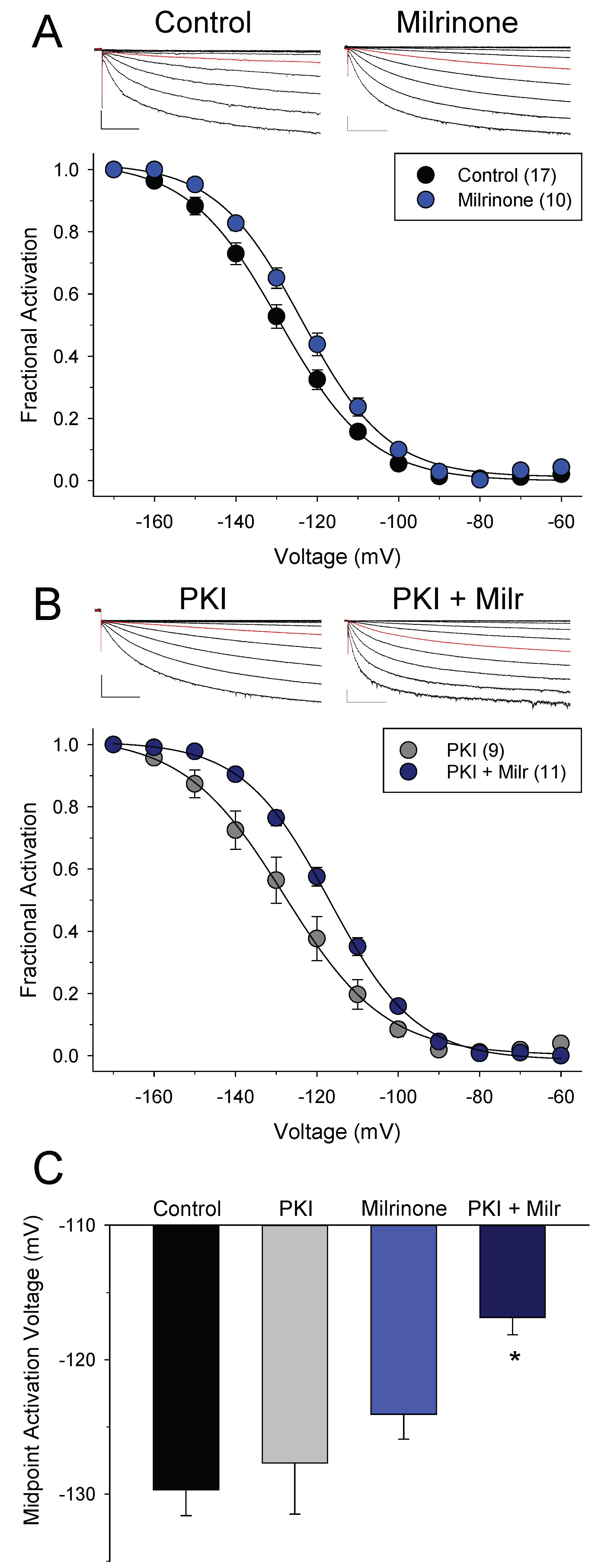
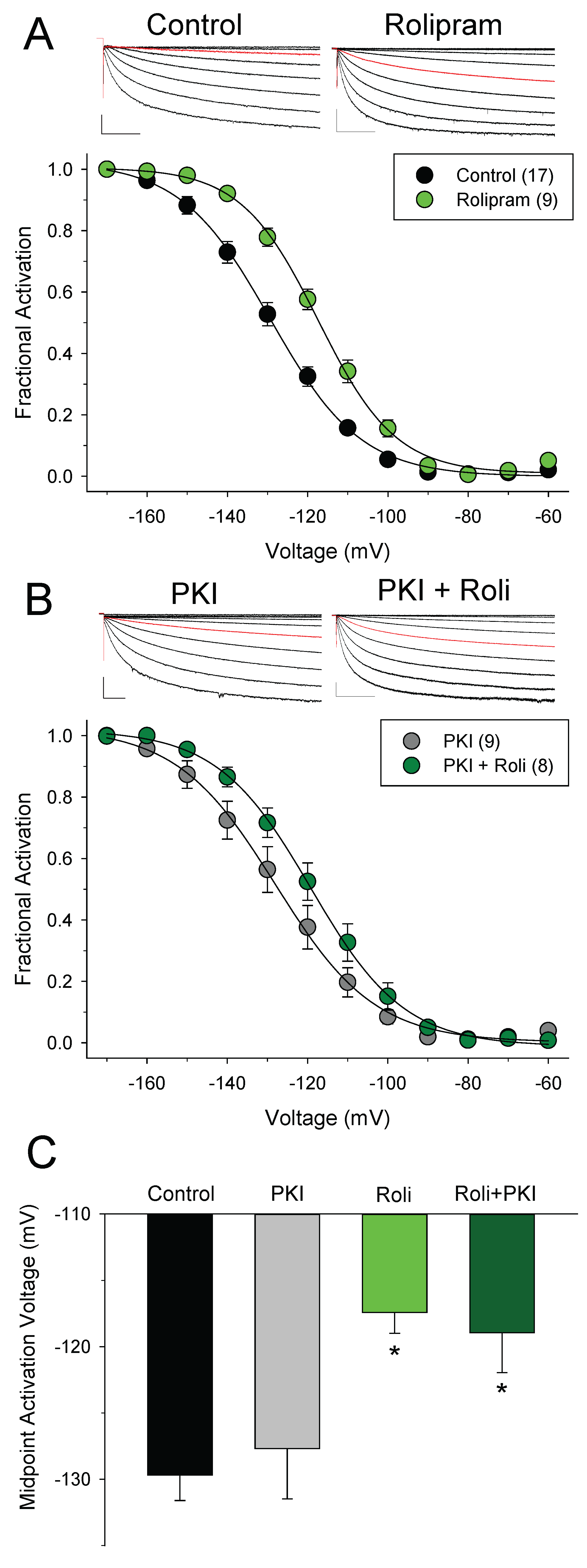
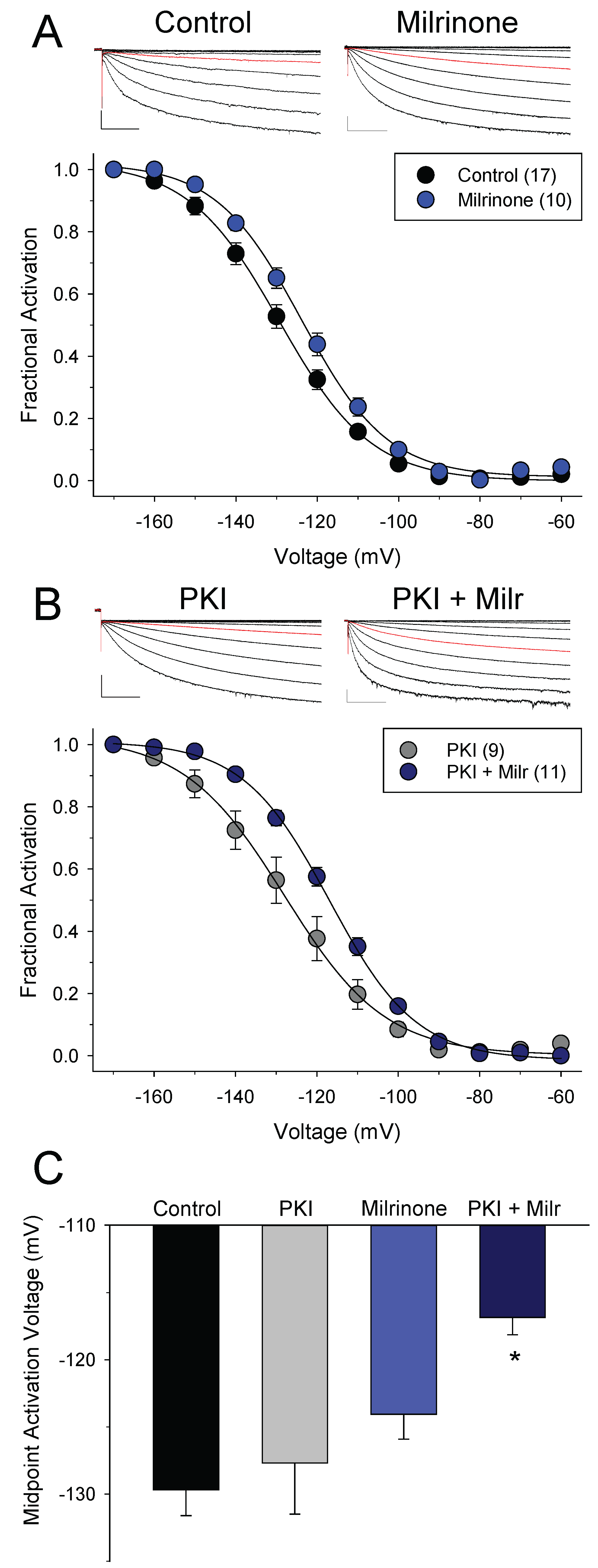
3.2. Different Effects of PDE4 and PDE3 on If under Basal Conditions
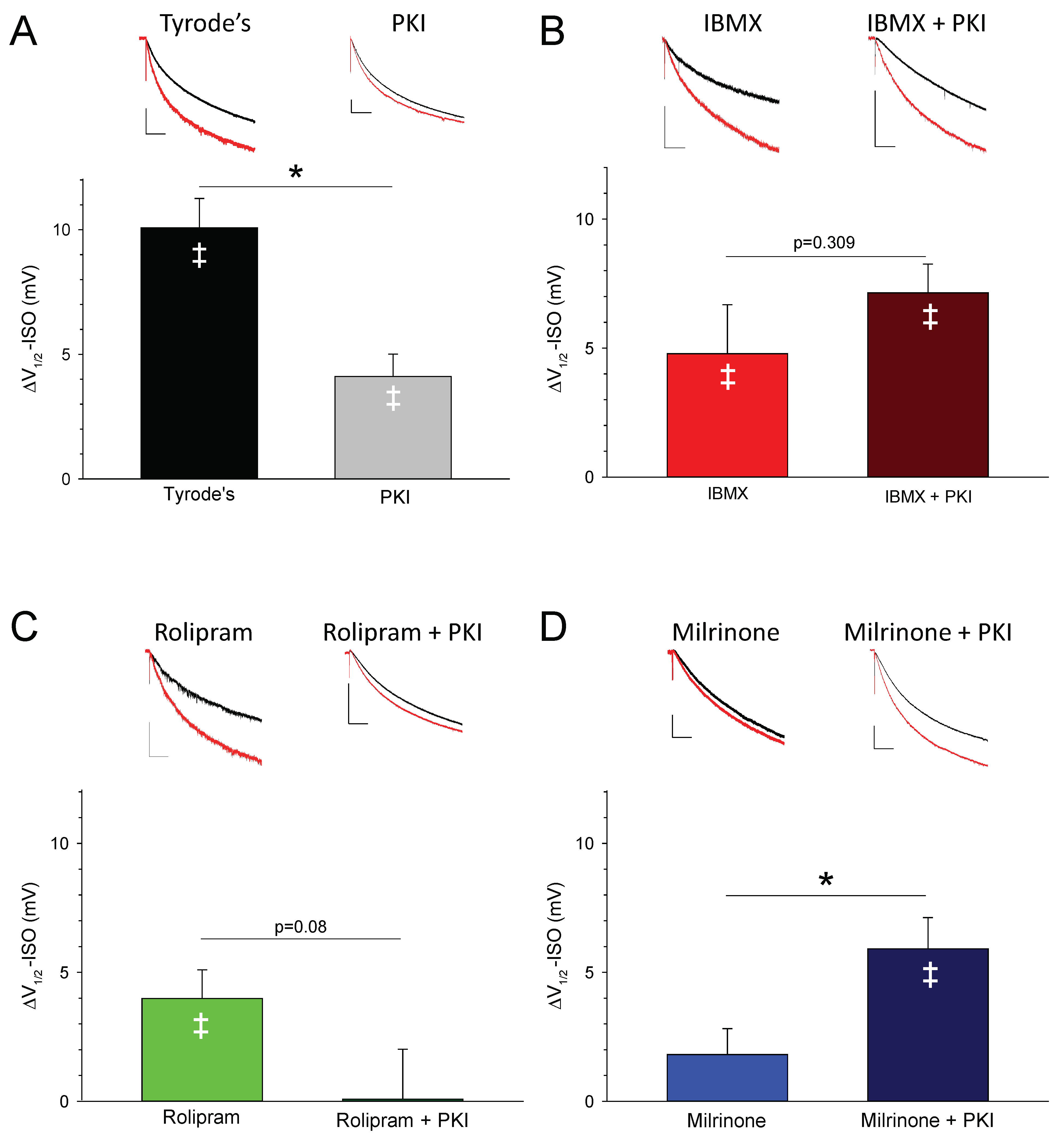
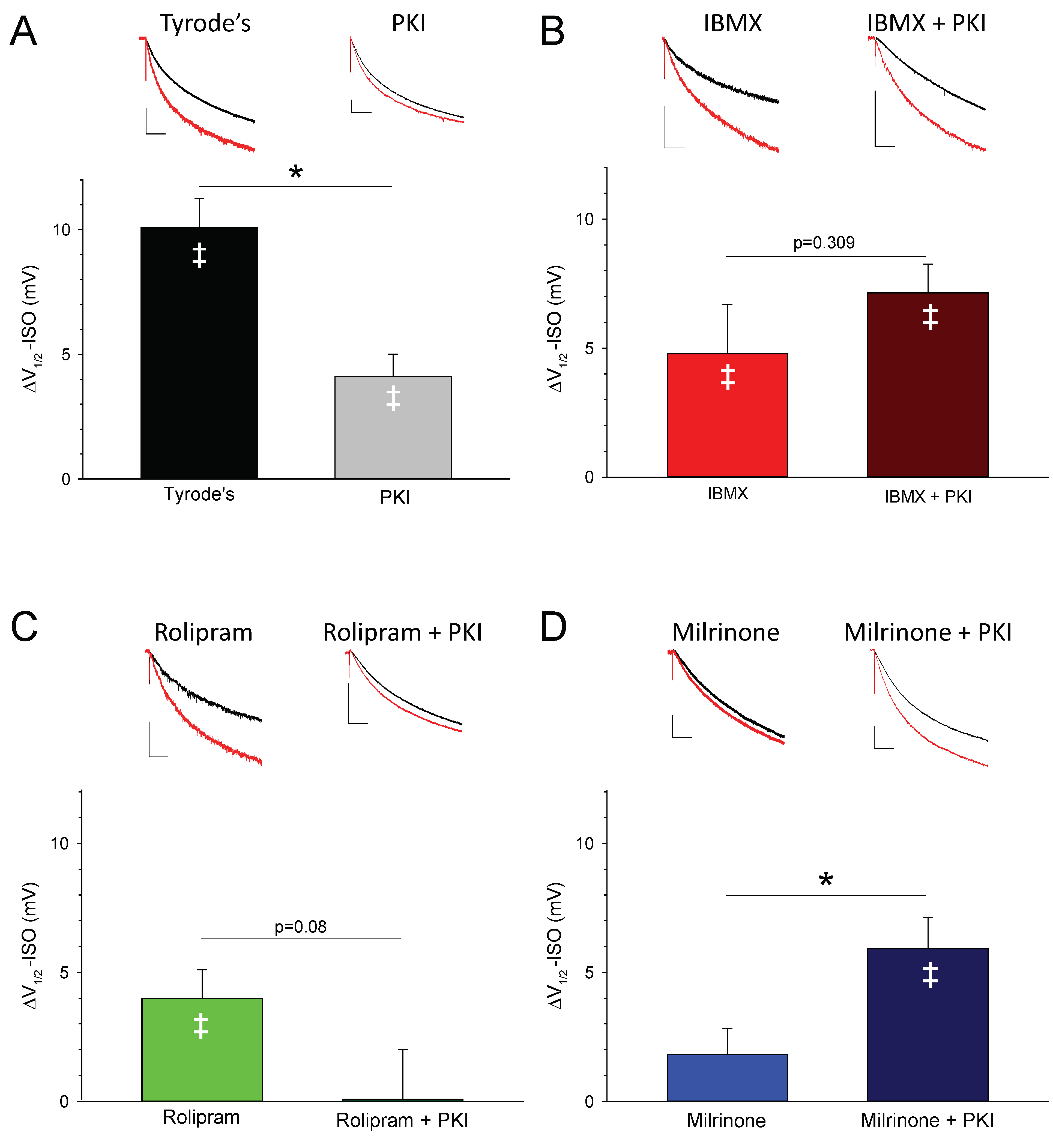
3.3. PDEs Establish the Requirement for PKA Activity in Signaling between βARs and HCN Channels in SAMs
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AP | action potential |
| βAR | adrenergic receptor |
| cAMP | 3’,5’-cyclic adenosine monophosphate |
| HCN channel | hyperpolarization-activated, cyclic nucleotide-sensitive ion channel |
| IBMX | 3-isobutyl-1-methylxanthine |
| If | funny current |
| ISO | isoproterenol |
| milr | milrinone |
| PDE | phosphodiesterase |
| PKA | cAMP-dependent protein kinase |
| PKI | PKA inhibitory peptide |
| roli | rolipram |
| SAM | sinoatrial node myocyte |
| V1/2 | midpoint activation voltage |
References
- Mangoni, M.; Nargeot, J. Genesis and regulation of the heart automaticity. Physiol. Rev. 2008, 88, 919–982. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; DiFrancesco, D. What keeps us ticking: A funny current, a calcium clock, or both? J. Mol. Cell. Cardiol. 2009, 47, 157–170. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D. The role of the funny current in pacemaker activity. Circ. Res. 2010, 106, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Maltsev, V.A.; Vinogradova, T.M. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res 2010, 106, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, T.; Lyashkov, A.; Zhu, W.; Ruknudin, A.; Sirenko, S.; Yang, D.; Deo, S.; Barlow, M.; Johnson, S.; Caffrey, J.; et al. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ. Res. 2006, 98, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dobrzynski, H.; Yanni, J.; Boyett, M.R.; Lei, M. Organisation of the mouse sinoatrial node: Structure and expression of HCN channels. Cardiovasc. Res. 2007, 73, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Marionneau, C.; Couette, B.; Liu, J.; Li, H.; Mangoni, M.E.; Nargeot, J.; Lei, M.; Escande, D.; Demolombe, S. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J. Physiol. 2005, 562, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Moosmang, S.; Stieber, J.; Zong, X.; Biel, M.; Hofmann, F.; Ludwig, A. Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur. J. Biochem. 2001, 268, 1646–1652. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Wymore, R.; Yu, H.; Wu, J.; Wymore, R.T.; Pan, Z.; Robinson, R.B.; Dixon, J.E.; McKinnon, D.; Cohen, I.S. Distribution and Prevalence of Hyperpolarization-Activated Cation Channel (HCN) mRNA Expression in Cardiac Tissues. Circ. Res. 1999, 85, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.E.; Nargeot, J. Properties of the hyperpolarization-activated current (I(f)) in isolated mouse sino-atrial cells. Cardiovasc. Res. 2001, 52, 51–64. [Google Scholar] [CrossRef]
- Liao, Z.; Lockhead, D.; Larson, E.; Proenza, C. Phosphorylation and modulation of hyperpolarization-activated HCN4 channels by protein kinase A in the mouse sinoatrial node. J. Gen. Physiol. 2010, 136, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Wilders, R. Pacemaker activity of the human sinoatrial node: An update on the effects of mutations in HCN4 on the hyperpolarization-activated current. Int. J. Mol. Sci. 2015, 16, 3071–3094. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, M.; Bucchi, A.; Milanesi, R.; Paina, M.; Barbuti, A.; Gnecchi-Ruscone, T.; Bianco, E.; Vitali-Serdoz, L.; Cappato, R.; DiFrancesco, D. A gain-of-function mutation in the cardiac pacemaker HCN4 channel increasing cAMP sensitivity is associated with familial Inappropriate Sinus Tachycardia. Eur. Heart J. 2017, 38, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, S.; Hofmann, F.; Stieber, J.; Ludwig, A. HCN channels in the heart: Lessons from mouse mutants. Br. J. Pharmacol. 2012, 166, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Borer, J.S. Drug insight: If inhibitors as specific heart-rate-reducing agents. Nat. Clin. Pract. Cardiovasc. Med. 2004, 1, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Roubille, F.; Tardif, J.-C. New Therapeutic Targets in Cardiology Heart Failure and Arrhythmia: HCN Channels. Circulation 2013, 127, 1986–1996. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D.; Tortora, P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 1991, 351, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Zagotta, W.N.; Olivier, N.B.; Black, K.D.; Young, E.C.; Olson, R.; Gouaux, E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature 2003, 425, 200–205. [Google Scholar] [CrossRef] [PubMed]
- St. Clair, J.R.; Liao, Z.; Larson, E.D.; Proenza, C. PKA-independent activation of I(f) by cAMP in mouse sinoatrial myocytes. Channels 2013, 7, 318–321. [Google Scholar]
- Conti, M.; Mika, D.; Richter, W. Cyclic AMP compartments and signaling specificity: Role of cyclic nucleotide phosphodiesterases. J. Gen. Physiol. 2014, 143, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.L.; Langeberg, L.; Scott, J.D. Compartmentation of Cyclic Nucleotide Signaling in the Heart the Role of A-Kinase Anchoring Proteins. Circ. Res. 2006, 98, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Karpen, J.W. Perspectives on: Cyclic nucleotide microdomains and signaling specificity. J. Gen. Physiol. 2014, 143, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.K.; Nikolaev, V.O. Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol. 2013, 207, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.; Lomas, O.; Jalink, K.; Ford, K.L.; Vaughan-Jones, R.D.; Lefkimmiatis, K.; Swietach, P. Intracellular tortuosity underlies slow cAMP diffusion in adult ventricular myocytes. Cardiovasc. Res. 2016, 110, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Warrier, S.; Ramamurthy, G.; Eckert, R.L.; Nikolaev, V.O.; Lohse, M.J.; Harvey, R.D. cAMP microdomains and L-type Ca2+ channel regulation in guinea-pig ventricular myocytes. J. Physiol. 2007, 580, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y. Compartmentalization of beta-adrenergic signals in cardiomyocytes. Circ. Res. 2011, 109, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M. cAMP signal transduction in the heart: Understanding spatial control for the development of novel therapeutic strategies. Br. J. Pharmacol. 2009, 158, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Jurevicius, J.; Fischmeister, R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc. Natl. Acad. Sci. USA 1996, 93, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F.; Brunton, L.L. Compartmentation of G Protein-Coupled Signaling Pathways in Cardiac Myocytes. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 751–773. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Jurevicius, J.; Skeberdis, V.A.; Fischmeister, R. Role of cyclic nucleotide phosphodiesterase isoforms in cAMP compartmentation following beta2-adrenergic stimulation of ICa,L in frog ventricular myocytes. J. Physiol. 2003, 551, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; McSorley, T.; Evellin, S.; Sood, A.; Lissandron, V.; Terrin, A.; Huston, E.; Hannawacker, A.; Lohse, M.J.; Pozzan, T.; et al. Fluorescence Resonance Energy Transfer-Based Analysis of cAMP Dynamics in Live Neonatal Rat Cardiac Myocytes Reveals Distinct Functions of Compartmentalized Phosphodiesterases. Circ. Res. 2004, 95, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Hua, R.; Adamczyk, A.; Robbins, C.; Ray, G.; Rose, R. Distinct patterns of constitutive phosphodiesterase activity in mouse sinoatrial node and atrial myocardium. PLoS ONE 2012, 7, e47652. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Tovar, A.; Kaumann, A.J. Phosphodiesterase-4 blunts inotropism and arrhythmias but not sinoatrial tachycardia of (−)-adrenaline mediated through mouse cardiac β1-adrenoceptors. Br. J. Pharmacol. 2008, 153, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Shakur, Y.; Fong, M.; Hensley, J.; Cone, J.; Movsesian, M.; Kambayashi, J.; Yoshitake, M.; Liu, Y. Comparison of the effects of cilostazol and milrinone on cAMP-PDE activity, intracellular cAMP and calcium in the heart. Cardiovasc. Drugs Ther. Spons. Int. Soc. Cardiovasc. Pharmacother. 2002, 16, 417–427. [Google Scholar] [CrossRef]
- Zaccolo, M.; Movsesian, M. cAMP and cGMP signaling cross-talk: Role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res. 2007, 100, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; St. Clair, J.R.; Larson, E.D.; Proenza, C. Myristoylated peptides potentiate the funny current (I(f)) in sinoatrial myocytes. Channels 2011, 5, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Groenke, S.; Larson, E.D.; Alber, S.; Zhang, R.; Lamp, S.T.; Ren, X.; Nakano, H.; Jordan, M.C.; Karagueuzian, H.S.; Roos, K.P.; et al. Complete atrial-specific knockout of sodium-calcium exchange eliminates sinoatrial node pacemaker activity. PLoS ONE 2013, 8, e81633. [Google Scholar] [CrossRef] [PubMed]
- St. Clair, J.R.; Sharpe, E.J.; Proenza, C. Culture and adenoviral infection of sinoatrial node myocytes from adult mice. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H490–H498. [Google Scholar] [CrossRef] [PubMed]
- Larson, E.D.; St. Clair, J.R.; Sumner, W.A.; Bannister, R.A.; Proenza, C. Depressed pacemaker activity of sinoatrial node myocytes contributes to the age-dependent decline in maximum heart rate. Proc. Natl. Acad. Sci. USA 2013, 110, 18011–18016. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, E.J.; St. Clair, J.R.; Proenza, C. Methods for the Isolation, Culture, and Functional Characterization of Sinoatrial Node Myocytes from Adult Mice. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, E.J.; Larson, E.D.; Proenza, C. Cyclic AMP reverses the effects of aging on pacemaker activity and If in sinoatrial node myocytes. J. Gen. Physiol. 2017, 149, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.H. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J. Neurosci. Methods 1994, 51, 107–116. [Google Scholar] [CrossRef]
- Kaumann, A.J.; Galindo-Tovar, A.; Escudero, E.; Vargas, M.L. Phosphodiesterases do not limit beta1-adrenoceptor-mediated sinoatrial tachycardia: Evidence with PDE3 and PDE4 in rabbits and PDE1–5 in rats. Naunyn. Schmiedebergs Arch. Pharmacol. 2009, 380, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, T.; Sirenko, S.; Lyashkov, A.; Younes, A.; Li, Y.; Zhu, W.; Yang, D.; Ruknudin, A.; Spurgeon, H.; Lakatta, E. Constitutive phosphodiesterase activity restricts spontaneous beating rate of cardiac pacemaker cells by suppressing local Ca2+ releases. Circ. Res. 2008, 102, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Kerfant, B.-G.; Zhao, D.; Lorenzen-Schmidt, I.; Wilson, L.S.; Cai, S.; Chen, S.R.W.; Maurice, D.H.; Backx, P.H. PI3Kγ Is Required for PDE4, not PDE3, Activity in Subcellular Microdomains Containing the Sarcoplasmic Reticular Calcium ATPase in Cardiomyocytes. Circ. Res. 2007, 101, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Mattick, P.; Parrington, J.; Odia, E.; Simpson, A.; Collins, T.; Terrar, D. Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the I(f) pacemaker current. J. Physiol. 2007, 582, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Brahmajothi, M.V.; Campbell, D.L. Heterogeneous expression of NO-activated soluble guanylyl cyclase in mammalian heart: Implications for NO- and redox-mediated indirect versus direct regulation of cardiac ion channel function. Channels 2007, 1, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Abramochkin, D.V.; Konovalova, O.P.; Kamkin, A.; Sitdikova, G.F. Carbon monoxide modulates electrical activity of murine myocardium via cGMP-dependent mechanisms. J. Physiol. Biochem. 2015, 71, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Stangherlin, A.; Gesellchen, F.; Zoccarato, A.; Terrin, A.; Fields, L.A.; Berrera, M.; Surdo, N.C.; Craig, M.A.; Smith, G.; Hamilton, G.; et al. cGMP Signals Modulate cAMP Levels in a Compartment-Specific Manner to Regulate Catecholamine-Dependent Signaling in Cardiac Myocytes. Circ. Res. 2011, 108, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Tovar, A.; Vargas, M.L.; Kaumann, A.J. Inhibitors of phosphodiesterases PDE2, PDE3, and PDE4 do not increase the sinoatrial tachycardia of noradrenaline and prostaglandin PGE1 in mice. Naunyn. Schmiedebergs Arch. Pharmacol. 2016, 389, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Kaumann, A.J. Phosphodiesterases reduce spontaneous sinoatrial beating but not the “fight or flight” tachycardia elicited by agonists through Gs-protein-coupled receptors. Trends Pharmacol. Sci. 2011, 32, 377–383. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | V1/2 before ISO (mV) | V1/2 after ISO (mV) | ΔV1/2-ISO (mV) | n | p Value Control vs. ISO (Paired t-Test) |
|---|---|---|---|---|---|
| CONTROL | −129.7 ± 1.9 | −119.6 ± 1.5 | 10.1 ± 1.2 | 17 | 0.000000237 |
| +PKI | −127.7 ± 3.8 | −123.6 ± 3.5 | 4.1 ± 0.7 | 9 | 0.00188 |
| IBMX | −114.9 ± 2.2 | −110.1 ± 2.8 | 4.8 ± 1.9 | 10 | 0.0328 |
| +PKI | −117.7 ± 2.7 | −110.6 ± 2.7 | 7.1 ± 1.1 | 10 | 0.000128 |
| ROLIPRAM | −117.4 ± 1.6 | −113.4 ± 2.2 | 4.0 ± 1.1 | 9 | 0.00712 |
| +PKI | −118.9 ± 3.0 | −118.8 ± 2.8 | 0.1 ± 1.9 | 7 | 0.967 |
| MILRINONE (10 μM) | −124.0 ± 4.3 | −121.1 ± 4.7 | 2.9 ± 1.7 | 8 | 0.123 |
| +PKI | −119.3 ± 4.7 | −112.5 ± 4.8 | 6.8 ± 2.4 | 10 | 0.0206 |
| MILRINONE (50 μM) | −124.1 ± 1.9 | −122.2 ± 2.0 | 1.8 ± 1.0 | 10 | 0.104 |
| +PKI | −116.8 ± 1.3 | −110.9 ± 1.8 | 5.9 ± 1.2 | 11 | 0.000648 |
| MDL-12,330A | −131.1 ± 1.9 | −132.0 ± 2.2 | −0.9 ± 1.1 | 7 | 0.469 |
| 1 mM cAMP | −112.0 ± 1.6 | - | - | 10 | - |
| 3 mM cAMP | −114.1 ± 1.9 | - | - | 7 | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
St. Clair, J.R.; Larson, E.D.; Sharpe, E.J.; Liao, Z.; Proenza, C. Phosphodiesterases 3 and 4 Differentially Regulate the Funny Current, If, in Mouse Sinoatrial Node Myocytes. J. Cardiovasc. Dev. Dis. 2017, 4, 10. https://doi.org/10.3390/jcdd4030010
St. Clair JR, Larson ED, Sharpe EJ, Liao Z, Proenza C. Phosphodiesterases 3 and 4 Differentially Regulate the Funny Current, If, in Mouse Sinoatrial Node Myocytes. Journal of Cardiovascular Development and Disease. 2017; 4(3):10. https://doi.org/10.3390/jcdd4030010
Chicago/Turabian StyleSt. Clair, Joshua R., Eric D. Larson, Emily J. Sharpe, Zhandi Liao, and Catherine Proenza. 2017. "Phosphodiesterases 3 and 4 Differentially Regulate the Funny Current, If, in Mouse Sinoatrial Node Myocytes" Journal of Cardiovascular Development and Disease 4, no. 3: 10. https://doi.org/10.3390/jcdd4030010
APA StyleSt. Clair, J. R., Larson, E. D., Sharpe, E. J., Liao, Z., & Proenza, C. (2017). Phosphodiesterases 3 and 4 Differentially Regulate the Funny Current, If, in Mouse Sinoatrial Node Myocytes. Journal of Cardiovascular Development and Disease, 4(3), 10. https://doi.org/10.3390/jcdd4030010